Productive Dengue Virus Infection of Human Endothelial Cells Is Directed by Heparan Sulfate-Containing Proteoglycan Receptors (original) (raw)
Abstract
Dengue virus causes leakage of the vascular endothelium, resulting in dengue hemorrhagic fever and dengue shock syndrome. The endothelial cell lining of the vasculature regulates capillary permeability and is altered by immune and chemokine responses which affect fluid barrier functions of the endothelium. Our findings indicate that human endothelial cells are highly susceptible to infection by dengue virus (type 4). We found that dengue virus productively infects ∼80% of primary human endothelial cells, resulting in the rapid release of ∼105 virions 1 day postinfection. Analysis of potential inhibitors of dengue virus entry demonstrated that antibodies and ligands to integrins and cellular receptors were unable to inhibit dengue virus infection of endothelial cells. In contrast, pretreating cells with heparin or heparan sulfate resulted in a 60 to 80% reduction in dengue virus-infected cells, and pretreatment of endothelial cells with heparinase III or protease reduced dengue infectivity by >80%. Dengue virus bound specifically to resin immobilized heparin, and binding was competitively inhibited by excess heparin but not other ligands. Collectively, these findings suggest that dengue virus specifically attaches to heparan sulfate-containing proteoglycan receptors on endothelial cells. Following attachment to human endothelial cell receptors, dengue virus causes a highly productive infection that has the potential to increase viral dissemination and viremia. This provides the potential for dengue virus-infected endothelial cells to directly alter barrier functions of the endothelium, contribute to enhancement of immune cell activation, and serve as potential targets of immune responses which play a central role in dengue pathogenesis.
INTRODUCTION
Dengue viruses (DV) are members of the flavivirus family that are primarily transmitted to humans by the Aedes aegypti mosquito. An estimated 50 million people contract dengue virus annually, and approximately 500,000 to 1,000,000 infections result in dengue hemorrhagic fever (DHF) or dengue shock syndrome (DSS), with 5 to 30% mortality rates (31–33). There are four dengue virus serotypes, and infection by one serotype predisposes individuals to more severe disease following a subsequent infection by a different dengue serotype. While the mechanisms of dengue virus pathogenesis are still being resolved, preexisting nonneutralizing antibodies to dengue virus proteins enhance infection of immune cells, increase the potential for DHF and DSS following dengue virus infection, and contribute to immune-mediated pathogenesis (1, 19, 34, 38, 61, 67, 74).
The endothelium is the primary fluid barrier of the vasculature, and dengue virus-induced responses resulting in edema or hemorrhagic disease ultimately cause changes in endothelial cell permeability. Dengue viruses infect a number of cell types, including peripheral leukocytes, dendritic cells, liver cells, and endothelial cells, in patients, murine models, and in vitro (6–8, 13, 14, 20, 39, 62, 71, 74). Patient blood samples have permitted the analysis of immune cells, released factors, and antibodies during dengue virus infections (9, 17, 48, 50, 59, 60, 67, 69); however, the role of dengue virus infection of endothelial cells is difficult to study in patients. Very little is known about the role of dengue virus-infected endothelial cells in disease or the kinetics, timing, and replication of dengue viruses within patient endothelial cells. And yet, changes in the vascular endothelium are central to understanding dengue virus-induced capillary permeability.
Endothelial cells respond to and elicit a myriad of cellular, platelet-associated, and secreted factors that affect vascular permeability (10, 22, 52, 56, 57, 63, 73, 82, 83). Autopsy samples suggest that a fraction of endothelial cells are infected (6, 39); however, it remains unknown whether dengue virus-infected endothelial cells play a more prominent role at earlier times postinfection. Although the role of dengue virus infection of endothelial cells in pathogenesis remains obscure, the presence of dengue virus-infected endothelial cells in patients rationalizes their likely role in DSS and DHF via several potential mechanisms (9, 47, 50, 61, 62). In fact, dengue virus infection of the endothelium has the potential to directly alter endothelial cell barrier functions, permit immune cell targeting of dengue virus antigens expressed by endothelial cells, elicit immune cell-enhancing chemokine and cytokine responses, and contribute to the production and spread of infectious virus (5, 42).
Dengue virus reportedly attaches to a variety of receptors on immune, dendritic, and liver cells; however, consensus dengue virus receptors have not been defined. The dengue virus envelope protein reportedly binds to Fc receptors, DC-SIGN, ICAM3, CD14, HSP70/90, GRP78, laminin receptor, and the mannose receptor (13, 16, 40, 54, 55, 58, 72). In addition, heparan sulfate proteoglycans (HSPGs) are also implicated as dengue virus attachment targets of Vero E6 and liver cell lines (15, 29, 36, 45). In contrast, dengue virus receptors on primary endothelial cells have not been revealed, and primary human endothelial cells contain a unique set of cell surface receptors that are lost upon continuous passage. One report, using a continuous ECV304 cell line, suggested that dengue virus interacted with 3 undefined cellular proteins (78). However, these interactions have not been studied further or analyzed in primary human endothelial cells (2, 12, 41, 44).
The means by which dengue virus attaches to and enters endothelial cells, as well as the effects of dengue virus on endothelial cell responses and functions, remain to be defined and are central to understanding the role of the endothelium in dengue virus pathogenesis. Here, we analyzed dengue virus type 4 (DV4) infection and entry of primary human endothelial cells. We found that ∼80% of endothelial cells contained dengue virus antigen 24 h after infection and that infection was rapidly productive, with ∼105 focus-forming units (FFU)/ml of dengue virus released into the medium 1 day postinfection. While antibodies to a library of cell surface receptors failed to block dengue virus infection, we found that heparin, heparan sulfate, heparinase, and protease blocked dengue virus infection of primary human endothelial cells. In addition, dengue virus bound specifically to immobilized heparin and binding was competitively inhibited by heparin but not other ligands. These findings indicate that dengue virus efficiently and productively infects human endothelial cells via interactions with heparan sulfate-containing cell surface receptors. This suggests that dengue virus-infected endothelial cells likely contribute to viremia and viral dissemination during dengue virus infections and provide a potential target for inhibiting infection of the endothelium and reducing dengue virus pathogenesis.
MATERIALS AND METHODS
Cells and virus.
C6/36 cells (Aedes albopictus) were maintained in M199 medium (5% fetal bovine serum [FBS]) at 32°C. Vero E6 cells were grown in DMEM (Dulbecco's modified Eagle's medium) with 10% FBS at 37°C. Human umbilical vein endothelial cells (HUVECs; passages 3 to 8) were purchased from Cambrex and grown in supplemented EBM2 medium (Cambrex) at 37°C in the presence of gentamicin (50 μg/ml), amphotericin B (50 μg/ml), and 10% FBS (Sigma). Dengue virus type 4 (DV4) was provided by C.-J. Lai (NIH, NIAID, LID) and propagated in C6/36 cells for 5 days in M199 medium (5% FBS) at 32°C. Viral titers were determined by infecting C6/36 cells with serial dilutions of DV4-containing medium and quantitating the number of DV4-infected cell foci (FFU) 18 to 24 h postinfection. Infected cells were detected using DV4 hyperimmune mouse ascites fluid (HMAF) at a dilution of 1:4,000 and immunoperoxidase staining as described below.
Dengue virus infection of human endothelial cells.
DV4 was diluted in DMEM and adsorbed for 1.5 h to ∼70% confluent primary human endothelial cell monolayers. Following adsorption, cell monolayers were washed three times with DMEM to remove unbound virus, and the medium replaced with supplemented EBM2 (10% FBS). Vero E6 cells were similarly infected and, after washing, incubated in supplemented DMEM (10% FBS). All cells were incubated at 37°C and 5% CO2 for the times indicated.
Detection of dengue virus antigen in infected endothelial cells.
Methods for the detection of viral antigen in infected cells by immunoperoxidase staining have been described previously (26, 28, 68). Briefly, cells were fixed with 100% methanol (20 min, −20°C) at times indicated, washed with PBS, and blocked in PBS–1% bovine serum albumin (BSA). Cells were incubated for 1 h with anti-DV4 HMAF polyclonal sera (1:4,000 in PBS–1% BSA) to detect dengue virus antigen. Subsequently, cells were washed and incubated with an anti-mouse horseradish peroxidase-labeled antibody (Amersham, 1:4,000 in PBS–1% BSA) for 1 h. After 3 washes, cells were incubated with 3-amino-9-ethylcarbazole (AEC, 0.026%) in 0.1 M sodium acetate (pH 5.2) and 0.03% H2O2 (26, 28, 68). AEC-stained, dengue virus antigen-containing cells were quantitated visually on an Olympus IX51 microscope.
Infectious focus assay for determining the titer of dengue virus.
For determination of DV4 titers, cellular supernatants containing infectious DV4 were serially diluted in M199 and adsorbed on C6/36 monolayers in duplicate for 1 h. Unbound virus was removed by washing three times with medium. Supplemented M199 (5% FBS) medium was added, and 20 h later, the C6/36 cells were fixed with methanol (20 min, −20°C). Infected cells were detected by immunoperoxidase staining with 1:4,000 anti-DV4 HMAF as above. The number of dengue virus-infected cells was determined by quantitating AEC-stained cells and reported as FFUs.
Ligands, peptides, and antibodies.
Heparin, sialic acid, glycophorin A, heparan sulfate, chondroitin sulfate A, dermatan sulfate, laminin, BSA, and collagen were purchased from Sigma. Vitronectin and fibrinogen were purchased from Chemicon. Antibodies to vascular endothelial cadherin (VE-cadherin), platelet and endothelial cell adhesion molecule (PECAM), intercellular adhesion molecule-1 (ICAM), green fluorescent protein (GFP), E-selectin, Syndecan-1, vascular endothelial cell growth factor receptor 2 (VEGFR2), platelet-activating factor receptor (PAFR), platelet-derived growth factor receptor (PDGFR), Tie-2, Edg-1, and vascular cell adhesion molecule (VCAM) were purchased from Santa Cruz Biotechnologies. C-X-C chemokine receptor type 3 (CXCR3) antibody was purchased from R&D Systems. Integrin antibodies to αVβ3 and α5β1 were purchased from Chemicon. Rabbit polyclonal antibodies to DV4 NS1 and capsid peptides were from Pacific Immunology. Monoclonal β-actin antibody was purchased from Sigma. Goat anti-mouse immunoglobulin G (H+L) is from Kirkegaard & Perry Laboratories, Inc. DV4-specific hyperimmune mouse ascites fluid (α-DV4 HMAF) was obtained from C.-J. Lai at NIH.
Ligand and antibody inhibition of dengue virus infection.
Potentially competitive ligands and compounds were diluted in DMEM (0.1 to 100 μg/ml) and incubated with DV4 (150 FFU) for 1 h at 25°C. DV4 and competitors were subsequently applied to endothelial cells grown in supplemented EBM2 on 96-well plates for 1 h at 25°C. After adsorption, cell monolayers were washed three times with PBS or DMEM and incubated at 37°C in supplemented EBM2 for 24 h prior to methanol fixation (20 min, −20°C) and immunoperoxidase staining of infected cells (26, 28, 68). For antibody inhibition assays, endothelial cell monolayers were pretreated with antibodies (0.5 or 5 μg/ml) diluted in DMEM for 1 h at 4°C. Primary antibodies were removed and replaced with anti-mouse, anti-rabbit, or anti-goat sera at 1:2,000 for 1 h at 4°C. Subsequently, 100 FFU of dengue virus was adsorbed to endothelial cell monolayers for 1 h at 4°C. Control cells were treated with anti-GFP antibody and secondary antibody as described above. Endothelial cells were washed, incubated at 37°C (5% CO2) for 24 h in supplemented EBM2, and immunoperoxidase stained as described above to detect dengue virus antigen (25, 28, 68). The number of dengue virus antigen-positive cells was quantitated from duplicate samples and compared to the number in control cells, set as 100% infected cells, that were pretreated with identical amounts of BSA (ligand inhibition assay) or anti-GFP antibody (antibody inhibition assays). Experiments were performed at least 2 times with similar results.
Enzyme pretreatment of endothelial cells.
Endothelial cells were grown on 96-well plates and incubated with increasing concentrations (0.5 to 3 U/ml) of heparinase III (Sigma) or chondroitin ABC lyase (Sigma) diluted in serum-free medium or controls containing no enzyme for 1 h at 37°C (15, 36). For proteinase K (Roche) digestion, endothelial cells were incubated with increasing concentrations of proteinase K (0.01 to 10 μg/ml) diluted in serum-free medium or no-enzyme control for 30 min at 37°C. After enzyme treatment, cells were washed twice in EBM2 (2% FBS). Under these conditions, >90% of the cell monolayer remained intact after treatment. Subsequently, dengue virus (150 FFU) was adsorbed to endothelial cells for 1 h at 25°C. Cells were washed and incubated for 24 h in supplemented EBM2 (10% FBS) prior to immunoperoxidase staining of DV4 antigen to quantitate the number of infected cells. Experiments were performed at least 2 times with similar results.
Dengue virus binding to immobilized heparin.
Viral binding assays were performed as previously described with modifications (66). Heparin immobilized on Sepharose resin or Sepharose resin alone was used to determine whether dengue virus specifically binds to heparin. Dengue virus (8 × 108 FFU) was incubated with heparin, vitronectin, or sialic acid (1 mg for 1 h at 4°C) or left untreated (no competitor). Subsequently, dengue virus was bound to heparin-Sepharose resin (Sigma) or unmodified Sepharose resin (100 μl/sample) in duplicate. Virus and resin were incubated for 1 h (4°C with rotation), and subsequently, Sepharose was pelleted (1,000 × g 1 min) and washed three times in DMEM (10% FBS). Bound dengue virus was eluted in DMEM containing 1.5 M NaCl and diluted 3-fold in DMEM (10% FBS), and the titer determined on C6/36 cells as described above. The numbers of dengue virus-infected cells bound to heparin-Sepharose versus Sepharose alone were determined by immunoperoxidase staining as described above and compared to virus binding in the presence of excess soluble heparin, vitronectin, or sialic acid ligands. This experiment was performed at least 3 times with similar results.
Immunoblot analysis of dengue virus proteins in endothelial cells.
Cells were lysed in 0.1% SDS lysis buffer (150 mM NaCl, 40 mM Tris, 2 mM EDTA, 5 mM NaF, 1 mM Na4P2O7, 1 mM Na3VO4, 0.1% NP-40, 10% glycerol, 0.1% SDS) and clarified by centrifugation at 40,000 rpm for 40 min at 4°C. Total protein levels in cell lysates were quantitated by bicinchoninic acid (BCA) assay (Pierce), and equivalent amounts of total protein were separated on SDS–10% polyacrylamide gels. Proteins were transferred to nitrocellulose and incubated with a 1:1,000 dilution of anti-DV4 capsid peptide sera, 1:300 anti-DV4 NS1 peptide sera, or 1:1,000 anti-actin followed by horseradish peroxidase-conjugated sheep anti-mouse or goat anti-rabbit immunoglobulin G (GE Healthcare). Proteins were detected by fluorography using Pierce enhanced chemiluminescence (ECL) reagents (Thermo Scientific) as previously described (49).
RESULTS
Dengue virus efficiently infects human endothelial cells.
Dengue virus infection leads to DHF and DSS, which are diseases characterized by increased vascular permeability, edema, thrombocytopenia, and hemorrhage. These changes stem from the diminished endothelial cell barrier of capillaries, and dengue virus-infected endothelial cells are present in patient autopsy samples, murine models, and cell culture (6, 14, 37, 39, 47). However, little information is available on the entry or productive infection of primary human endothelial cells by dengue virus. We infected Vero E6 cells, which are highly susceptible to dengue virus infection (35), and primary human endothelial cells at a multiplicity of infection (MOI) of 6 with type 4 dengue virus and detected viral antigens within cells using anti-DV4 HMAF sera and immunoperoxidase staining 24 h postinfection. The results shown in Fig. 1A demonstrate that 70 to 80% of both primary human endothelial cells and Vero E6 cells were infected with dengue virus 24 h postinfection. Consistent with these results, dengue virus NS1 and capsid proteins were detected in both endothelial and Vero E6 cell lysates (Fig. 1B). Similar to viral replication in Vero E6 cells, dengue virus infection of human endothelial cells rapidly resulted in titers of 105 FFU/ml in cell supernatants 1 day postinfection (Fig. 1C). These findings demonstrate that dengue efficiently and productively infects human endothelial cells and results in the rapid replication and release of infectious virions.
Fig. 1.
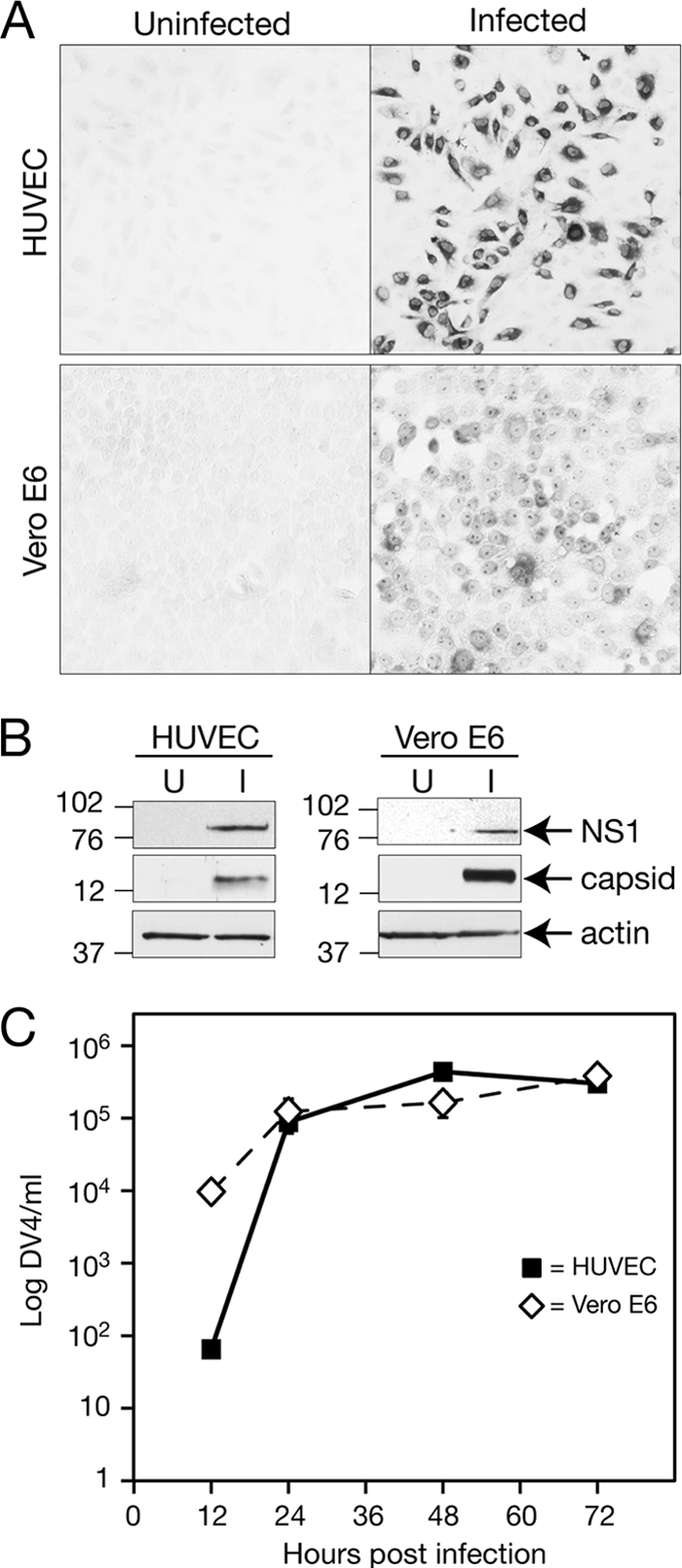
Productive infection of primary human endothelial cells by dengue virus. (A) Dengue virus type 4 (DV4) was adsorbed to primary human endothelial cells or Vero E6 cells at an MOI of 6 or mock infected for 1.5 h at room temperature. Inocula were removed, and cell monolayers washed with PBS three times. Cells were grown in complete medium for 24 h, the medium removed, and monolayers fixed with 100% ice-cold methanol for 10 min. Dengue virus antigen in infected cells was detected using DV4-specific HMAF antibody (1:4,000) followed by binding to a horseradish peroxidase-tagged secondary antibody (1:4,000) and immunoperoxidase staining using amino-ethyl carbazole (26, 28, 68). (B) Uninfected (U) or dengue virus-infected (I) endothelial or Vero E6 cells were treated as described for panel A, and cell lysates were collected 24 h postinfection. Proteins were separated by SDS-PAGE (10%) and Western blotted using anti-DV4 NS1 peptide (1:300), anti-DV4 capsid peptide (1:1,000), or anti-actin (1:1,000) antibodies. (C) Endothelial and Vero E6 cells were infected in duplicate as described for panel A with a constant amount of dengue virus (MOI of 6), washed with PBS, and incubated (37°C, 5% CO2) for various lengths of time. The titers of dengue virus present in supernatants of endothelial and Vero E6 cells (12, 24, 48, and 72 h postinfection) were determined on C6/36 cells using DV4 HMAF antibody and immunoperoxidase staining as described for panel A. Dengue virus antigen-containing infected-cell foci were quantitated and reported as focus forming units (FFU).
Heparin blocks dengue virus infection of endothelial cells.
Dengue virus receptors on endothelial cells have yet to be defined, although several immune and dendritic cell receptors have been reported to mediate dengue virus attachment and entry (13). In addition, heparan sulfate-containing proteoglycans (HSPGs) reportedly direct dengue virus attachment to Vero, Huh7, and HepG2 cells (29, 36, 45). In order to identify endothelial cell receptors for dengue virus, we pretreated human endothelial cells with potentially inhibitory antibodies and quantitated their ability to inhibit dengue virus infection of endothelial cells. We found that pretreating primary human endothelial cells with 0.5 to 5 μg/ml of antibodies to PECAM, VCAM, ICAM, E-selectin, VE-cadherin, PAFR, PDGFR, syndecan-1, CXCR3, VEGFR2, Edg-1, Tie-2, and integrins αVβ3 and α5β1 had little or no effect (<25%) on dengue virus infectivity (Fig. 2). Even using up to 20 μg/ml of antibody failed to specifically inhibit infection (not shown). A similar approach was used to determine whether pretreating dengue virus with potentially competitive ligands inhibited dengue virus infection of endothelial cells. We found that dengue virus infection of endothelial cells in the presence of ligands to integrin receptors (vitronectin, collagen, fibrinogen, and laminin; 1 to 100 μg/ml) had little or no effect (Fig. 3). In contrast, pretreating dengue virus with heparin prior to adsorption inhibited infection of endothelial cells by >60% compared to the level of infection in BSA-treated control cells (Fig. 3). The addition of glycophorin A or sialic acid failed to block dengue virus infection at any concentration. These findings demonstrate that soluble heparin blocks dengue virus infection of primary human endothelial cells.
Fig. 2.

Effect of endothelial cell receptor blockade on dengue virus infection. Primary human endothelial cells were pretreated with antibodies (0.5 or 5 μg/ml) to the indicated cellular receptors or a control antibody to green fluorescent protein (GFP) in duplicate (1 h at 4°C), followed by species-specific secondary antibody (1:2,000, 1 h, 4°C) (26, 28, 68). Subsequently, 100 FFU of DV4 was adsorbed to endothelial cell monolayers (1 h), which were then washed 3 times with PBS and incubated for 24 h (37°C, 5% CO2). Dengue virus antigen-positive infected cells were quantitated as described for Fig. 1A using HMAF antibody and an immunoperoxidase staining assay. The number of dengue virus-infected cells following receptor blockade are reported as the percentage of infected cells treated with a control GFP antibody (100%).
Fig. 3.
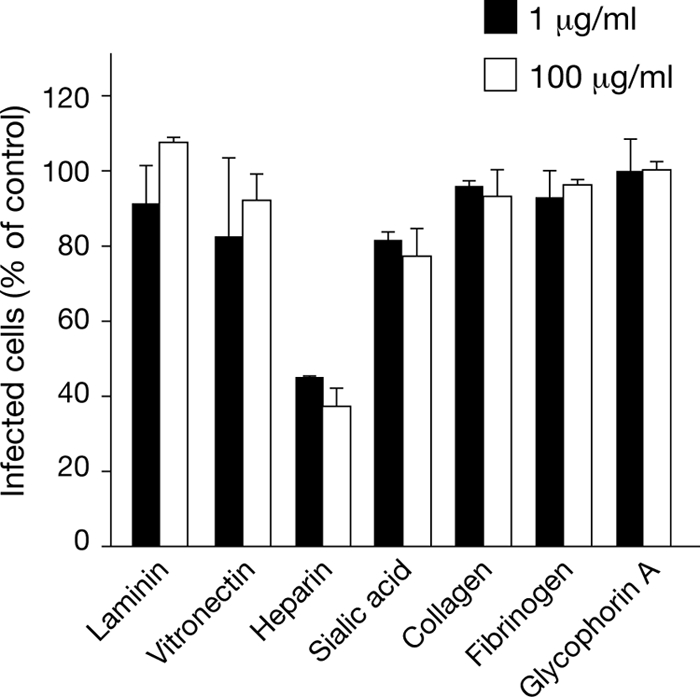
Ligands inhibit endothelial cell infection by dengue virus. Dengue virus (150 FFUs) was prebound to potentially competitive ligands (1 and 100 μg/ml, 1 h) prior to infection of endothelial cells. Following adsorption, endothelial cell monolayers were fixed 24 h postinfection and the number of dengue virus-infected cells was determined by the DV4-HMAF antibody immunoperoxidase staining assay described for Fig. 1A. The number of endothelial cells infected in the presence of a specific ligand is presented as the percentage of infected cells observed in control BSA-treated samples (100%).
Heparin and the related ligands heparan sulfate, dermatan sulfate, and chondroitin sulfate were analyzed further to determine their ability to inhibit dengue virus infection of human endothelial cells. Similar to the inhibitory effects of heparin, we found that high concentrations (>10 μg/ml) of heparan sulfate, chondroitin sulfate, and dermatan sulfate were able to inhibit dengue virus infection (50 to 70%) compared to the levels of infection in controls treated with identical amounts of BSA (Fig. 4). However, at a 10-fold-lower concentration (≥1 μg/ml), only heparin was capable of inhibiting dengue virus infection of endothelial cells by >60% (Fig. 4). This indicates that dengue virus infection of human endothelial cells is specifically inhibited by heparin and heparan sulfate-containing derivative ligands.
Fig. 4.

Heparin inhibits dengue virus infection of endothelial cells. Dengue virus (150 FFU) was prebound to increasing concentrations (0.1 to 100 μg/ml, 1 h) of heparin, heparan sulfate, dermatan sulfate, chondroitin sulfate A, or BSA diluted in DMEM. Following adsorption of the DV4 samples onto endothelial cells, the monolayers were washed and incubated at 37°C for 24 h. The number of dengue virus-infected cells was determined by the DV4-HMAF antibody immunoperoxidase staining assay described for Fig. 1A. The number of endothelial cells infected in the presence of a specific ligand is presented as the percentage of infected cells observed in control BSA-treated endothelial cells (100%).
The inhibition of dengue virus infection by heparin-like ligands suggested a role for cell surface heparan sulfate-containing proteoglycans (HSPGs) in mediating dengue virus attachment. To analyze the potential role of HSPGs in dengue virus attachment and entry, endothelial cells were treated with heparinase III, chondroitinase, or proteinase K enzymes or mock treated prior to dengue virus adsorption. We found that pretreating cells with increasing concentrations of heparinase III and proteinase k but not chondroitinase reduced the number of dengue virus-infected endothelial cells by >80% compared to the number in mock-treated cells (Fig. 5). In contrast, chondroitinase treatment resulted in only a 10 to 25% decrease in dengue virus-infected endothelial cells compared to the level of infection in control cells. Analogous results using similar enzyme concentrations have been used to show that dengue virus binds to both Vero E6 and Huh7 cells via HSPG-mediated cell surface components (15, 36). These results suggest that HSPGs direct dengue virus infection of human endothelial cells.
Fig. 5.

Enzymatic treatment of human endothelial cells. Endothelial cell monolayers were incubated with heparinase III (0.5 to 3 U/ml) (white bars) or chondroitinase (0.5 to 3 U/ml) (black bars) (A) or proteinase K (0.01 to 10 μg/ml, striped bars) (B) or were mock treated (gray bar; A, B) for 30 to 60 min at 37°C. Monolayers were washed with PBS, and dengue virus (150 FFU) was adsorbed to cells (1 h). Following adsorption, endothelial cell monolayers were washed and incubated at 37°C for 24 h. The number of dengue virus-infected cells was determined by the DV4-HMAF antibody immunoperoxidase staining assay described for Fig. 1A. The number of endothelial cells infected following enzymatic treatment is reported as the percentage of infected cells observed in mock-treated, no-enzyme-added controls (100%).
Dengue virus binds immobilized heparin.
In order to determine whether dengue virus specifically binds heparin, we analyzed dengue virus binding to resin containing immobilized heparin in the presence or absence of potentially competitive ligands. Dengue virus was bound to Sepharose resin alone or heparin conjugated Sepharose resin, and after washing, the amount of dengue virus bound to the resin was determined by quantitating the amount of infectious virus using an infectivity assay. We found that dengue virus specifically bound to resin containing immobilized heparin compared to its binding to Sepharose alone (Fig. 6). Further, binding of dengue virus to the immobilized heparin resin was specifically inhibited by incubating dengue virus with an excess of heparin (80% reduction in binding) but not of vitronectin or sialic acid (Fig. 6). These findings demonstrate that dengue virus binds to immobilized heparin and that this binding is specific to heparin, since it is inhibited by the presence of excess competitive ligand. These findings demonstrate that dengue virus binds to a heparin component of HSPGs and, in combination with the above-described data, suggest that heparin-containing HSPGs are human endothelial cell receptors for dengue virus.
Fig. 6.

Dengue virus binds to immobilized heparin. Binding of dengue virus to heparin-Sepharose or unmodified Sepharose was used to determine whether dengue virus was capable of binding heparin. Dengue virus (8 × 108 FFU) was pretreated with 1 mg of soluble heparin, vitronectin, or sialic acid (in DMEM, 1 h, 4°C) in duplicate and subsequently incubated with heparin-Sepharose resin or Sepharose resin alone (1 h, 4°C with rotation). Resin was pelleted and washed 3 times with DMEM, and bound virus was eluted with DMEM containing 1.5 M NaCl. Eluates were diluted 3-fold with medium, and the titer of infectious DV4 bound to resin was determined on C6/36 cells as described for Fig. 1C. Dengue virus bound to resin in the presence of excess ligand is reported as the percentage of virus bound to heparin-conjugated Sepharose alone (100%).
DISCUSSION
DHF and DSS are characterized by increased vascular permeability resulting in hemorrhagic and edematous disease. Observations from dengue patients and murine models of dengue virus disease show that vascular endothelial cells are infected in animals and individuals with DHF and DSS symptoms (6–8, 14, 39, 62, 71, 81). The endothelium regulates vascular leakage and responds to external signals which modify interendothelial adherence and vascular permeability. As a result, the means by which dengue virus infects endothelial cells is likely to be of vital importance to endothelial cell responses that contribute to pathogenesis.
Here, we demonstrate that dengue virus efficiently infects primary endothelial cells (70 to 80%). Importantly, we found that dengue virus infection of endothelial cells was highly permissive and resulted in the rapid production of progeny virus (4 × 105 FFU/ml) 1 day postinfection (Fig. 1C). Interestingly, the timing of dengue virus emergence from both endothelial cells and interferon (IFN) locus-deficient Vero E6 cells (23) was nearly identical and resulted in similar titers. This suggests the potential for dengue virus to regulate early IFN responses of endothelial cells. Prior reports indicated that only a small fraction (<1 to 20%) of endothelial cells or endothelial-like cell lines are infected by dengue virus 1 to 2 days postinfection (1, 2, 14, 43, 65, 70, 77). Our findings may be the result of synchronously infecting low-passage-number primary human endothelial cells using an MOI of 6. In fact, the initial infection of only a small fraction of endothelial cells in prior studies may have induced paracrine IFN responses that reduced dengue virus spread (21). Our findings demonstrate that human endothelial cells are both highly susceptible to dengue virus infection and likely to be a substantial source of progeny virus which contribute to acute viremia and viral spread to macrophages and dendritic cells (19, 34, 38, 75, 79). Since endothelial cells form a fluid barrier within the vasculature, productive dengue virus infection of endothelial cells is likely to be an important contributor to dengue virus pathogenesis.
Endothelial cells are susceptible to infection by a number of viruses that utilize cell surface-expressed β1 or β3 integrins as entry receptors (11, 18, 24, 28, 51, 76, 80). Pathogenic hantavirus interaction with endothelial cell αVβ3 integrins sensitizes cells to the permeabilizing effects of VEGF and increases vascular permeability (27). This rationalized integrins as potential endothelial cell receptors for dengue virus, an idea which was bolstered by studies showing that antibodies to αVβ3 block West Nile virus, Japanese encephalitis virus, and to a lesser extent, dengue virus infection of Vero E6 cells (18). However, antibody and ligand inhibition assays demonstrate that αVβ3 antibodies, as well as antibodies and ligands to other integrin receptors, did not significantly inhibit dengue virus infection of human endothelial cells (Fig. 2 and 3).
Interestingly, dengue virus bound specifically to immobilized heparin and both heparin and heparan sulfate ligands blocked dengue virus infection, suggesting that heparan sulfate proteoglycans (HSPGs) are dengue virus receptors on endothelial cells. These findings are consistent with dengue virus attachment to Vero E6 cells and hepatocyte cell lines where HSPGs have been shown to mediate attachment (29, 36, 45). Additionally, purified dengue virus envelope protein domains reportedly interact with heparan sulfate (15, 46, 64). Our data indicate that heparin is an effective inhibitor of dengue virus infection of endothelial cells. This finding is supported by the loss of infectivity following endothelial cell treatment with heparinase III, which cleaves both heparin and heparan sulfate side chains from cell surface HSPGs (Fig. 5). Syndecans and glypicans are the most abundant HSPGs on cell surfaces, with syndecan-3 and glypican-1 being highly expressed on vascular endothelial cells (30, 53). Although syndecan antibodies failed to block dengue virus infection of endothelial cells, additional studies are needed to define HSPGs that mediate dengue virus infection of endothelial cells and are involved in dengue virus regulation of additional endothelial cell functions.
While emphasis has been put on dengue virus infection of immune cells, the importance of infecting endothelial cells, which line the vasculature and control capillary fluid barrier functions, should not be underestimated. Our studies demonstrate that dengue virus attachment to heparan sulfate-containing endothelial cell receptors permits efficient infection of endothelial cells. It is obvious that direct dengue virus infection of endothelial cells may contribute to endothelial barrier dysfunction; however, the mechanisms by which dengue viruses alter endothelial cell functions have not been defined. Dengue virus-infected endothelial cells may contribute to the induction of chemokines and cytokines that activate immune cells, as well as immune cell adherence to the infected endothelium (9, 48, 50, 67). Dengue virus-infected endothelial cells could have a significant impact on the severity of dengue disease and serve as critical targets for controlling disease outcome. In addition, dengue virus-infected endothelial cells provide a vascular target for enhanced immune responses to dengue virus antigens which contribute to DSS and DHF (3, 4). Although specific HSPGs need to be defined to selectively inhibit dengue virus infection of endothelial cells, our data suggest a potential means for reducing viremia during dengue virus infections through an endothelial cell receptor blockade. Blocking endothelial cell infection could reduce immune targeting of dengue virus-infected endothelial cells and dengue virus-induced changes to endothelial cells which contribute to vascular leakage. These findings also suggest the need for studies to define the role of endothelial cells in dengue virus disease models.
ACKNOWLEDGMENTS
We thank Irina Gavriloskaya and Elena Gorbunova for technical support with antibody inhibition assays and Zoe Speed for help with dengue virus propagation. We also thank C.-J. Lai (NIH, NIAID, LID) for providing us with dengue virus (type 4) and DV4-specific HMAF.
This work was supported by National Institutes of Health grants R01AI47873, PO1AI055621, R21AI1080984, and U54AI57158 (Northeast Biodefense Center [director, W. I. Lipkin]).
Footnotes
▿
Published ahead of print on 6 July 2011.
REFERENCES
- 1.Arevalo M. T., Simpson-Haidaris P. J., Kou Z., Schlesinger J. J., Jin X. 2009. Primary human endothelial cells support direct but not antibody-dependent enhancement of dengue viral infection. J. Med. Virol. 81:519–528 [DOI] [PubMed] [Google Scholar]
- 2.Avirutnan P., Malasit P., Seliger B., Bhakdi S., Husmann M. 1998. Dengue virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J. Immunol. 161:6338–6346 [PubMed] [Google Scholar]
- 3.Avirutnan P., et al. 2006. Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J. Infect. Dis. 193:1078–1088 [DOI] [PubMed] [Google Scholar]
- 4.Avirutnan P., et al. 2007. Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog. 3:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azizan A., et al. 2006. Differential proinflammatory and angiogenesis-specific cytokine production in human pulmonary endothelial cells, HPMEC-ST1.6R infected with dengue-2 and dengue-3 virus. J. Virol. Methods 138:211–217 [DOI] [PubMed] [Google Scholar]
- 6.Balsitis S. J., et al. 2009. Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am. J. Trop. Med. Hyg. 80:416–424 [PubMed] [Google Scholar]
- 7.Balsitis S. J., Harris E. 2010. Animal models of dengue virus infection and disease: applications, insights and frontiers, p. 103–115_In_Hanley K. A., Weaver S. C.(ed.), Frontiers in dengue virus research. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 8.Balsitis S. J., et al. 2010. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog. 6:e1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basu A., Chaturvedi U. C. 2008. Vascular endothelium: the battlefield of dengue viruses. FEMS Immunol. Med. Microbiol. 53:287–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bates D. O., Harper S. J. 2002. Regulation of vascular permeability by vascular endothelial growth factors. Vascul. Pharmacol. 39:225–237 [DOI] [PubMed] [Google Scholar]
- 11.Berinstein A., Roivainen M., Hovi T., Mason P. W., Baxt B. 1995. Antibodies to the vitronectin receptor (integrin alpha V beta 3) inhibit binding and infection of foot-and-mouth disease virus to cultured cells. J. Virol. 69:2664–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosch I., et al. 2002. Increased production of interleukin-8 in primary human monocytes and in human epithelial and endothelial cell lines after dengue virus challenge. J. Virol. 76:5588–5597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabrera-Hernandez A., Smith D. R. 2005. Mammalian dengue virus receptors. Dengue Bull. 29:119–135 [Google Scholar]
- 14.Chen H. C., Hofman F. M., Kung J. T., Lin Y. D., Wu-Hsieh B. A. 2007. Both virus and tumor necrosis factor alpha are critical for endothelium damage in a mouse model of dengue virus-induced hemorrhage. J. Virol. 81:5518–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y., et al. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3:866–871 [DOI] [PubMed] [Google Scholar]
- 16.Chen Y., Maguire T., Marks R. M. 1996. Demonstration of binding of dengue virus envelope protein to target cells. J. Virol. 70:8765–8772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y. C., Wang S. Y., King C. C. 1999. Bacterial lipopolysaccharide inhibits dengue virus infection of primary human monocytes/macrophages by blockade of virus entry via a CD14-dependent mechanism. J. Virol. 73:2650–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu J. J., Ng M. L. 2004. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J. Biol. Chem. 279:54533–54541 [DOI] [PubMed] [Google Scholar]
- 19.Dejnirattisai W., et al. 2010. Cross-reacting antibodies enhance dengue virus infection in humans. Science 328:745–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diamond M. S., Edgil D., Roberts T. G., Lu B., Harris E. 2000. Infection of human cells by dengue virus is modulated by different cell types and viral strains. J. Virol. 74:7814–7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diamond M. S., et al. 2000. Modulation of dengue virus infection in human cells by alpha, beta, and gamma interferons. J. Virol. 74:4957–4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dvorak H. F. 2010. Vascular permeability to plasma, plasma proteins, and cells: an update. Curr. Opin. Hematol. 17:225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emeny J. M., Morgan M. J. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43:247–252 [DOI] [PubMed] [Google Scholar]
- 24.Feire A. L., Koss H., Compton T. 2004. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. U. S. A. 101:15470–15475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavrilovskaya I., et al. 1999. New York 1 and Sin Nombre viruses are serotypically distinct viruses associated with hantavirus pulmonary syndrome. J. Clin. Microbiol. 37:122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gavrilovskaya I. N., Brown E. J., Ginsberg M. H., Mackow E. R. 1999. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J. Virol. 73:3951–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavrilovskaya I. N., Gorbunova E. E., Mackow N. A., Mackow E. R. 2008. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 82:5797–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gavrilovskaya I. N., Shepley M., Shaw R., Ginsberg M. H., Mackow E. R. 1998. beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 95:7074–7079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Germi R., et al. 2002. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology 292:162–168 [DOI] [PubMed] [Google Scholar]
- 30.Gotte M. 2003. Syndecans in inflammation. FASEB J. 17:575–591 [DOI] [PubMed] [Google Scholar]
- 31.Gubler D. J. 1998. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 11:480–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gubler D. J. 2006. Dengue/dengue haemorrhagic fever: history and current status. Novartis Found. Symp. 277:3–16 [DOI] [PubMed] [Google Scholar]
- 33.Gubler D. J. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 10:100–103 [DOI] [PubMed] [Google Scholar]
- 34.Halstead S. B. 2003. Neutralization and antibody-dependent enhancement of dengue viruses. Adv. Virus Res. 60:421–467 [DOI] [PubMed] [Google Scholar]
- 35.Henchal E. A., Putnak J. R. 1990. The dengue viruses. Clin. Microbiol. Rev. 3:376–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hilgard P., Stockert R. 2000. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology 32:1069–1077 [DOI] [PubMed] [Google Scholar]
- 37.Huang K. J., et al. 2000. Manifestation of thrombocytopenia in dengue-2-virus-infected mice. J. Gen. Virol. 81:2177–2182 [DOI] [PubMed] [Google Scholar]
- 38.Huang K. J., et al. 2006. The dual-specific binding of dengue virus and target cells for the antibody-dependent enhancement of dengue virus infection. J. Immunol. 176:2825–2832 [DOI] [PubMed] [Google Scholar]
- 39.Jessie K., Fong M. Y., Devi S., Lam S. K., Wong K. T. 2004. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 189:1411–1418 [DOI] [PubMed] [Google Scholar]
- 40.Kaufmann B., Rossmann M. G. 2011. Molecular mechanisms involved in the early steps of flavivirus cell entry. Microbes Infect. 13:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiessling F., Kartenbeck J., Haller C. 1999. Cell-cell contacts in the human cell line ECV304 exhibit both endothelial and epithelial characteristics. Cell Tissue Res. 297:131–140 [DOI] [PubMed] [Google Scholar]
- 42.Krishnamurti C., Peat R. A., Cutting M. A., Rothwell S. W. 2002. Platelet adhesion to dengue-2 virus-infected endothelial cells. Am. J. Trop. Med. Hyg. 66:435–441 [DOI] [PubMed] [Google Scholar]
- 43.Liao H., Xu J., Huang J. 2010. FasL/Fas pathway is involved in dengue virus induced apoptosis of the vascular endothelial cells. J. Med. Virol. 82:1392–1399 [DOI] [PubMed] [Google Scholar]
- 44.Liew K. J., Chow V. T. 2004. Differential display RT-PCR analysis of ECV304 endothelial-like cells infected with dengue virus type 2 reveals mRNA expression profiles of multiple human genes involved in known and novel roles. J. Med. Virol. 72:597–609 [DOI] [PubMed] [Google Scholar]
- 45.Lin Y. L., et al. 2002. Heparin inhibits dengue-2 virus infection of five human liver cell lines. Antiviral Res. 56:93–96 [DOI] [PubMed] [Google Scholar]
- 46.Marks R. M., et al. 2001. Probing the interaction of dengue virus envelope protein with heparin: assessment of glycosaminoglycan-derived inhibitors. J. Med. Chem. 44:2178–2187 [DOI] [PubMed] [Google Scholar]
- 47.Martina B. E., Koraka P., Osterhaus A. D. 2009. Dengue virus pathogenesis: an integrated view. Clin. Microbiol. Rev. 22:564–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathew A., Rothman A. L. 2008. Understanding the contribution of cellular immunity to dengue disease pathogenesis. Immunol. Rev. 225:300–313 [DOI] [PubMed] [Google Scholar]
- 49.Matthys V., Gorbunova E. E., Gavrilovskaya I. N., Pepini T., Mackow E. R. 2011. The C-terminal 42 residues of the Tula virus Gn protein regulate interferon induction. J. Virol. 85:4752–4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McBride W. J., Bielefeldt-Ohmann H. 2000. Dengue viral infections; pathogenesis and epidemiology. Microbes Infect. 2:1041–1050 [DOI] [PubMed] [Google Scholar]
- 51.McMillan N. A., Payne E., Frazer I. H., Evander M. 1999. Expression of the alpha6 integrin confers papillomavirus binding upon receptor-negative B-cells. Virology 261:271–279 [DOI] [PubMed] [Google Scholar]
- 52.McVerry B. J., Garcia J. G. 2004. Endothelial cell barrier regulation by sphingosine 1-phosphate. J. Cell. Biochem. 92:1075–1085 [DOI] [PubMed] [Google Scholar]
- 53.Mertens G., Cassiman J. J., Van den Berghe H., Vermylen J., David G. 1992. Cell surface heparan sulfate proteoglycans from human vascular endothelial cells. Core protein characterization and antithrombin III binding properties. J. Biol. Chem. 267:20435–20443 [PubMed] [Google Scholar]
- 54.Miller J. L., et al. 2008. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 4:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mondotte J. A., Lozach P. Y., Amara A., Gamarnik A. V. 2007. Essential role of dengue virus envelope protein N glycosylation at asparagine-67 during viral propagation. J. Virol. 81:7136–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukherjee S., Tessema M., Wandinger-Ness A. 2006. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ. Res. 98:743–756 [DOI] [PubMed] [Google Scholar]
- 57.Mukhopadhyay D., Zeng H., Bhattacharya R. 2004. Complexity in the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF)-receptors signaling. Mol. Cell. Biochem. 264:51–61 [DOI] [PubMed] [Google Scholar]
- 58.Navarro-Sanchez E., et al. 2003. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 4:723–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navarro-Sanchez E., Despres P., Cedillo-Barron L. 2005. Innate immune responses to dengue virus. Arch. Med. Res. 36:425–435 [DOI] [PubMed] [Google Scholar]
- 60.Noisakran S., et al. 2009. Detection of dengue virus in platelets isolated from dengue patients. Southeast Asian J. Trop. Med. Public Health 40:253–262 [PubMed] [Google Scholar]
- 61.Noisakran S., Perng G. C. 2008. Alternate hypothesis on the pathogenesis of dengue hemorrhagic fever (DHF)/dengue shock syndrome (DSS) in dengue virus infection. Exp. Biol. Med. (Maywood) 233:401–408 [DOI] [PubMed] [Google Scholar]
- 62.Oishi K., Saito M., Mapua C. A., Natividad F. F. 2007. Dengue illness: clinical features and pathogenesis. J. Infect. Chemother. 13:125–133 [DOI] [PubMed] [Google Scholar]
- 63.Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. 2006. VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell Biol. 7:359–371 [DOI] [PubMed] [Google Scholar]
- 64.Pattnaik P., Babu J. P., Verma S. K., Tak V., Rao P. V. 2007. Bacterially expressed and refolded envelope protein (domain III) of dengue virus type-4 binds heparan sulfate. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 846:184–194 [DOI] [PubMed] [Google Scholar]
- 65.Peyrefitte C. N., et al. 2006. Dengue virus infection of human microvascular endothelial cells from different vascular beds promotes both common and specific functional changes. J. Med. Virol. 78:229–242 [DOI] [PubMed] [Google Scholar]
- 66.Raymond T., Gorbunova E., Gavrilovskaya I. N., Mackow E. R. 2005. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent alphavbeta3 integrin conformers. Proc. Natl. Acad. Sci. U. S. A. 102:1163–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rothman A. L. 2010. Cellular immunology of sequential dengue virus infection and its role in disease pathogenesis. Curr. Top. Microbiol. Immunol. 338:83–98 [DOI] [PubMed] [Google Scholar]
- 68.Shaw R. D., Vo P. T., Offit P. A., Coulson B. S., Greenberg H. B. 1986. Antigenic mapping of the surface proteins of rhesus rotavirus. Virology 155:434–451 [DOI] [PubMed] [Google Scholar]
- 69.Srikiatkhachorn A., et al. 2007. Virus-induced decline in soluble vascular endothelial growth receptor 2 is associated with plasma leakage in dengue hemorrhagic fever. J. Virol. 81:1592–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Talavera D., Castillo A. M., Dominguez M. C., Gutierrez A. E., Meza I. 2004. IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J. Gen. Virol. 85:1801–1813 [DOI] [PubMed] [Google Scholar]
- 71.Tan G. K., et al. 2010. A non mouse-adapted dengue virus strain as a new model of severe dengue infection in AG129 mice. PLoS Negl. Trop. Dis. 4:e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tassaneetrithep B., et al. 2003. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197:823–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tiruppathi C., Minshall R. D., Paria B. C., Vogel S. M., Malik A. B. 2002. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul. Pharmacol. 39:173–185 [DOI] [PubMed] [Google Scholar]
- 74.Ubol S., Phuklia W., Kalayanarooj S., Modhiran N. 2010. Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J. Infect. Dis. 201:923–935 [DOI] [PubMed] [Google Scholar]
- 75.van der Schaar H. M., Wilschut J. C., Smit J. M. 2009. Role of antibodies in controlling dengue virus infection. Immunobiology 214:613–629 [DOI] [PubMed] [Google Scholar]
- 76.Wang X., Huang D. Y., Huong S. M., Huang E. S. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat. Med. 11:515–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warke R. V., et al. 2003. Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. J. Virol. 77:11822–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei H. Y., Jiang L. F., Fang D. Y., Guo H. Y. 2003. Dengue virus type 2 infects human endothelial cells through binding of the viral envelope glycoprotein to cell surface polypeptides. J. Gen. Virol. 84:3095–3098 [DOI] [PubMed] [Google Scholar]
- 79.Whitehead S. S., Blaney J. E., Durbin A. P., Murphy B. R. 2007. Prospects for a dengue virus vaccine. Nat. Rev. Microbiol. 5:518–528 [DOI] [PubMed] [Google Scholar]
- 80.Wickham T. J., Mathias P., Cheresh D. A., Nemerow G. R. 1993. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73:309–319 [DOI] [PubMed] [Google Scholar]
- 81.Yen Y. T., Chen H. C., Lin Y. D., Shieh C. C., Wu-Hsieh B. A. 2008. Enhancement by tumor necrosis factor alpha of dengue virus-induced endothelial cell production of reactive nitrogen and oxygen species is key to hemorrhage development. J. Virol. 82:12312–12324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yuan S. Y. 2006. New insights into eNOS signaling in microvascular permeability. Am. J. Physiol. Heart Circ. Physiol. 291:H1029–H1031 [DOI] [PubMed] [Google Scholar]
- 83.Yuan S. Y. 2002. Protein kinase signaling in the modulation of microvascular permeability. Vascul. Pharmacol. 39:213–223 [DOI] [PubMed] [Google Scholar]