Anti-Hepatitis C Virus Activity and Toxicity of Type III Phosphatidylinositol-4-Kinase Beta Inhibitors (original) (raw)
Abstract
Type III phosphatidylinositol-4-kinase beta (PI4KIIIβ) was previously implicated in hepatitis C virus (HCV) replication by small interfering RNA (siRNA) depletion and was therefore proposed as a novel cellular target for the treatment of hepatitis C. Medicinal chemistry efforts identified highly selective PI4KIIIβ inhibitors that potently inhibited the replication of genotype 1a and 1b HCV replicons and genotype 2a virus in vitro. Replicon cells required more than 5 weeks to reach low levels of 3- to 5-fold resistance, suggesting a high resistance barrier to these cellular targets. Extensive in vitro profiling of the compounds revealed a role of PI4KIIIβ in lymphocyte proliferation. Previously proposed functions of PI4KIIIβ in insulin secretion and the regulation of several ion channels were not perturbed with these inhibitors. Moreover, PI4KIIIβ inhibitors were not generally cytotoxic as demonstrated across hundreds of cell lines and primary cells. However, an unexpected antiproliferative effect in lymphocytes precluded their further development for the treatment of hepatitis C.
INTRODUCTION
Chronic hepatitis C virus (HCV) infection, a major cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma, afflicts approximately 3% of the world's population (24). The current standard of care for treating hepatitis C is pegylated interferon and ribavirin, which shows poor tolerability and is capable of achieving a sustained viral response in only half of genotype 1 patients (7). Two NS3 protease inhibitors, telaprevir and boceprevir, have been approved recently, and additional direct-acting antivirals are in clinical development. While triple therapy with interferon, ribavirin, and a protease inhibitor increases the percentage of patients showing a sustained viral response to 75% and can shorten the treatment time, it still has limitations: only genotype 1 patients are responsive, side effects (such as anemia) prevent the use in transplant patients, and the inconvenient dosing schedule (three times a day) might cause noncompliance. Development of viruses resistant to direct antivirals occurs very rapidly and leads to relapse and viral breakthrough. A possible exception might be nucleoside inhibitors, since viruses with resistance mutations are not viable. We therefore executed high-throughput small interfering RNA (siRNA) screens in order to identify novel cellular targets for the treatment of HCV. Type III phosphatidylinositol-4-kinases (PI4KIIIs) were identified from these studies and in screens performed in other laboratories (3, 4, 20–22).
Mammalian cells express a large number of lipid kinases, including four enzymes that phosphorylate phosphatidylinositol at position four of the inositol ring, the phosphatidylinositol-4-kinases (PI4Ks). Lipid kinases are involved in multiple functions of the cell, of which phosphatidylinositol 3,4,5-trisphosphate (PIP3) signaling is the most thoroughly investigated process. The four PI4Ks (type II α and β and type III α and β) are localized to different sites in the cell by protein-protein interactions and are regulated by different proteins. Thus, the four enzymes are likely to have individual roles other than synthesis of the PIP3 or PI(4,5)P2 precursors. In the past it has been difficult to analyze specific roles of each enzyme for several reasons. Most importantly, no specific inhibitors existed for the individual PIKs. A classification of class II and III PI4Kα or -β is based on the sensitivity to adenosine, phenylarsine oxide, and wortmannin (2). Second, many studies were performed in yeast for ease of genetically manipulating the cells. There are, however, only three PI4Ks expressed in yeast, and only two are enzymatically active (2). Last, quantifying the enzymatic product [PI(4)P] generated by any one specific PI4K is quite challenging, as separation of PI(4)P from other monophosphates such as PI(5)P is technically very difficult (17). Indeed, the phosphorylated lipids are kept in a balance by phosphatases and may exist for only short periods of time in the cell (14). Taken together, the cellular functions of the PI4IIIKs have not been thoroughly characterized, and thus determining the safety of inhibiting these host enzymes was essential to evaluate their therapeutic potential.
A small-molecule high-throughput screen was initiated (Novartis compound library, ∼1.5 million compounds) in order to identify small molecules that inhibit PI4KIIIβ. Subsequent medicinal chemistry on the screen hits and on previously disclosed PI3K inhibitors produced highly specific and potent small-molecule inhibitors suitable for in vitro and in vivo characterization (data not shown). This report describes the in vitro and in vivo characterization of the antiviral activity and host cell effects associated with inhibition of PI4KIIIβ.
MATERIALS AND METHODS
Compounds and siRNAs.
All PI4KIIIβ inhibitors were synthesized at Novartis. The compounds employed in the mode-of-action studies are listed in Table 1. Pools of four siRNAs for PI4KIIIα, PI4KIIIβ, an siRNA control, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were purchased from Dharmacon. The final siRNA concentration was 25 nM.
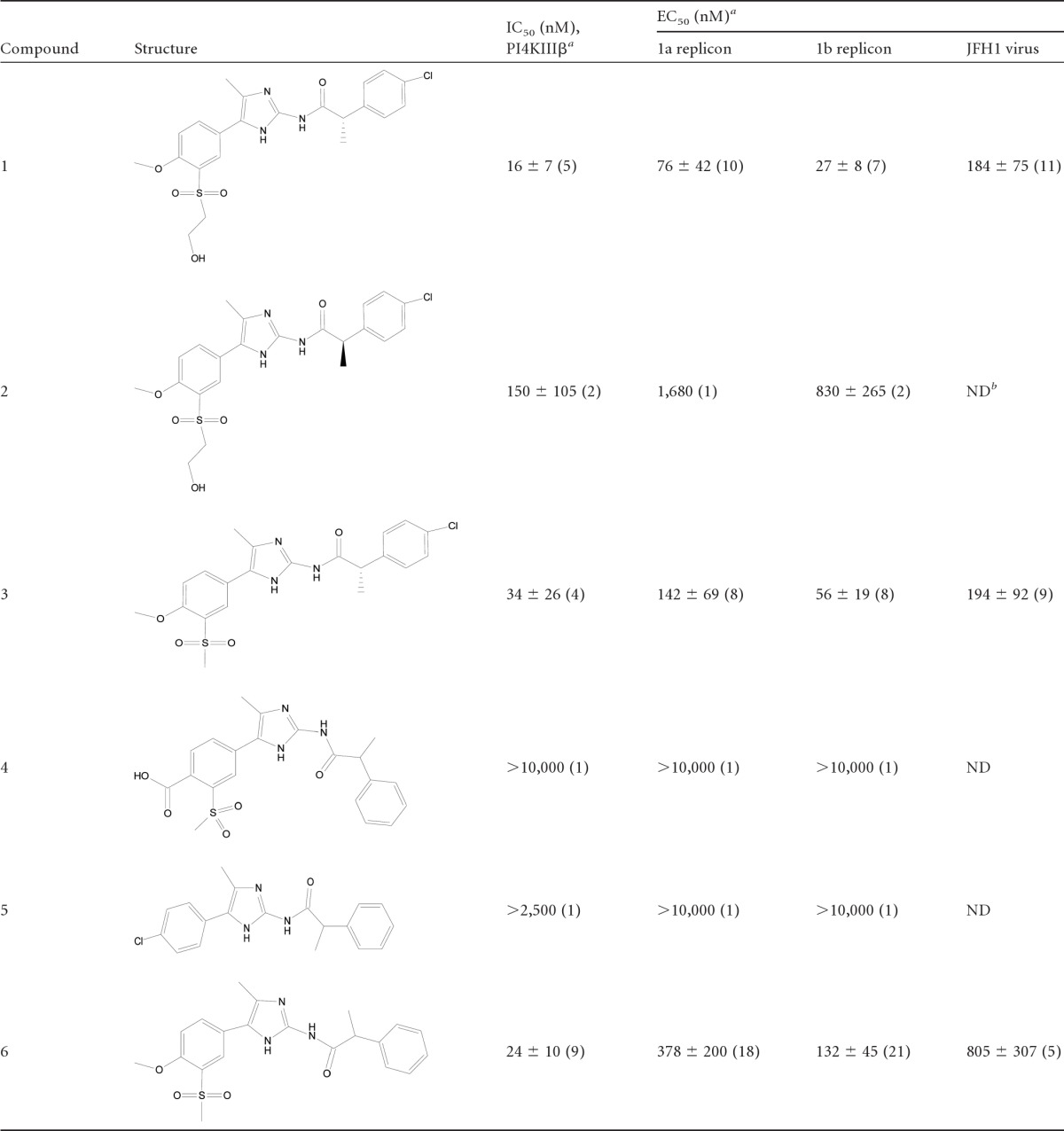
Table 1.
Structures, PI4KIIIβ enzyme activities, and potencies in replicon and virus assays of the compounds described in this study
Enzyme assay.
PI4KIIIβ activity was analyzed as described previously (4). The Novartis kinase panel includes 59 protein and 8 lipid kinase assays.
Analysis of anti-HCV activity.
Huh7 cells containing subgenomic genotype 1b replicons were licensed (ReBLicon GmbH, Germany). Huh7 cells containing subgenomic genotype 1a replicons and JFH1 HCV in cell culture (HCVcc) were constructed in-house. Compound activity (50% effective concentration [EC50]) was measured as firefly or Renilla luciferase activity for replicons and JFH1 HCVcc, respectively, using commercial kits (Promega) as described previously (4).
Analysis of cytotoxicity.
Viability of cells has been analyzed in parallel to the compound susceptibility test as described previously (4). Dehydrogenase activity of viable cells was estimated using the Cell Titer 96 AQueous nonradioactive cell proliferation assay (Promega) following the instructions of the manufacturer.
The concentration-dependent effects of compounds on the viabilities of a broad range of cell lines (∼450 transformed cell lines) and primary human foreskin fibroblasts were tested using the CellTiterGlow assay (Promega) as described previously (23).
Selection of resistant replicon cell lines.
Huh7 cells containing subgenomic genotype 1b replicons (ReBLicon GmbH, Germany) were treated in 6-well plates with 1×, 2×, 5×, and 10× the EC50 of compound 6. After several passages, cells in the well which allowed growth of single colonies were further amplified in T25 flasks using the same compound concentration followed by an increased concentration until the cells grew consistently. The susceptibility to compound 6 was analyzed when the cells were able to grow in medium with ∼75× the EC50 (10 μM) and ∼150 the EC50 (20 μM) of the compound.
In vitro insulin secretion assays. (i) Insulin secretion in MIN6 cells after siRNA-mediated depletion of PI4K.
Two independent experiments were conducted. Min6 cells were seeded in 24-well culture plates (3 × 105 cells/well). After 24 h, the cells were transfected with 100 nM siRNA using Lipofectamine 2000 in Dulbecco modified Eagle medium (DMEM)–12.5 mM glucose–1.5% fetal bovine serum (FBS). After 72 h, the cells were washed three times with Krebs-Ringer bicarbonate buffer containing 109 mM NaCl, 4.6 mM KCl, 5 mM NaHCO3, 2 mM CaCl2, 0.15 mM Na2HPO4, 0.4 mM KH2PO4, 1 mM MgSO4, 20 mM HEPES (pH 7.4), and 0.05% fatty-acid-free (FAF) bovine serum albumin (BSA) and preincubated in the identical buffer for 30 min at 37°C. One aliquot of cells was further incubated in Krebs-Ringer buffer, and a second aliquot was stimulated with 20 mM glucose in the same buffer for 30 min at 37°C. Supernatants were collected and centrifuged for 2 min at 2,000 rpm. The cell-free supernatants were transferred to a new vial and diluted 5-fold prior to insulin secretion measurements using an Ultra-Sensitive Rat Insulin enzyme-linked immunosorbent assay (ELISA) kit (Crystal Chem, Inc.). Cellular mRNA was isolated by using a TurboCapture mRNA kit (Ambion), and cDNA was generated by using a high-capacity cDNA archive kit (Applied Biosystems). Knockdown of target mRNA was confirmed by using a TaqMan gene expression assay (Applied Biosystems) according to the manufacturer's instructions. The cDNA was quantified using premixed Applied Biosystems probes and primers (PI4KIIIα, Mm01344908_m1; PI4KIIIβ, Mm00660064_m1; and GAPDH, Mm03302249_g1) and a real-time PCR system (Applied Biosystems 7900HT).
(ii) Insulin secretion in INS-E1 cells treated with PI4KIIIβ inhibitors.
A selective PI4KIIIβ inhibitor and a structurally similar compound lacking PI4KIIIβ and replicon activity were tested at 1 and 10 μM in a glucose-stimulated insulin secretion (GSIS) assay. Mean values and standard errors of the means SEMs were calculated from triplicate samples.
INS-1E cells (passage 71) were plated in poly-d-lysine-coated 96-well plates at 50,000 cells per well in RPMI medium (Invitrogen) containing 11 mM glucose and 5% FBS. All incubations were done at 37°C and 5% CO2. After 3 days, compounds were added to the cells in fresh medium. Forty-eight hours later, the medium was changed to contain 5 mM glucose and the compounds were added again. After overnight incubation, the GSIS assay was performed. Cells were washed twice with HEPES-buffered salt solution (HBSS) containing 3 mM glucose and incubated for 2 h with HBSS and 3 mM glucose plus compounds. The medium was replaced with HBSS containing 2.8, 7.0, or 16.7 mM glucose and incubated for 2 h. The medium was aspirated from cells, and insulin levels were estimated using a rat insulin ELISA kit (Mercodia).
IPGTT in rats.
Eight-week-old male Sprague-Dawley (SD) rats were fasted for 4.5 h prior to the intraperitoneal (i.p.) glucose tolerance test (IPGTT). Animals were divided into groups administered vehicle (n = 8) or compound (n = 8), with their mean baseline plasma glucose values matched prior to the treatment. The rats were orally dosed with vehicle or compound (30 mg/kg) 2.5 h prior to IPGTT. Compounds were dissolved in vehicle containing 6% 1 N HCl, 18% polyethylene glycol (PEG) 300, 9% Cremophor EL, 1% 1 N NaOH, and 100 mM citrate (pH 5). The rats were injected i.p. with glucose solution at 2 g/kg. Tail blood samples were collected at basal time, at 2 h after vehicle or compound treatment before the IPGTT, and at 3, 8, 15, 30, 60, and 90 min after the glucose load for plasma glucose and insulin measurements. Tail blood samples were collected at 2 and 4 h after compound dose, and pancreas, liver, epididymal fat, and gastrocnemius muscle samples were harvested after the 4-h blood sample collection to analyze compound levels.
The animal study was conducted under a protocol approved by Novartis Animal Care and Use Committee. All procedures in this study were in compliance with Animal Welfare Act regulations 9 CFR parts 1, 2, and 3 and other guidelines.
hMLR assay.
The human mixed-lymphocyte reaction (hMLR) assay was performed according to standard procedures (8). Briefly, peripheral blood mononuclear cells (PBMC) were isolated on Ficoll from buffy coat with unknown HLA type (Kantonspital, Basel, Switzerland). Cells were kept at 2 × 107 cells/ml (90% fetal calf serum, 10% dimethyl sulfoxide [DMSO]) in cryotubes in liquid nitrogen until use. Cells from three different donors (A, B, and C) were thawed, washed, and counted. In each experiment, three individual two-way reactions (A-B, A-C, and B-C) were set up by mixing cells from two donors at a 1:1 ratio. Triplicates of the cell mixtures (4 × 105 cells/0.2 ml) were cultured in the presence of compound serial dilutions for 6 days at 37°C and 5% CO2 in RPMI medium supplemented with 1 mM Na pyruvate, minimal essential medium (MEM) nonessential amino acids, 0.5% 2-mercaptoethanol, 2 mM l-glutamine, 100 μg/ml kanamycin, 20 μg/ml Bacto asparagine, 5 μg/ml human insulin, 40 μg/ml human transferrin, and 10% fetal calf serum. During the last 16 h, [3H]thymidine (1 μCi/0.2 ml) was added, and proliferation was determined by measuring incorporation of radioactive nucleotide into DNA after breaking the cells and washing with water using a harvester.
Murine bone marrow proliferation (BMP) assay.
Bone marrow cells were isolated from femurs and tibiae of both hind legs of CBA mice (BRL, Switzerland) by flushing with ∼3 ml of Hanks' balanced salt solution. The cells were washed in cold RPMI medium supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 μM 2-mercaptoethanol and resuspended to 5 × 105 cells/ml in the same medium plus 1 ng/ml interleukin-3 (IL-3). Duplicates of seven 3-fold compound dilutions were prepared in 50 μl in a 96-well plate, and 50-μl cell suspensions (2.5 × 104 cells) were added to each well. Cells without compounds were used as high controls, and cells without IL-3 and compounds were used as low controls. The cells were incubated for 4 days at 37°C and 5% CO2. During the last 4 h, [3H]thymidine (1 μCi/well) was added, and proliferation was determined by measuring incorporation of radioactive nucleotide into DNA in a Trilux 1450 instrument (Perkin-Elmer, Schwarzenbach, Switzerland) after harvesting and washing of cells using a TomTec 9600 MACH III harvester (TomTec, Hamden, CT).
Distinction between proliferation and signaling.
The effects of PI4KIIIβ inhibitors on proliferation, cytokine production, and activation markers (CD25, CD69, and CD86) was analyzed in murine spleen T cells and BALB/c mouse B cells after activation. Cells were grown in RPMI 1640 (Gibco) supplemented with 1:100-diluted Zell Shield (Minerva Biolabs), 50 μM 2-mercaptoethanol (Gibco), 1:100-diluted ITS (insulin-transferrin-selenium supplement; Gibco), 1:100-diluted nonessential amino acids (Gibco), and 10% fetal clone I serum (HyClone). Cells were isolated from 8- to 12-week-old female BALB/c mice (IFFA Credo). The spleens were taken out and homogenized with a gentleMACS dissociator. After three washes with RPMI 1640, T and B cells were purified with magnetic beads by negative selection according to the manufacturer's protocol using the EasySep mouse T-cell enrichment kit and EasySep negative-selection mouse B-cell enrichment kit (Stemcell). The cells were washed with medium and counted, and the cell number was adjusted to 106 cells/ml or 107 cells/ml RPMI for T and B cells, respectively.
For T-cell stimulation, 96-well plates (Costar) were coated overnight at room temperature either with 50 μl/well anti-mouse CD3 145-2C11 (10 μg/ml) for T-cell receptor (TCR) triggering alone or with anti-mouse CD3 145-2C11 (1 μg/ml) and anti-mouse CD28 PV-1 (5 μg/ml) (both from SBA) for costimulation. The plates were washed 3 times with 200 μl phosphate-buffered saline (PBS) under sterile conditions before addition of compounds and cells. Compound dilutions were done in duplicates in precoated plates starting at a concentration of 2 × 10−5 M, and then with 1:3 dilutions in RPMI. Thereafter, 100 μl of the cell suspension was added and incubated in a humidified incubator at 37°C and 10% CO2. After 20 h of stimulation, cells were taken for CD25 and CD69 surface marker analysis by fluorescence-activated cell sorting (FACS). IL-2 and gamma interferon were analyzed in culture supernatants using commercial ELISA kits (R&D Systems) after 48 h.
For the analysis of B-cell markers, 100 μl of B-cell suspension was added to the compound dilutions and then stimuli were added: either rat anti-mouse IgM b-7-6 (5 μg/ml; Novartis) plus 250 U/ml mouse IL-4 (CHO-mouse IL-4 Kl-5; Novartis) or mouse CD40L (chimeric mouse CD40L-C8α fusion protein) and 10% supernatant from cell line 8-40-19 plus IL-4 or lipopolysaccharide (LPS) (100 μg/ml; Difco). The cells were incubated in a humidified incubator at 37°C and 10% CO2. After 20 h, cells were taken for CD86 and CD69 surface marker analysis by FACS. IL-6 and IL-10 were analyzed in culture supernatants using commercial kits (R&D Systems) after 48 h.
Proliferation of T and B cells was analyzed on day 3 by treatment with [3H]thymidine (Amersham; 1 μCi in 50 μl RPMI 1640) for 6 h. Thereafter, the plates were harvested and the thymidine incorporated into DNA estimated by measuring radioactivity in a β-plate counter (Wallac).
Fifty percent inhibitory concentrations (IC50s) were extrapolated from titration curves generated with the means of each concentration point (n = 2) using Xlfit.
Rat SRBC assay.
T-cell-mediated B-cell activation and antibody formation in response to an injection of sheep red blood cells (SRBCs) into rats (n = 4 for each group) were assessed ex vivo via plaque formation in an antigen-antibody-agar mix (15). Compound 1 was dosed in 10% 0.1 N NaOH, 20% PEG 300, 10% Solutol HS15, and 100 mM citrate buffer to rats at 50 mg/kg twice a day (BID) and 100 mg/kg four times a day (QD) for 4 days. Blood was drawn and the plasma level of compound analyzed 2 h after the last compound was dosed by liquid chromatography-mass spectrometry (LC/MS). Data are presented as means ± SEM. Analysis of data was done using analysis of variance (ANOVA) followed by Dunnett's multiple comparison. Graph Pad Prism was used for graphics and statistical analysis.
The animal experiment was done in strict adherence to the Swiss law for animal protection and was approved by the Veterinary Office, Basel (animal license no. 1182).
RESULTS
A high-throughput screen of the Novartis compound library was performed, and the lead molecules were optimized. Small molecules of several different chemotypes were identified as selective inhibitors of type III PI4Kβ with strong inhibitory effects on genotype 1a and b replicons and the JFH1 virus. PI4KIIIβ inhibition generally correlated with anti-HCV activities (i.e., replicon EC50). This study concentrated on aminoimidazole inhibitors exemplified by compounds shown in Table 1, which varied in potency (16 nM to >10,000 nM). Compound 1, with the strongest PI4KIIIβ-inhibitory activity, also remained the most potent HCV inhibitor, whereas compounds 4 and 5, of the same scaffold, which were inactive in the kinase assay, also lost activity in the replicon assay. Cell penetration and protein binding can affect the correlation of enzymatic and cellular activities. The correlation became, for instance, weaker when protein binding was taken into account by adding 40% human serum to the replicon assay mixture (data not shown). The compounds were inactive on other lipid kinases, including type III PI4Kα, the type II PI4Ks, and type I to III PI3Ks, and on 59 protein kinases (see Table S1 in the supplemental material). Compounds 1 and 3 were additionally screened in the Ambit (San Diego, CA) kinase panel at a 10 μM compound concentration (binding competition assay), and no significant activity on any other kinases was observed (data not shown). Thus, the compounds were potent inhibitors of type III PI4Kβ and selective over all other kinases measured.
Since cellular targets are reported to provide a higher barrier for the development of resistance, we characterized the resistance to PI4K inhibition (12, 18). A genotype 1b replicon-containing cell line was treated with increasing amounts of compound 6. The cells grew as well as wild-type cells at 10 μM and 20 μM compound 6 after 38 and 53 days of treatment, respectively. The 50% cytotoxic concentration (CC50) of compound 6 was estimated to be >31.6 μM (48-h assay). The EC50 shifts were small (approximately 3-fold and 5-fold) (Fig. 1).
Fig 1.
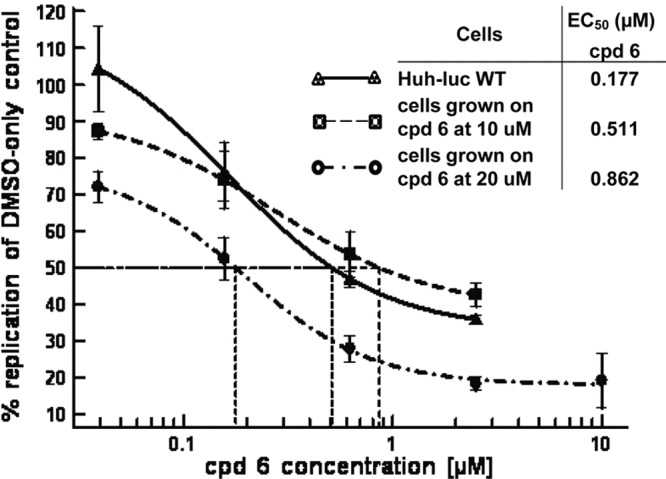
Incremental development of resistance to compound (cpd) 6 causes low EC50 shifts. Susceptibility curves were plotted and EC50s (means) calculated using XlFit from triplicate samples for the wild type (WT) and five replicas for the resistant cell lines.
PI4KIIIβ had also previously been implicated in secretion processes, in particular in insulin secretion (6, 9). Since such an effect could be counterindicative for HCV treatment, siRNA depletion and compound-mediated inhibition of PI4KIIIβ were characterized. First, the effect of siRNA depletion was tested in a murine pancreatic beta cell line (MIN6). No effect was observed in two independent experiments (Fig. 2). Next, insulin levels were analyzed in a rat insulinoma cell line (INS-E1) after compound treatment. Again, there was no significant effect observed for the PI4KIIIβ inhibitor (Fig. 3). The activity at 10 mM of a close analog lacking PI4KIIIβ activity is either an artifact or an off-target effect of this compound. Finally, an in vivo glucose tolerance test was conducted in rats at plasma compound levels that were inhibitory for replicon and virus (i.e., the EC50). Neither insulin secretion nor glucose metabolism was changed upon treatment with compound 2 (Fig. 4). Taken together, the results of the three studies described above indicate that type III PI4Kβ is not involved in the regulated secretion of insulin in rodents.
Fig 2.

siRNA-mediated depletion of PI4KIIIβ has no effect on basal or glucose-stimulated insulin secretion in Min6 cells. The insulin levels were not significantly changed when the PI4KIIIβ mRNA concentration was reduced to 10 to 20%. The x axis values indicate the glucose concentration in mM and the siRNA targets. Shown are mean values of triplicates ± SEMs from a representative experiment.
Fig 3.

Inhibition of PI4KIIIβ does not change glucose-stimulated insulin secretion in INS-E1 cells. Compound 6, a potent PI4KIIIβ inhibitor, and compound 5, a structural analog lacking PI4KIIIβ activity, were used to treat INS-E1 cells stimulated to different degrees with glucose. Mean secreted insulin concentrations and SEMs were calculated from triplicate samples.
Fig 4.

PI4KIIIβ inhibition does not affect insulin secretion or glucose metabolism in an in vivo glucose tolerance test in rats. Fasted rats (n = 8 per group) were given an oral dose of compound or vehicle at 2.5 h before an intraperitoneal injection of glucose. Blood was drawn from the tails to analyze glucose, insulin, and compound levels. (A and B) Glucose and insulin levels in compound- and vehicle-treated rats were measured at 0 h (basal) and 2 h (A) and at short time intervals (B). (C and D) The area under the curve (AUC) for glucose and insulin during the glucose tolerance test was calculated using integrated area of excursion above the baseline (C), and the compound concentration was estimated at 0, 2, and 4 h in plasma and at 4 h in tissues (D). Graphs and columns show mean values for 8 animals ± SEMs.
In parallel to the screen for replicon activity, cytotoxicity was analyzed for each compound in the replicon-containing Huh7 cells using a commercial MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)] assay (CellTiter 96 Aqua 5000 ASYS; Promega) which proved to be more sensitive than the commercial Cell TiterGlow assay (Promega). Potent PI4KIIIβ inhibitors (e.g., compound 6 [IC50 = 23 nM]) did not affect ATP production at 30 μM, indicating that they were not cytotoxic in these cells. Nine compounds were also tested in a concentration-dependent manner in ∼450 transformed cell lines and, remarkably, showed no cytotoxic potential (23). Finally, to exclude an effect on cell viability in nontransformed cells, a set of ∼150 compounds was tested in primary human foreskin fibroblasts (ATCC SCRC-1041), again without detection of cytotoxicity as measured by ATP levels (data not shown). In conclusion, PI4Kβ selective inhibitors did not appear to be generally cytotoxic. However, when the potent compounds were evaluated in the human mixed-lymphocyte reaction (MLR) assay and murine bone marrow proliferation (BMP) assay, surprisingly, the compounds were quite active in both assays (Table 2). The data also correlated with PI4Kβ inhibition and the antiproliferative effect in lymphocytes, as compound 1 exhibited the strongest activities in both assays whereas compound 4 was inactive. Furthermore, to determine if the growth inhibition occurs on the level of signal transduction (e.g., PI4KIIIβ could create the precursor molecule for the PI3K cascade) or directly on proliferation, activation markers and cytokine production were analyzed in mouse splenic T cells and B cells. Levels of CD69/CD25 and CD69/CD86 in T cells and B cells, respectively, were unchanged while activation markers were strongly reduced (Tables 3 to 5). This indicated that proliferation and not signaling was affected by PI4KIIIβ inhibition.
Table 2.
Correlation of compound activity in the PI4KIIIβ enzymatic assay and the mixed-lymphocyte reaction and bone marrow proliferation assays_a_
| Compound | IC50 (nM)a | ||
|---|---|---|---|
| PI4KIIIβ | MLR | BMP | |
| Cyclosporine | ND_b_ | 11/11 (1,011) | 1,900/671 (14) |
| 1 | 16 ± 7 (5) | 96 ± 49 (3) | 43 (1) |
| 2 | 150 ± 105 (2) | ND | 1,390 ± 625 (2) |
| 3 | 34 ± 26 (4) | 77 ± 22 (3) | 159 ± 56 (6) |
| 4 | >10,000 (1) | >10,000 (5) | >10,000 (3) |
| 6 | 24 ± 10 (9) | 63 ± 31 (2) | 589 ± 59 (2) |
Table 3.
PI4KIIIβ inhibition inhibits proliferation of T cells and secretion of chemokines but not the production of signaling molecules_a_
| Compound or assay specification parameter | IC50 (nM) with: | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Anti-CD3 (10 μg/ml) | Anti-CD3 (1 μg/ml) + anti-CD28 (5 μg/ml) | |||||||||
| Proliferation | IL-2 | IFN-γ | CD69 | CD25 | Proliferation | IL-2 | IFN-γ | CD69 | CD25 | |
| Compounds | ||||||||||
| 3 | 325 | 531 | 253 | >5,000 | 5,113 | 146 | 88 | 62 | >5,000 | >5,000 |
| 4 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | 5,381 | 4,769 |
| Cyclosporine | 13 | 7 | 3 | 2,100 | 136 | 6 | <1.4 | <1.4 | >1,000 | >1,000 |
| DMSO | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 |
| Assay specification parameters | ||||||||||
| Low control (cpm/ratio) | 460 | 0.02 | 0.05 | 9 | 14 | 110 | 0.05 | 0.04 | 9 | 14 |
| High control (cpm/ratio) | 5,862 | 0.10 | 0.20 | 18 | 21 | 23,423 | 0.23 | 0.14 | 11 | 17 |
| S/I | 10 | 5 | 4 | 2 | 1.5 | 213 | 5 | 3.5 | 1.2 | 1.3 |
Table 5.
PI4KIIIβ inhibition does not inhibit production of signaling molecules on B cells_a_
| Compound | IC50 (nM) | |||||
|---|---|---|---|---|---|---|
| B cells, % CD69+ | B cells, % CD86+ | |||||
| Anti-IgM/IL-4 | CD40/IL-4 | LPS | Anti-IgM/IL-4 | CD40/IL-4 | LPS | |
| 3 | >5,000 | 3,209/>5,000 | 747/2,309 | >5,000 | >5,000 | 492/2,758 |
| 4 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 | >5,000 |
| MMF | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 | 9,832 |
Table 4.
PI4KIIIβ inhibition inhibits proliferation of B cells and secretion of chemokines_a_
| Compound | B-cell proliferation (IC50, nM) with: | ||
|---|---|---|---|
| Anti-IgM/IL-4 | CD40L/IL-4 | LPS | |
| 3 | 312 | 75 | 108 |
| 4 | >5,000 | >5,000 | >5,000 |
| MMF | 132 | 198 | 178 |
To assess the correlation of in vitro assay results with in vivo effects and the magnitude of the therapeutic window, compound 1 was tested in a rat sheep red blood cell (SRBC) assay. The SRBC assay is routinely used to measure effects on B-cell proliferation and antibody production (15). Compound pharmacokinetics were first evaluated in rats (escalating doses of 10, 30, and 100 mg/kg), and oral doses (as predicted by modeling) which yielded plasma levels with _C_trough ∼5-fold above the serum adjusted replicon EC50 (50 mg/kg BID) and _C_trough at the serum adjusted replicon EC50 (100 mg/kg QD) were selected. The replicon EC50 was adjusted for serum binding by addition of 40% human serum to the regular replicon assay mixture. Compound 1 was orally dosed at 50 mg/kg BID and at 100 mg/kg QD for 4 days, and the spleen cells were harvested and tested in vitro for their ability to generate antibodies to the antigen (sheep rat blood cells). The results shown in Fig. 5 demonstrate that compound 1 at 100 mg/kg reduced the antibody production to a similar extent as the positive control, cyclosporine. This result, together with the evidence shown in Tables 3 to 5, indicates that compound 1 inhibits B-cell proliferation in vivo, which leads to a reduced antibody titer.
Fig 5.
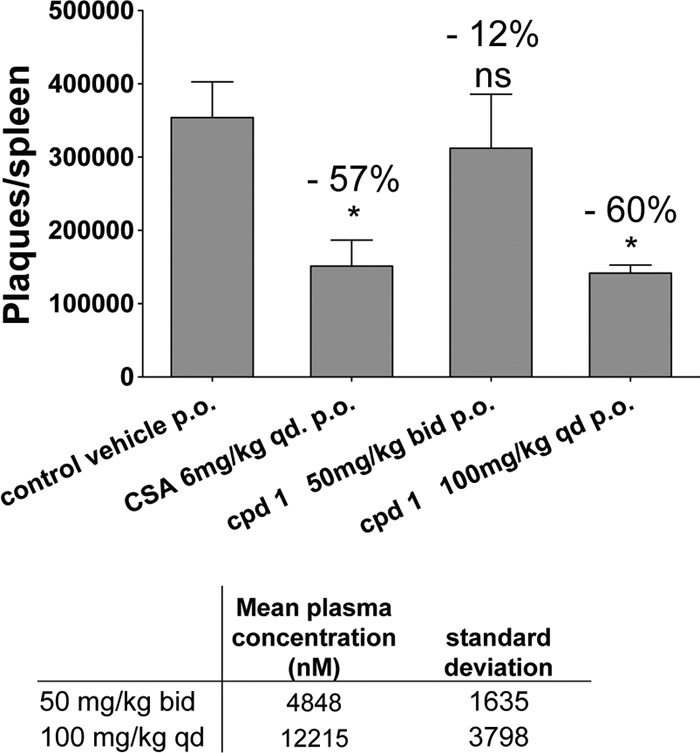
PI4KIIIβ inhibition over 4 days reduces the T-cell-mediated B-cell activation and antibody formation in response to an injection of sheep red blood cells into rats. Treatment of rats with 100 mg/kg over 4 days resulted in a 12 μM compound concentration in plasma 2 h after last dose and reduced the plaque-forming antibody titer per spleen to the same extent as the control compound cyclosporine (CSA). Plaques per spleen and compound concentrations in blood are mean values from 4 animals ± SEMs analyzed using Graph Pad Prism software. The decrease of antibody production is shown as the percentage of the value for the vehicle control above each treatment. The asterisk indicates a significant change and ns a nonsignificant change as determined by ANOVA and Dunnett's multiple-comparison test.
DISCUSSION
Several laboratories had identified a role of PI4KIIIα in HCV propagation: an interaction with the viral protein NS5A (1, 16, 19). A possible function for PI4KIIIβ had been only implied by siRNA experiments and the use of nonspecific inhibitors (4, 21). Here we present specific PI4KIIIβ inhibitors which potently inhibited genotype 1 replicons and the genotype 2a virus. Resistant cell lines developed after several weeks of treatment with increasing amounts of compound in the growth medium, and the resistance level was low (a 3- to 5-fold increase in EC50 compared to that for the wild type). This finding suggests a higher barrier to resistance for cellular targets, as recently reported for cyclophilin inhibitors (18). Characterization of the resistant clones is ongoing to evaluate if the low resistance is an artifact of the in vitro assay where the cells are kept under antibiotic selection pressure.
To characterize the safety of inhibition of PI4KIIIβ and assess the therapeutic potential, we characterized the inhibition of this lipid kinase in cells. Deciphering the roles of the individual PI4Ks was previously hampered by the lack of specific inhibitors and by the lack of a suitable LC-MS method that could separate the products of the four PI4 kinases expressed in human cells from other monophosphates. Many functions are attributed to PI4KIIIβ in the literature based on siRNA depletion studies, genetic manipulation of yeast cells, or inhibition studies with promiscuous inhibitors such as wortmannin. Therefore, the selective inhibitors shown in this study are excellent tools to examine the cellular roles of PI4KIIIβ.
Following reports on PI4KIIIβ regulation of insulin secretion, insulin levels were quantified after siRNA depletion or inhibition of the enzyme (6, 9). PI4KIIIβ RNA was depleted to 80 to 90% of the level for a nontargeting siRNA control, but this did not inhibit insulin secretion (Fig. 2). Likewise, inhibition of PI4KIIIβ with compound 6 did not affect insulin secretion at different rates of stimulation through increasing amounts of glucose (Fig. 3). The effect seen with 10 μM compound 5 is most likely off-target related, since this compound is a structural analog lacking PI4KIIIβ activity. The lack of function in insulin secretion was confirmed in an intraperitoneal glucose tolerance test in rats with the selective PI4KIIIβ inhibitor compound 3 at a dose that yielded plasma and tissue levels well above the replicon EC50. The different results obtained here compared to published results may be rationalized by the use of endogenous PI4KIIIβ here, while other studies overexpressed PI4KIIIβ in either pancreatic beta or neuronal cells and measured release of growth hormone also expressed from a vector.
Treatment of HCV patients with the current standard of care lasts at least 12 weeks. The recently approved protease inhibitor telaprevir may shorten the treatment time, but several weeks of therapy will still continue to be required (11). Therefore, due to the extended length of dosing, safety is of utmost importance with any potential HCV drug. Thus, cytotoxicity was monitored during the discovery and optimization of PI4KIIIβ inhibitors in replicon-containing Huh7 cells. Compounds were also profiled in primary fibroblasts and ∼450 transformed cell lines and tested in proliferating bone marrow cells and lymphocytes. While the PI4KIIIβ inhibitors were not cytotoxic in primary fibroblast or transformed tumor cell lines, they prevented proliferation in bone marrow cells and lymphocytes in vitro. A correlation of PI4KIIIβ, MLR, and BMP activities was observed for all compounds made in this drug development program, including different chemotypes (data not shown), indicating a direct effect on proliferation rather than an off-target effect. PI4KIIIβ might therefore play a more pronounced role in highly proliferating cells.
Ca2+ release-activated Ca2+ (CRAC) channels play a role in T-cell activation and have been shown to be regulated by PI4P (5, 10). Korzeniowski et al. (13) proposed a role for PI4KIIIα but did not rule out PI4KIIIβ as a player in PI4P production. Compounds 3 and 4 were tested in an in-house CRAC assay and did not have an effect on calcium flux (data not shown), strengthening the data of Korzeniowski et al. obtained with the nonspecific inhibitors wortmannin, PIK93, and LY294002. PI4KIIIβ inhibitors also did not affect the sodium ion channel Nav 1.5, the calcium ion channel Cav 1.2, or the potassium ion channels hERG, and KCNQ/MIK (data not shown).
Compound 1 was tested in an _in vivo_-specific antibody production assay, the rat sheep red blood cell (SRBC) assay, and inhibited specific antibody production. Together with the observation of no effects on the production of signaling molecules (Tables 3 to 5), this suggests that B-cell proliferation was prevented, leading to the lowering of antibody levels produced ex vivo. The dose chosen for compound 1 was expected to be efficacious based on pharmacokinetic modeling and cellular potency. This is, to our knowledge, the first description of PI4KIIIβ being involved in lymphocyte proliferation, and further studies are necessary to unravel the exact mode of action of PI4KIIIβ in this process.
Host factors are potentially attractive antiviral targets because of their high barrier to the development of resistance. The use of cyclophilin inhibitors such as NIM811 and Deb025 has proven this hypothesis in vitro (18). However, the potential toxicity or side effects associated with inhibition of normal cellular functions of host targets necessitate careful studies and consideration. In the case of the PI4KIIIβ inhibitors presented here, their antiproliferative effect precludes the usage of PI4KIIIβ as a target for the treatment of hepatitis C virus. Their strong effect on bone marrow proliferation in addition to lymphocytes makes them also not suitable to prevent transplant rejection.
Supplementary Material
Supplemental material
Footnotes
Published ahead of print 23 July 2012
REFERENCES
- 1.Ahn J, et al. 2004. Systematic identification of hepatocellular proteins in interacting with NS5A of the hepatitis C virus. J. Biochem. Mol. Biol. 37:741–748 [DOI] [PubMed] [Google Scholar]
- 2.Balla A, Balla T. 2006. Phosphatidylinositol4-kinases: old enzymes with emerging functions. Trends Cell Biol. 16:351–361 [DOI] [PubMed] [Google Scholar]
- 3.Berger KL, et al. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:7577–7582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borawski J, et al. 2009. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 83:10058–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cahalan MD, et al. 2007. Molecular basis of the CRAC channel. Cell Calcium 42:133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBarry J, et al. 2006. Functional implication of neuronal calcium sensor-1 and phosphoinositol 4-kinase-beta interaction in regulated exocytosis of PC12 cells. J. Biol. Chem. 281:18098–18111 [DOI] [PubMed] [Google Scholar]
- 7.Feld JJ, Hoofnagle JH. 2005. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436:967–972 [DOI] [PubMed] [Google Scholar]
- 8.Goeken NE, Melton ZJ. 1983. Immunoregulatory activity of T-cell subsets activated in human mixed lymphocyte reaction. Hum. Immunol. 6:79–90 [DOI] [PubMed] [Google Scholar]
- 9.Gromada J, et al. 2005. Neuronal calcium sensor-1 potentiates glucose-dependent exocytosis in pancreatic beta cells through activation of phosphatidylinositol 4-kinase beta. Proc. Natl. Acad. Sci. U. S. A. 102:10303–10308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halfon P, Locarnini S. 2011. Hepatitis C virus resistance to protease inhibitors. J. Hepatol. 55:192–206 [DOI] [PubMed] [Google Scholar]
- 11.Jacobson IM, et al. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 12.Khattab MA. 2009. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J. Gastroenterol. 15:3472–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korzeniowski MK, Manjarres IM, Varnai P, Balla T. 2010. Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal. 3:ra82 doi:10.1126/scisignal.2001122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krauss M, Haucke V. 2007. Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep. 8:241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ladics GS. 2007. Use of SRBC antibody responses for immunotoxicity testing. Methods 41:9–19 [DOI] [PubMed] [Google Scholar]
- 16.Lim Y, Hwang SB. 2011. Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type IIIα and regulates viral propagation. J. Biol. Chem. 286:11290–11298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mallo GV, et al. 2008. SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J. Cell Biol. 182:741–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puyang X, et al. 2010. Mechanism of resistance of hepatitis C virus replicons to structurally distinct cyclophilin inhibitors. Antimicrob. Agents Chemother. 54:1981–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiss S, et al. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai AW, et al. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trotard M, et al. 2009. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 23:3780–3789 [DOI] [PubMed] [Google Scholar]
- 22.Vaillancourt FH, et al. 2009. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387:5–10 [DOI] [PubMed] [Google Scholar]
- 23.Venkatesan K, et al. 2010. Prediction of drug response using genomic signatures from the Cancer Cell Line Encyclopedia, abstr. PR2. Abstr. AACR Meet., September 2010, Denver, CO [Google Scholar]
- 24.Wasley A, Alter MJ. 2000. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin. Liver Dis. 20:1–16 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material