DNMT3A Mutations and Response to the Hypomethylating Agent Decitabine in Acute Myeloid Leukemia (original) (raw)
. Author manuscript; available in PMC: 2013 Jul 1.
Published in final edited form as: Leukemia. 2011 Nov 29;26(5):1106–1107. doi: 10.1038/leu.2011.342
To the editor
Acute myeloid leukemia (AML) is a disease of the elderly, affecting patients with a median age of 65 to 70 years. Cytotoxic chemotherapy remains the mainstay of treatment in younger patients (often defined as those below the age of 60 years), but is associated with lower complete remission (CR) rates and greater toxicity in older patients (≥60 years), resulting in poor long-term outcomes and a median overall survival (OS) of less than one year.1 Thus, there is a need for novel, effective and well-tolerated treatment approaches, especially for older patients.
Epigenetic changes, including aberrant DNA methylation, can result in silencing of tumor suppressor genes and likely contribute to the pathogenesis of AML.2,3 Aberrant DNA methylation can be reversed pharmacologically by inhibitors of DNA methyltransferase (DNMT) enzymes.2,3 Two azanucleotide DNMT inhibitors, azacitidine and decitabine, are approved for the treatment of patients with myelodysplastic syndromes, and are also being studied in AML.4,5 Our group has recently reported promising results with a well-tolerated 10-day schedule of low-dose decitabine, including a CR rate of 47% and a median OS of one year in older patients with untreated AML who were considered unfit for or refused intensive chemotherapy.6
Recurrent DNMT3A gene mutations were recently discovered in AML,7–9 and are associated with worse outcomes.8–10 It has been reported that at least some DNMT3A mutations, including those at the mutational hotspot codon R882, impair the protein’s methyltransferase activity.7,9 Although the biologic mechanisms linking DNMT3A mutations to leukemogenesis are not yet fully understood, an obvious question from the clinical standpoint is whether DNMT3A mutations influence the response to treatment with hypomethylating agents. We therefore evaluated DNMT3A mutation status in a cohort of 46 predominantly older patients with previously untreated AML, who were enrolled in two clinical trials and received decitabine at an initial dose of 20 mg/m2 i.v. over 1h for 10 days, as described in detail earlier.6 Patients received decitabine either as a single agent [n=39; clinicaltrials.gov identifier NCT00492401 (ref. 6)] or in combination with bortezomib (0.7–1.3 mg/m2 i.v. on days 5, 8, 12 and 15; n=7; NCT00703300). All patients who had genomic DNA from pretreatment bone marrow specimens available were included in this analysis. DNMT3A exons 18, 19, 21, 22, and 24–26 (GeneBank accession number NM_175629), where approximately 90% of mutations are located,8 were studied using polymerase chain reaction and direct sequencing. Patients were also characterized for _FLT3_-internal tandem duplications (_FLT3_-ITD) and tyrosine kinase domain mutations (_FLT3_-TKD) and mutations in NPM1, IDH1, IDH2, TET2 and CEBPA as reported previously.11–15
The median age of the entire cohort was 74 years (range, 32–85 years; only one patient was aged younger than 60 years). Seventeen of the 46 patients had cytogenetically normal (CN-) AML, based on the analysis of at least 20 metaphases from bone marrow, and 29 had aberrant karyotypes. According to the European LeukemiaNet (ELN) reporting system for genetic changes in AML,16 10 patients fell into the ELN favorable genetic category. This included two patients with t(8;21), and eight patients with CN-AML who had CEBPA mutations or mutated NPM1 without _FLT3_-ITD. Nine patients belonged to the ELN intermediate-I category (ie, CN-AML with wild-type CEBPA, and wild type NPM1 with or without _FLT3_-ITD or mutated NPM1 with _FLT3_-ITD). Eleven patients fell into the ELN intermediate-II category (ie, had cytogenetic abnormalities not classified as favorable or adverse). Sixteen had adverse cytogenetic aberrations as classified by the ELN, including 11 with a complex karyotype.
We found eight DNMT3A mutations among the 46 patients (17%). Six were missense mutations affecting the mutational hot spot codon R882 [3 patients with c.2645G>A; p.(R882H), and 3 with c.2644C>T; p.(R882C)]. One patient had a nonsense mutation [c.1729A>T; p.(K577*)], and one had a splice-site mutation [c.2322+1G>A; p.0?]. All eight mutations were predicted to be “disease causing” by the MutationTaster algorithm, which evaluates the disease-causing potential of gene mutations based on analyses of evolutionary conservation, splice-site changes, loss of protein features and changes affecting the amount of mRNA.17
The associations of DNMT3A mutations with baseline patient characteristics are reported in Table 1. _DNMT3A-_mutated patients had a higher median white blood cell count (WBC) than _DNMT3A_-wild type patients (_P_=.04). DNMT3A mutations occurred in all ELN genetic groups. DNMT3A mutations were significantly associated with NPM1 mutations (_P_=.004), and we observed a trend towards a higher prevalence of _FLT3_-ITD among _DNMT3A_-mutated patients (_P_=.07). None of the other gene mutations we studied was significantly associated with DNMT3A mutations.
Table 1.
Association of DNMT3A mutation status with patient characteristics
| _DNMT3A_-mutated | _DNMT3A_-wild type | P | |
|---|---|---|---|
| n=8 | n=38 | ||
| Age (years), median (range) | 70.5 (68 – 83) | 74 (32 – 85)* | .49 |
| Female sex, no. (%) | 1 (13) | 11 (29) | .66 |
| WBC (×103/μl), median (range) | 26.1 (1.7 – 120) | 2.3 (0.6 – 150) | .04 |
| Bone marrow blasts (%), median (range) | 59 (20 – 92) | 35 (18 – 90) | .27 |
| ELN genetic category,16 no. | .62 | ||
| Favorable | 3 | 7 | |
| Intermediate-I | 1 | 8 | |
| Intermediate-II | 1 | 10 | |
| Adverse | 3 | 13 | |
| NPM1 mutated, no. (%) | 5 (63) | 4 (11) | .004 |
| IDH1/IDH2 mutated, no. (%) | 1 (13) | 6 (16) | 1.0 |
| IDH1 R132 mutated, no. | 1† | 3 | |
| IDH2 R140 mutated, no. | 1† | 3 | |
| TET2 mutated, no. (%) | 1 (13) | 7 (18) | .64 |
| _FLT3_-ITD, no. (%) | 2 (25) | 1 (3) | .07 |
| _FLT3-_TKD, no. (%) | 0 | 1(3) | 1.0 |
| CEBPA mutated, no. (%) | 1 (13) | 4 (11) | 1.0 |
| Single mutation, no. | 0 | 1 | |
| Double mutation, no. | 1 | 3 |
Overall, the CR rate in this cohort was 41% (19/46). Six of eight _DNMT3A_-mutated patients (75%) achieved CR, compared to 13 of 38 with wild type DNMT3A (34%; _P_=.05; Figure 1). Notably, all five patients with mutated DNMT3A and mutated NPM1 achieved CR, a significantly better response rate than among the remaining patients with other genotypes [14/41 (34%); _P_=.008]. No other pretreatment patient characteristics or gene mutations were associated with response to decitabine (Table 2). This included TET2 or IDH1/IDH2 mutations (considered separately or in combination), which were recently linked to aberrant DNA methylation patterns in AML.18,19
Figure 1.
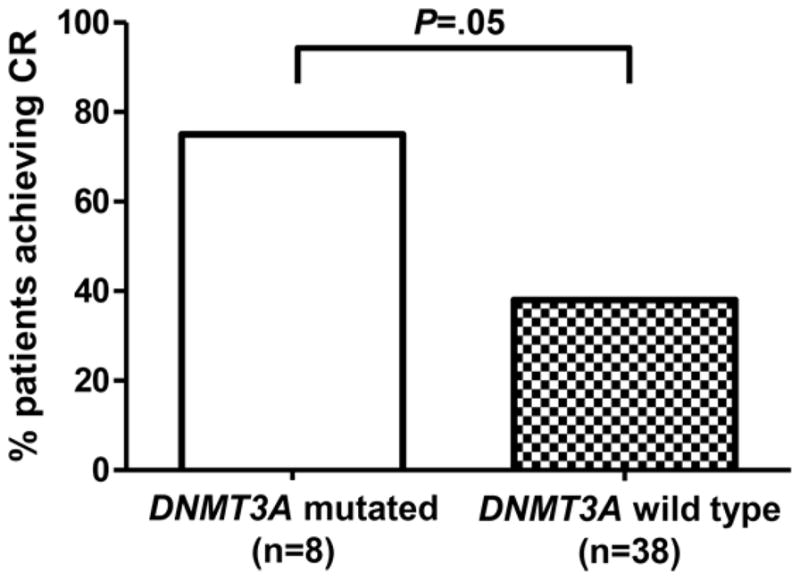
Complete remission rate according to DNMT3A mutational status in AML patients treated with decitabine.
Table 2.
Patient characteristics and response to decitabine treatment
| Complete remission | No complete remission | P | |
|---|---|---|---|
| n=19 | n=27 | ||
| Age (years), median (range) | 72 (62 – 85) | 76 (32 – 84) | .09 |
| Female sex, no. | 7 | 5 | .19 |
| WBC (×103/μl), median (range) | 2.7 (1.1 – 119.5) | 2.2 (0.6 – 150) | .74 |
| Bone marrow blasts (%), median (range) | 35 (20 – 87) | 39 (18 – 92) | .23 |
| ELN genetic category,16 no. | .77 | ||
| Favorable | 5 | 5 | |
| Intermediate-I | 4 | 5 | |
| Intermediate-II | 3 | 8 | |
| Adverse | 7 | 9 | |
| DNMT3A mutated, no. | 6 | 2 | .05 |
| NPM1 mutated, no. | 6 | 3 | .13 |
| DNMT3A mutated & NPM1 mutated, no. | 5 | 0 | .008 |
| IDH1/IDH2 mutated, no. | 3 | 4 | 1.0 |
| TET2 mutated, no. | 3 | 5 | 1.0 |
| _FLT3_-ITD, no. | 2 | 1 | .56 |
| _FLT3-_TKD, no. | 0 | 1 | 1.0 |
| CEBPA mutated, no. | 1 | 4 | .39 |
The median OS of patients with DNMT3A mutations was 15.2 months, compared to 11.0 months for patients with _DNMT3A-_wild type. This comparison is limited by the small number of _DNMT3A_-mutated patients, and the difference did not reach statistical significance.
Our findings are based on a relatively small and heterogeneous patient cohort, and therefore must be interpreted cautiously. Nevertheless, we believe that our observations are provocative, because they support a potential link between DNMT3A gene mutations, altered DNMT3A activity, and sensitivity to hypomethylating drugs. Our group has shown before that high levels of microRNA miR-29b, which targets DNMT3A, are significantly associated with response to decitabine in AML (_P_=.02). Lower DNMT3A expression levels also showed a borderline (_P_=.06) association with higher CR rate in decitabine-treated patients.6 Taken together, our results suggest that AML patients whose leukemic blasts have low DNMT3A activity, either due to loss-of-function mutations or due to low gene expression, may benefit from treatment with hypomethylating agents. If further studies confirm that DNMT3A mutations identify a subgroup of AML patients who are likely to respond to decitabine treatment, this would represent an important advance for risk-adapted stratification of AML patients to epigenetics-targeting therapy.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Estey E. Acute myeloid leukemia and myelodysplastic syndromes in older patients. J Clin Oncol. 2007;25:1908–1915. doi: 10.1200/JCO.2006.10.2731. [DOI] [PubMed] [Google Scholar]
- 2.Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ, Baylin SB. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res. 1997;57:837–841. [PubMed] [Google Scholar]
- 3.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 4.Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the Cancer and Leukemia Group B. J Clin Oncol. 2002;20:2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 6.Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Nat Acad Sci USA. 2010;107:7473–7478. doi: 10.1073/pnas.1002650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamashita Y, Yuan J, Suetake I, Suzuki H, Ishikawa Y, Choi YL, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010;29:3723–3731. doi: 10.1038/onc.2010.117. [DOI] [PubMed] [Google Scholar]
- 8.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 10.Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011;29:2889–2896. doi: 10.1200/JCO.2011.35.4894. [DOI] [PubMed] [Google Scholar]
- 11.Whitman SP, Maharry K, Radmacher MD, Becker H, Mrózek K, Margeson D, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. Blood. 2010;116:3622–3626. doi: 10.1182/blood-2010-05-283648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker H, Marcucci G, Maharry K, Radmacher MD, Mrózek K, Margeson D, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol. 2009;28:596–604. doi: 10.1200/JCO.2009.25.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcucci G, Maharry K, Radmacher MD, Mrózek K, Vukosavljevic T, Paschka P, et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B study. J Clin Oncol. 2008;26:5078–5087. doi: 10.1200/JCO.2008.17.5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 18.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 Function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]