Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies (original) (raw)
Abstract
Background During the past 5 years, high-throughput technologies have been successfully used by epidemiology studies, but almost all have focused on sequence variation through genome-wide association studies (GWAS). Today, the study of other genomic events is becoming more common in large-scale epidemiological studies. Many of these, unlike the single-nucleotide polymorphism studied in GWAS, are continuous measures. In this context, the exercise of searching for regions of interest for disease is akin to the problems described in the statistical ‘bump hunting’ literature.
Methods New statistical challenges arise when the measurements are continuous rather than categorical, when they are measured with uncertainty, and when both biological signal, and measurement errors are characterized by spatial correlation along the genome. Perhaps the most challenging complication is that continuous genomic data from large studies are measured throughout long periods, making them susceptible to ‘batch effects’. An example that combines all three characteristics is genome-wide DNA methylation measurements. Here, we present a data analysis pipeline that effectively models measurement error, removes batch effects, detects regions of interest and attaches statistical uncertainty to identified regions.
Results We illustrate the usefulness of our approach by detecting genomic regions of DNA methylation associated with a continuous trait in a well-characterized population of newborns. Additionally, we show that addressing unexplained heterogeneity like batch effects reduces the number of false-positive regions.
Conclusions Our framework offers a comprehensive yet flexible approach for identifying genomic regions of biological interest in large epidemiological studies using quantitative high-throughput methods.
Keywords: Epigenetic epidemiology, DNA methylation, genome-wide analysis, bump hunting, batch effects
Introduction
Identification of biologically relevant regions of the genome in epidemiological studies frequently involves measurements from a large number of genomic loci.1,2 As the cost of microarray technologies has rapidly decreased over the past several years, large epidemiological studies have begun to measure thousands to millions of genomic markers on thousands of people. Searching for association between disease outcomes and genomic sequence variation, marked by single-nucleotide polymorphisms (SNPs), has been the most common genomics application, referred to as genome-wide association studies (GWAS).3–5 In these studies, measurements from SNPs are categorical with three possible genotypes (AA, Aa or aa). Today, other genomic measurements, such as DNA methylation, are becoming common in large-scale epidemiological studies. Many of these, unlike SNPs, are continuous measurements, are more susceptible to measurement error, are more densely spaced across the genome, and have more complicated correlation structures.6–8 The goal of these additional types of genomic studies is similar to GWAS—screen genome-scale data to identify contiguous regions for which a genomic event, such as methylation, is associated with an outcome of interest. Yet, the differences between these newer technologies and GWAS require new analysis techniques to accomplish this goal.
The methodology presented here is motivated by genome-scale array-based DNA methylation data. DNA methylation is a chemical modification of DNA that can be inherited during cell division but is not contained in the DNA sequence itself. DNA methylation involves the modification of a cytosine base (C) to form methyl-cytosine. In adult cells of mammals, this modification occurs almost exclusively at _C_s that are immediately followed by a guanine (G) in the 5′ to 3′ direction, denoted CpG. Since DNA methylation is inherited during cell division, yet is dynamic enough to vary across cells with the same genome, it is considered an important developmental mechanism that helps explain phenotypic variability across cell types.9,10
The health implications of deciphering the DNA methylation code have recently received much attention both in the scientific literature and in the media.11–14 In cancer biology, aberrations in DNA methylation accompany the initiation and progression of cancers.15,16 Much of the excitement surrounding epigenetics relates to the promise of therapies that alter the epigenetic code by activating or silencing disease-related genes. In fact, two epigenetic drugs that reactivate tumour suppressor genes by removing methylation marks (Vidaza and decitabine) have received United States Food and Drug Administration (FDA) approval,17,18 highlighting the medical promise of mapping and understanding the role of DNA methylation. Furthermore, DNA methylation is of particular interest to epidemiologists because it is more susceptible to environmental insults than DNA sequence, and may be a mechanism for environmental risk factors for disease.
Currently, the DNA methylation data produced by large epidemiology studies are mostly microarray based. For each individual, DNA methylation levels are measured for thousands to millions of CpGs. Although at the cellular level, DNA methylation is binary on each strand (methylated or not), these technologies require millions of cells, and therefore report continuous measurements related to the proportion of cells methylated at the site in question. The general task in studies performing genome-wide DNA methylation scans is to identify genomic regions exhibiting an association between DNA methylation levels and the outcome of interest (Figure 1A). Various authors have noted that methylation levels are strongly correlated across the genome.6,7 Furthermore, reported functionally relevant findings have been generally associated with genomic regions rather than single CpGs, either CpG islands,19 CpG island shores,20 genomic blocks16 or generic 2-kb regions.21 Therefore, we propose a search for association at the region level as opposed to the single CpG level and demonstrate that this approach greatly improves specificity. From a statistical perspective, this task amounts to finding spatial intervals in which an estimated function (e.g. average difference between outcome groups, or correlation with a continuous trait) is different from 0 (Figure 1B). We propose a method to accomplish this that borrows from a topic widely discussed in the statistical literature referred to as ‘bump hunting’.22–25
Figure 1.

Example of a differentially methylation region (DMR). (A) The points show methylation measurements from the colon cancer dataset plotted against genomic location from illustrative region on chromosome 2. Eight normal and eight cancer samples are shown in this plot and represented by eight blue points and eight red points at each genomic location for which measurements were available. The curves represent the smooth estimate of the population-level methylation profiles for cancer (red) and normal (blue) samples. The green bar represents a region known to be a cancer DMR.20 (B) The black curve is an estimate of the population-level difference between normal and cancer. We expect the curve to vary due to measurement error and biological variation but to rarely exceed a certain threshold, for example those represented by the red horizontal lines. Candidate DMRs are defined as the regions for which this black curve is outside these boundaries. Note that the DMR manifests as a bump in the black curve
In genomics, bump hunting has been referred to as ‘peak detection’ in the context of finding transcription factor binding sites with chromatin immunoprecipitation onto microarray (ChIP-chip) data.26,27 However, a key difference between the epigenomic data, for which our method is developed, and previous bump hunting problems, is that the number of individuals is relatively large (we are now analysing data sets as large as 320 individuals, and anticipate thousands). Furthermore, the correlation observed in epigenomic data is substantially different than previously published applications. For example, we observe measurement error correlations between adjacent probes genome-wide ranging from 0.064 to 0.26, whereas most existing approaches are developed for independent data. Epigenomic bumps are expected to have greater variability in size and shape than in previous applications as well. For example, while ChIP data (used to find, for example, transcription factor binding sites) peaks are expected to be triangle shapes spanning several hundred base pairs,26 regions of differential DNA methylation range from several hundred base pairs to several megabases.16 In some situations, for example, in cancer studies, we also expect a larger number of bumps (thousands), leading to different approaches to correct for multiple testing comparisons. Finally, and perhaps most importantly, the fact that samples in large studies are acquired, and often measured, across long periods of time make them particularly susceptible to ‘batch effects’ – unobserved correlation structures between subgroups of samples run in high-throughput experiments.28 These effects are characterized by sub-groups of measurements that have qualitatively different behaviour across conditions and are unrelated to the biological or scientific variables in a study. The most common batch effect is introduced when subsets of experiments are run on different dates. Although processing date is commonly used to account for batch effects, in a typical experiment these are probably only surrogates for other unknown sources of variation, such as ozone levels, laboratory temperatures and reagent quality. Unfortunately, most possible sources of batch effects are not recorded during genomic data generation.
The problems outlined above for DNA methylation high-throughput data in epidemiological studies require a novel analysis strategy. Here, we introduce a generic method that combines surrogate variable analysis (SVA),29 a statistical method for modelling unexplained heterogeneity like batch effects in genomic measurements, with regression modelling, smoothing techniques and modern multiple comparison approaches to provide reliable lists of epigenomic regions of interest from epidemiological data. We highlight the strengths of our method and demonstrate the utility of combining batch correction with bump hunting in DNA methylation data.
Methods
Our goal is to identify genomic regions associated with disease via genome-scale microarray-based epigenomic data and epidemiological disease-related (covariate/exposure/phenotype) data.
Statistical methods
We formalize the relationship between methylation, disease phenotype, covariates and potential confounding due to batch effects via the following statistical model (Equation 1):
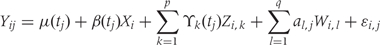
For the epigenomics data, let Yij be the epigenomic measurement (e.g. percentage DNA methylation), appropriately normalized and transformed, at the _j_-th genomic locus (e.g. each vertical scatter of points in Figure 1A) for individual i. The variable tj denotes the location on the genome of the _j_-th locus (i.e. ‘chromosome 2, position 42233500’), and the population baseline level of our epigenomic measurement is μ(tj). In a case–control setting, μ(tj) represents the population-level DNA methylation profile of the controls. Note that in Figure 1A the blue curve is an estimate of μ(t).
We let Xi represent the outcome of interest (like dichotomous cancer status in Figure 1, or a continuous outcome in later examples), and β(tj) measure the association between the outcome of interest Xi and the epigenomic measurement Yij at location tj. Genomic locations of interest are those in which outcome is associated with DNA methylation; i.e. locations tj for which 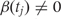 Note that in Figure 1B, the black curve is an estimate of β(t). Potential measured confounders (e.g. sex, age, race) are denoted by the Z_s, and the γ_k(tj) represents the effects of confounder k at locus tj, with each column of Z representing a different confounder. We let W represent potential unmeasured confounders or batch effects, estimated via SVA (described further below), and al,j is the effect of unmeasured confounder l on locus tj. The remaining unexplained variability is represented by
Note that in Figure 1B, the black curve is an estimate of β(t). Potential measured confounders (e.g. sex, age, race) are denoted by the Z_s, and the γ_k(tj) represents the effects of confounder k at locus tj, with each column of Z representing a different confounder. We let W represent potential unmeasured confounders or batch effects, estimated via SVA (described further below), and al,j is the effect of unmeasured confounder l on locus tj. The remaining unexplained variability is represented by 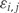 and includes both the variability associated with measurement error as well as natural biological variability. Because biological variance is known to depend on genomic location8,30 we permit the variance
and includes both the variability associated with measurement error as well as natural biological variability. Because biological variance is known to depend on genomic location8,30 we permit the variance 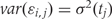 to depend on location tj. We further assume measurement error is a stationary random process with symmetric marginal distribution centered at 0 and allow a general correlation structure.
to depend on location tj. We further assume measurement error is a stationary random process with symmetric marginal distribution centered at 0 and allow a general correlation structure.
A formal definition of regions of biological interest can now be provided as the contiguous intervals 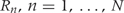 for which for all
for which for all  . These are the genomic regions in which methylation levels at consecutive measured locations are associated with the outcome of interest. Previous work and biological insight suggests that for DNA methylation, β(t) can be modeled as a smooth function of genomic position t since DNA methylation levels for CpGs within 1000 bases have been shown to be significantly correlated6. Since for most of the genome, β(t) = 0, β(t) can be thought of as a straight horizontal line with N bumps. Our goal is to find these bumps, i.e. detect the _Rn_s. We implement a modular approach (Figure 2) with the following four steps: (i) estimate the β(tj) for each tj; (ii) use these to estimate the smooth function β(t); (iii) use this to estimate the regions Rn; and (iv) use permutation tests to assign statistical uncertainty to each estimated region.
. These are the genomic regions in which methylation levels at consecutive measured locations are associated with the outcome of interest. Previous work and biological insight suggests that for DNA methylation, β(t) can be modeled as a smooth function of genomic position t since DNA methylation levels for CpGs within 1000 bases have been shown to be significantly correlated6. Since for most of the genome, β(t) = 0, β(t) can be thought of as a straight horizontal line with N bumps. Our goal is to find these bumps, i.e. detect the _Rn_s. We implement a modular approach (Figure 2) with the following four steps: (i) estimate the β(tj) for each tj; (ii) use these to estimate the smooth function β(t); (iii) use this to estimate the regions Rn; and (iv) use permutation tests to assign statistical uncertainty to each estimated region.
Figure 2.

Step-by-step illustration of our bump-hunting algorithm. (A) Logit-transformed methylation measurements are plotted against the outcome of interest (gestational age) for a specific probe j. A regression line obtained from fitting the model presented in Equation 1 is shown as well. The estimated slope 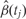 is retained for the next step. (B) For 48 consecutive probes, the estimated are plotted against their genomic location tj. The specific estimated slope from the probe in (A) is indicated by ‘A’ and an arrow. The blue curve represents the smooth estimate
is retained for the next step. (B) For 48 consecutive probes, the estimated are plotted against their genomic location tj. The specific estimated slope from the probe in (A) is indicated by ‘A’ and an arrow. The blue curve represents the smooth estimate 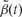 obtained using loess. (C) The smooth estimate from (b) is shown but here with predefined thresholds represented by red horizontal lines. The region for which exceeds the lower threshold is considered a candidate DMR. The area shaded in grey is used as a summary statistic. (D) A null distribution for the area summary statistic described in (c) is estimated by performing using permutations (as described in the text). The histogram summarizes the null areas obtained from permutations and estimates the null distribution. The area obtained from the region shown in (C) is highlighted with an arrow and the label ‘C’. Note that this DMR region is not statistically significant as it can easily happen by chance
obtained using loess. (C) The smooth estimate from (b) is shown but here with predefined thresholds represented by red horizontal lines. The region for which exceeds the lower threshold is considered a candidate DMR. The area shaded in grey is used as a summary statistic. (D) A null distribution for the area summary statistic described in (c) is estimated by performing using permutations (as described in the text). The histogram summarizes the null areas obtained from permutations and estimates the null distribution. The area obtained from the region shown in (C) is highlighted with an arrow and the label ‘C’. Note that this DMR region is not statistically significant as it can easily happen by chance
Note that if we fix j, Equation 1 is a linear regression model. However, because q and the _W_s are unknown, estimating the methylation association parameters β(tj) with the standard least squares approach is not appropriate. Generally, much of the variability observed in high-throughput data is associated with unwanted factors that affect groups of samples in ways which introduce artificial within-sample correlations, as described in a recent review article.28 For example, an unmeasured difference in temperature throughout a day in which samples were processed may result in correlation structures that generate distinct ‘batches’: morning, midday and afternoon samples. In Equation 1, we account for such sources of variability with columns of the W matrix. A well-known statistical technique that uncovers such structures is principal component analysis. In high-throughput experiments, the first few principal components are commonly associated with unwanted sources of variability.28 However, simply removing these may result in unwittingly discarding important biological signal. SVA uses an iterative procedure that simultaneously estimates biological signal of interest, e.g. preserves information on β(tj), as well as effects of unwanted sources of variability.29 Specifically, SVA estimates q (the number of unmeasured confounders) and the columns of W (the confounders themselves) in our model. SVA was originally designed to handle batch effects in gene expression data, although it can also be used with appropriately transformed DNA methylation data, as we,31 and others,32 have demonstrated. With the SVA estimates in place, we use least squares to fit Equation 1 for each tj to produce locus-specific estimates (Figure 2A). For most microarray data, this involves fitting thousands to millions of regression models (see our open source code for details available at rafalab.jhu.edu).
Although for each tj, is an unbiased estimate of β(tj) the assumption that β(tj) is smooth implies we can improve precision with smoothing techniques. We therefore smooth the s using loess,33 a smoother that is robust to outliers, with a smoothing window ranging from 300 to 900 bp and weighting each point based on the standard error obtained in the linear model fit (Figure 2B). We denote the smoothed estimate with (blue line in Figure 2B) to distinguish it from the point-wise estimate (points in Figure 2B). The smoothing window size was motivated by the epigenetics literature6 as well as our own statistical evaluation described in the ‘Simulation’ section.
We then generate candidate regions 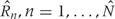 using contiguous runs of measurements for which
using contiguous runs of measurements for which 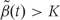 or
or 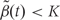 where K is a predetermined threshold (e.g. the 99th percentile of the ). To then assess the statistical uncertainty for each candidate region
where K is a predetermined threshold (e.g. the 99th percentile of the ). To then assess the statistical uncertainty for each candidate region 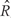 , we use permutation techniques to accommodate the correlated measurement errors, batch effects, and the high-throughput nature of data when estimating the probability that an observed R occurred by chance, given β(tj) = 0 across the genome. We propose two approaches below for generating data with β(tj) = 0 for all j but that retain all other statistical characteristics of the original data, such as batch effects and correlated errors. Thus, any resulting regions identified in these permuted data sets are actually ‘null’ candidate regions occurring by chance. We repeat these procedures hundreds of times to generate a distribution of null candidate regions. To work with scalars, we summarize the strength of evidence for each region with its area, computed with
, we use permutation techniques to accommodate the correlated measurement errors, batch effects, and the high-throughput nature of data when estimating the probability that an observed R occurred by chance, given β(tj) = 0 across the genome. We propose two approaches below for generating data with β(tj) = 0 for all j but that retain all other statistical characteristics of the original data, such as batch effects and correlated errors. Thus, any resulting regions identified in these permuted data sets are actually ‘null’ candidate regions occurring by chance. We repeat these procedures hundreds of times to generate a distribution of null candidate regions. To work with scalars, we summarize the strength of evidence for each region with its area, computed with 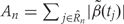 (Figure 2C). This area can be used to rank regions of interest for further investigation. Our two permutation procedures and associated metrics construct a null distribution of area statistics based on the observed data, but under the global null hypothesis. The first approach simply permutes the outcome variable Xi and re-runs the entire bump hunting procedure; all four steps. We do this _B_=1000 times, and for each permutation _b=_1, … , B, we produce a set of null areas
(Figure 2C). This area can be used to rank regions of interest for further investigation. Our two permutation procedures and associated metrics construct a null distribution of area statistics based on the observed data, but under the global null hypothesis. The first approach simply permutes the outcome variable Xi and re-runs the entire bump hunting procedure; all four steps. We do this _B_=1000 times, and for each permutation _b=_1, … , B, we produce a set of null areas 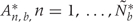 and define empirical _P_-values as the fraction of null areas greater than each observed area. For example, an observed area greater than 95% of the areas obtained from the permutation exercise will be assigned an empirical _P_-value of 0.05. To account for the multiplicity problem introduced by genome-wide screening, we computed false discovery rates (FDRs)34 based on these _P_-values, a standard approach in microarray data analysis. Namely, we use the _P_-values to estimate FDRs and for each candidate region define its _Q_-value as the minimum FDR at which the associated area may be called significant.35 We also report a more conservative uncertainty assessment based on family-wise error rate (FWER)36 protection that computes, for each observed area, the proportion of maximum area values per permutation that are larger than the observed area (Figure 2D).
and define empirical _P_-values as the fraction of null areas greater than each observed area. For example, an observed area greater than 95% of the areas obtained from the permutation exercise will be assigned an empirical _P_-value of 0.05. To account for the multiplicity problem introduced by genome-wide screening, we computed false discovery rates (FDRs)34 based on these _P_-values, a standard approach in microarray data analysis. Namely, we use the _P_-values to estimate FDRs and for each candidate region define its _Q_-value as the minimum FDR at which the associated area may be called significant.35 We also report a more conservative uncertainty assessment based on family-wise error rate (FWER)36 protection that computes, for each observed area, the proportion of maximum area values per permutation that are larger than the observed area (Figure 2D).
Since in this first approach we estimate unmeasured confounders (_W_s) 1000 times, this procedure was time-consuming (for the Tracking Health Related to Environmental Exposures, THREE, data each of the 1000 permutations took 2 h), we developed a second, faster approach based on the application of the bootstrap to linear models of Efron and Tibshirani37 that yielded practically equivalent results for _Q-_values and _P_-values (Supplementary Figure 1, available as Supplementary Data at IJE online). Note that neither procedure requires the model errors to follow a normal distribution to produce valid inference.
Study population
To demonstrate the utility of our method, we applied it to both quantitative and qualitative outcomes. For utility with a binary outcome, we applied it to a published colon cancer dataset20 including eight tumours matched with eight normal tissue samples. To show the method for a quantitative phenotype, we examined the relationship between gestational age at birth and DNA methylation data among 141 newborn cord blood DNA samples from Johns Hopkins Hospital. This study, THREE, has previously been used to correlate carefully measured environmental exposures with anthropomorphic and maternal characteristics.38 The description of this study and the particular methods for this epigenomics project can be found in our companion paper.31
Comprehensive high-throughput arrays for relative methylation microarray
Both data sets presented here contain DNA methylation measurements from the comprehensive high-throughput arrays for relative methylation (CHARM) microarray design. This array has been used to successfully identify regions of differential methylation for cancer20 and stages of differentiation.39,40 Methods for preprocessing this data type have been previously described.8,20,41 The THREE data set used the CHARM 2.0 array design,31 whereas the colon cancer data set used the CHARM 1.0 design.
Pyrosequencing data
DNA methylation levels across three regions identified via our bump hunting method in the THREE study (see our companion paper, Lee et al.31) were also measured via pyrosequencing, the gold standard for validating DNA methylation measurements generated by microarrays. These served as ‘positive controls’ in the assessments of our method. Control probes included in the CHARM array served as ‘negative controls’. These control probes are from regions without CpGs and therefore no methylation which implies we know β(t) = 0.
Simulations
To systematically assess accuracy and precision of our approach to microarray data, we generated DNA methylation data following Equation 1. We created data sets of 100 000 probes in 1000 probe groups (100 probes per group) with similar statistical properties (e.g. autocorrelated DNA methylation profiles) as the observed THREE data on 141 newborns. To emulate the observed correlation in the THREE data, we used an autoregressive, lag 1 [AR(1)] process with coefficient 0.21 and a standard deviation of 0.5. To emulate the presence of outliers we used a _t_-distribution with 5 degrees of freedom to generate the AR(1) innovations.42 The actual gestational ages of the THREE study samples were used as our outcome of interest so that simulated effect sizes were realistic. We emulated 10 genomic regions of interest by letting β(t) > 0 in 10 probe groups. We varied the magnitude β(t) from 0.005 to 0.05 and the region lengths from 5 to 50 consecutive probes.
We simulated 100 data sets per combination and applied our procedure with various choices for the threshold constant K [as a percentile of ]. We also ran our procedure with and without smoothing. All statistical analyses and simulations were performed in the R statistical environment (version 2.13).
Results
As a demonstration of our approach, after normalizing raw data41 and applying the logit transform, we applied our four-step bump hunting method to identify epigenomic regions associated with gestational age at birth. The residuals were symmetric and approximately _t_-distributed (Supplementary Figure 2, available as Supplementary Data at IJE online) complying with the necessary assumptions for Equation 1 and loess smoothing. The method identified three differentially methylated regions (DMRs) at a 5% FDR (and 10% FWER) (Supplementary Figure 3, available as Supplementary Data at IJE online). These regions are biologically interesting and provide insight into late gestational development as we report in our companion paper (see Lee et al.31). These regions were positively validated by bisulfite pyrosequencing, which we used to assess the accuracy of the results obtained by applying our method to the microarray data. To assess precision, we used the CHARM control probes, which measure background and technical signal. We therefore expected no differential DNA methylation at these probes, i.e. β(t) = 0. We compared our procedure with and without the smoothing step and found that smoothing improves precision substantially without affecting accuracy (Supplementary Figure 4, available as Supplementary Data at IJE online).
Simulation studies confirmed that, in general, smoothing is beneficial (Figure 3A and B) when associated epigenomic regions were analogous to the DMRs identified in the real THREE data. However, over-smoothing reduced our ability to detect shorter regions—for example, when β(t) = 0.01 and the width was 10 probes, the optimal smoothing span was in the range of 5–9 consecutive probes (Supplementary Figure 5A, available as Supplementary Data at IJE online), or ~375–675 bp for the CHARM design (there are a median 70 bp between consecutive probes). We also found that higher thresholds (K) for declaring an associated region are preferable, up to a point in which sensitivity decreases. Specifically, when changing the threshold level from the 99th percentile of to the 99.9th, we lost ability to detect true signals. We also confirmed that the estimated FWER agreed with the observed error rate (Supplementary Figure 5B, available as Supplementary Data at IJE online).
Figure 3.

Receiver operating characteristic curves obtained from Monte Carlo simulation. True positive rate is plotted against false-positive rate for various tuning parameters needed for the bump hunting procedure. We examined the performance of three choices for the threshold used to define candidate DMRs. The three choices are represented with line type (solid, dashed, dotted). Specifically we compared the performance of using the 95th, 99th and 99.9th percentile of the . We also compared three choices of smoothing parameters used by loess: no smoothing and smoothing windows of 9 probes (675 bp) and 15 probes (1125 bp). These are represented by colour. We assessed performance in two scenarios. (A) We inserted 10 true DMRs each 10 probes long (~750 bp) with true effect size β = 0.01. (B) As in (A), but true DMRs were 20 probes long (~1500 bp) with the same effect size
The importance of explicitly investigating potential batch effects is best motivated with dichotomous outcome data. For the colon cancer data we computed the distance between each sample based on the raw methylation measurements. We observed strong clustering, which was driven mostly by batch (Figure 4A). To demonstrate how the batch effect, if unaccounted for, can lead to false-positive regions, we applied our bump hunting procedure to the cancer dataset, with processing date (Day 1 vs Day 2) as the covariate of interest. We did not run SVA because we were explicitly looking for batch effects. We found regions as long as 1 kb where methylation differs as much as 30% between batches (Figure 4B). These effect sizes were similar to the effects found with cancer status as the covariate of interest (an example DMR in this dataset is shown in Figure 1A). However, when cancer was defined as the outcome of interest, SVA appropriately dealt with the batch effect by detecting and removing variation due to date (Figure 4C). However, because in this situation, batch and outcome were perfectly balanced, the results obtained by our method were practically the same as the ones previously published.20
Figure 4.

Illustration of batch effects. (A) A multidimensional scaling (MDS) plot of tumour (‘C’ label) and matched normal (‘N’ label) colon mucosa samples, processed during two different dates (green is batch 1 and orange is batch 2). Note the strong horizontal separation between the two batches. Note that the batch variability is stronger than the biological variability represented by the vertical separation between the disease states. (B) The points show methylation measurements from the colon cancer data set plotted against genomic location. Batches one and two are represented by 10 green and 6 orange points. The curves represent the smooth estimate of the batch-level methylation profiles for batch one (green) and two (orange). The horizontal lines represent a false DMR driven by batch. (C) As in (B) but after removing batch effects with SVA
Finally, we used the pyrosequencing data to evaluate the effect of batch removal on the results from THREE. We found that the correlation between the pyrosequencing and CHARM measurements improved with SVA, while precision, measured by the standard deviation of the 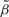 null regions, improved by 50% (Table 1). Note that for DMR #3, the difference was substantial. We also confirmed that unexplained DNA methylation heterogeneity was reduced using SVA in these DMRs (Supplementary Figure 6, available as Supplementary Data at IJE online). Our experience with batch effects is that they affect some regions more than others and this is confirmed here.
null regions, improved by 50% (Table 1). Note that for DMR #3, the difference was substantial. We also confirmed that unexplained DNA methylation heterogeneity was reduced using SVA in these DMRs (Supplementary Figure 6, available as Supplementary Data at IJE online). Our experience with batch effects is that they affect some regions more than others and this is confirmed here.
Table 1.
Batch correction on DNA methylation correlation and slope variability
Discussion
We have presented a general bump hunting framework for association studies based on high-throughput, genome-wide DNA methylation data. We begin with raw high-throughput data and end with regions of interest with appropriate measures of statistical uncertainty. This genome-wide bump hunting approach accommodates the features of quantitative microarray genomics data that have not been previously addressed in GWAS analysis, based on categorical genomic data. The method addresses batch effects, exploits the correlation structure of the microarray data to identify DMRs, and provides a genome-wide measure of uncertainty.
Although we illustrated our statistical methodology on CHARM data, our approach can be applied to other microarray platforms (Table 2). The only requirement is closely spaced measurements across all (or portions of) the genome to facilitate the smoothing process.7 Our approach can also be extended to data from next-generation sequencing (NGS) technology. We are pursuing extensions of this approach to account for binomially distributed data such as those produced by NGS. Furthermore, by using a linear model and modular approach, our method can be easily adapted to accommodate other epidemiological study designs.
Table 2.
Applicability of our approach to other microarray platforms
| Microarray Platform | Median distance between probes (1st and 3rd quartiles) [bp] | No. of probe clusters with seven or more probes | Total no. of probes (% of array) | Do we recommend use of our method? |
|---|---|---|---|---|
| Illumina 27 k | 3505 (542–76 371) | 23 | 0.178 K (<1) | No |
| Illumina 450 k | 300 (78–2395) | 12 502 | 133 K (27) | Yes |
| CHARM 1.0 (2.1M) | 36 (33–36) | All | All | Yes |
| CHARM 2.0 (2.1M) | 70 (66–76) | All | All | Yes |
| Nimblegen CGI+ Promoter (720 K) | 100 (92–114) | 37 238 | 670 K (94) | Yes |
| Nimblegen CGI+ Promoter (385 K) | 100 (95–115) | 30 430 | 365 K (95) | Yes |
While our method, applied to microarray data, successfully identifies epigenomic regions of biological interest, it cannot identify single base changes due to the smoothing step. Although there is some evidence that altered DNA methylation at a specific locus might affect biological processes like transcription factor binding,43,44 the strong correlation between neighbouring CpGs and the concern for many false positives resulting from technical artifacts suggests that smoothing provides statistically and biologically meaningful results.
In general, our framework offers a comprehensive yet flexible approach for identifying epigenetic regions of biological interest in epidemiological studies. While GWAS have been performed on dozens of diseases, few have been able identify a substantial amount of the estimated heritability. We therefore expect many of these studies to explore the possible role of epigenetics in these diseases.14 Since Illumina has recently released a comprehensive yet relatively inexpensive microarray product,45 we expect microarrays to be the technology of choice for these studies. In fact, we expect dozens of large epidemiological studies to use these arrays in the near future. Our bump-hunting approach can be applied to data from these arrays. The results presented here suggest that our approach will outperform the single CpG analyses that have been previously applied on Illumina arrays. Our methodology will therefore be indispensable for the necessary data analysis in the emerging field of epigenetic epidemiology.
Software availability
The R code is available from: http://rafalab.jhsph.edu/software.html
Supplementary Data
Supplementary Data are available at IJE online.
Funding
This work was partially funded by the National Institute of Health (grant numbers R01 GM083084, R01 RR021967, P50 HG003233; R01ES017646).
Supplementary Material
Supplementary Data
Acknowledgements
We thank Terry Speed for pointing out the connection between our method and bump hunting.
Conflict of interest: None declared.
KEY MESSAGES.
- Genome-wide DNA methylation measurements will be ubiquitous in the emerging field of epigenetic epidemiology.
- Systematic errors, unwanted variability and multiple testing issues make it necessary to apply rigorous statistical methodology.
- We have developed bump-hunting methodology useful for finding loci of biological interest in the context of DNA methylation studies. Our approach can be applied to a wide range of technologies including Illumina's Infinium HumanMethylation450 BeadChip.
References
- 1.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 2.Mockler TC, Chan S, Sundaresan A, Chen H, Jacobsen SE, Ecker JR. Applications of DNA tiling arrays for whole-genome analysis. Genomics. 2005;85:1–15. doi: 10.1016/j.ygeno.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Arking DE, Pfeufer A, Post W, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–51. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 4.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kottgen A, Glazer NL, Dehghan A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet. 2009;41:712–17. doi: 10.1038/ng.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eckhardt F, Lewin J, Cortese R, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irizarry RA, Ladd-Acosta C, Carvalho B, et al. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–90. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaffe AE, Feinberg AP, Irizarry RA, Leek JT. Significance analysis and statistical dissection of variably methylated regions. Biostatistics. 2012;13:166–78. doi: 10.1093/biostatistics/kxr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–10. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- 10.Doi A, Park IH, Wen B, et al. Differential methylation of tissue-and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–53. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.NOVA. Ghost in your Genes. c1996–2007. http://www.pbs.org/wgbh/nova/genes/ [Google Scholar]
- 12.Cloud J. Why your DNA isn't your destiny. Time Magazine. 18 January 2010. [Google Scholar]
- 13.Schübeler D. Epigenomics: methylation matters. Nature. 2009;462:296–97. doi: 10.1038/462296a. [DOI] [PubMed] [Google Scholar]
- 14.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 16.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma S, Kelly T, Jones P. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaminskas E, Farrell A, Abraham S, et al. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res. 2005;11:3604–08. doi: 10.1158/1078-0432.CCR-04-2135. [DOI] [PubMed] [Google Scholar]
- 19.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 20.Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo-and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wegman EJ. A note on the estimation of the mode. Ann Math Stat. 1971;42:1909–15. [Google Scholar]
- 23.Grund B, Hall P. On the minimisation of Lp error in mode estimation. Ann Stat. 1995;23:2264–84. [Google Scholar]
- 24.Poline J, Worsley K, Evans A, Friston K. Combining spatial extent and peak intensity to test for activations in functional imaging. Neuroimage. 1997;5:83–96. doi: 10.1006/nimg.1996.0248. [DOI] [PubMed] [Google Scholar]
- 25.Schwartzman A, Gavrilov Y, Adler R. Harvard University Biostatistics Working Paper Series; 2009. Peak detection as multiple testing; p. 120. [Google Scholar]
- 26.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11:733–39. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–35. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci USA. 2010;107(Suppl. 1):1757–64. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee H, Jaffe AE, Feinberg JI, et al. DNA Methylation shows genome-wide association of NFIX, RAPGEF2, and emph MSRB3 with Gestational Age at Birth. Int J Epidemiol. 2012;41:188–99. doi: 10.1093/ije/dyr237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics. 2011;27:1496–505. doi: 10.1093/bioinformatics/btr171. [DOI] [PubMed] [Google Scholar]
- 33.Cleveland WS. Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc. 1979;74:829–836. [Google Scholar]
- 34.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- 35.Storey JD. The optimal discovery procedure: a new approach to simultaneous significance testing. J Roy Stat Soc B. 2007;69:347–68. [Google Scholar]
- 36.Shaffer JP. Multiple hypothesis testing. Annu Rev Psychol. 1995;46:561–84. [Google Scholar]
- 37.Efron B, Tibshirani RJ. An Introduction to the Bootstrap. New York, NY: Chapman and Hall; 1993. p. 436. [Google Scholar]
- 38.Apelberg BJ, Goldman LR, Calafat AM, et al. Determinants of fetal exposure to polyfluoroalkyl compounds in Baltimore, Maryland. Environ Sci Technol. 2007;41:3891–97. doi: 10.1021/es0700911. [DOI] [PubMed] [Google Scholar]
- 39.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–90. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji H, Ehrlich LI, Seita J, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aryee MJ, Wu Z, Ladd-Acosta C, et al. Accurate genome-scale percentage DNA methylation estimates from microarray data. Biostatistics. 2011;12:197–210. doi: 10.1093/biostatistics/kxq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waller LA, Gotway CA. Applied Spatial Statistics for Public Health Data. Hoboken, NJ: John Wiley & Sons; 2004. xviii, 494. [Google Scholar]
- 43.Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 1991;64:1123–34. doi: 10.1016/0092-8674(91)90267-3. [DOI] [PubMed] [Google Scholar]
- 44.Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993;3:226–31. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 45.Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data