Identification and Biochemical Characterization of a Novel Mutation in DDX11 Causing Warsaw Breakage Syndrome (original) (raw)
. Author manuscript; available in PMC: 2015 Oct 9.
Published in final edited form as: Hum Mutat. 2012 Oct 17;34(1):103–107. doi: 10.1002/humu.22226
Abstract
Mutations in the gene encoding the iron–sulfur-containing DNA helicase DDX11 (ChlR1) were recently identified as a cause of a new recessive cohesinopathy, Warsaw breakage syndrome (WABS), in a single patient with severe microcephaly, pre- and postnatal growth retardation, and abnormal skin pigmentation. Here, using homozygosity mapping in a Lebanese consanguineous family followed by exome sequencing, we identified a novel homozygous mutation (c.788G>A [p.R263Q]) in DDX11 in three affected siblings with severe intellectual disability and many of the congenital abnormalities reported in the WABS original case. Cultured lymphocytes from the patients showed increased mitomycin C-induced chromosomal breakage, as found in WABS. Biochemical studies of purified recombinant DDX11 indicated that the p.R263Q mutation impaired DDX11 helicase activity by perturbing its DNA binding and DNA-dependent ATP hydrolysis. Our findings thus confirm the involvement of DDX11 in WABS, describe its phenotypical spectrum, and provide novel insight into the structural requirement for DDX11 activity. Hum Mutat 34:103-107, 2013.
Keywords: DDX11, Warsaw breakage syndrome, WABS, helicase
Warsaw breakage syndrome (WABS; MIM# 613398) is a new form of cohesinopathy showing defects in sister chromatid cohesion and hypersensitivity to chemicals that induce replication stress, thus combining distinct cytogenetic features seen in Roberts syndrome (MIM# 268300) and Fanconi anemia (MIM# 227650), respectively [van der Lelij et al., 2010b]. WABS has been reported in a single individual with several congenital abnormalities, including severe pre- and postnatal growth retardation, microcephaly, facial dysmorphy, deafness, ventricular septal defects, mild intellectual disability (ID), and abnormal skin pigmentation. This patient carried compound heterozygous mutations in DDX11 (MIM# 601150; NM_030653.3), including a splice site mutation (c.2271+2T>C, previously reported as IVS22+2T>C) and a 3-bp in-frame C-terminal deletion (c.2689_2691del [p.K897del]) that was recently shown to abrogate the DDX11 helicase activity [van der Lelij et al., 2010a; Wu et al., 2012]. So far, no other patients with DDX11 mutations have been identified.
DDX11 (ChlR1), an orthologue of the yeast Chl1, is a member of the superfamily 2 (SF2) of ATP-dependent DEAH-box DNA helicases [Hirota and Lahti, 2000; Skibbens, 2004]. DDX11 shares sequence homology with the related SF2 DNA helicases FANCJ (MIM# 609054), ERCC2 (XPD; MIM# 126340), and RTEL1 (MIM# 608833), which all contain an iron–sulfur (Fe–S) motif between helicase domains IA and II [Rudolf et al., 2006; Wu et al., 2009]. FANCJ and ERCC2 (also known as XPD) are also implicated with genetic instability disorders in humans, and RTEL1 had been suggested to play a role in the maintenance of telomere length and genome stability in mice [Ding et al., 2004; Lehmann, 2001; Levitus et al., 2005]. In human cells, DDX11 was shown to interact with components of the cohesin complex and play a role in sister chromatid cohesion [Parish et al., 2006].
Here, we describe the identification and biochemical characterization of a novel deleterious homozygous mutation in DDX11 in a consanguineous Lebanese family showing many of the symptoms associated with WABS. This family, which was recruited for this study with the approval of our institutional ethics committee, consists of two healthy first cousins once removed and their three affected children (Fig. 1). We first performed whole-genome SNP genotyping in two affected siblings (patients V-1 and V-3) by using the Il-lumina Human 610 Genotyping BeadChip panel, which interrogates 620,901 SNPs, and we used PLINK (v.1.06) to search for homozygosity regions (HR) containing >30 consecutive SNPs and extending over >1 MB. We identified 10 candidate HR shared by the two siblings (Supp. Table S1). To find potential causative mutations in these regions, we performed exome capture (Agilent SureSelect 50 MB kit) and sequencing (paired-end SOLiD4) on one of the affected siblings (patient V-1) and obtained an average per target base coverage of 25×, with at least 85% of the target region being covered at >= 3×. Exome capture, read mapping as well as variant calling and annotation were performed as previously described [Daoud et al., 2012] and as noted in the Supporting Information. In total, 148 homozygous nonsynonymous coding and splicing variants were identified in the HR, of which only two were not found in our in-house set of 198 control exomes (from 30 healthy individuals and from 168 patients with other rare diseases, including amyotrophic lateral sclerosis, essential tremor, aneurysm, and hereditary spastic paraplegia) or not reported at frequencies >0.5% in public SNP databases (dbSNP135 [http://www.ncbi.nlm.nih.gov/projects/SNP/], 1000 genomes [http://browser.1000genomes.org/index.html], and EVS Exome Variant Server [http://evs.gs.washington.edu/EVS/]). Sanger sequencing confirmed that only one of these two variants, a missense mutation in DDX11 (c.788G>A [p.R263Q]; NM_030653.3) present in the largest HR (chr12: 33.6 MB), was heterozygous in the parents and homozygous in the three affected siblings (Fig. 1; Supp. Table S2). This variant was submitted to a gene-specific database (http://www.lovd.nl/DDX11). We also genotyped 150 individuals from the Middle East, including 100 Lebanese and 50 Palestinians, and did not identify a single carrier of the DDX11 p.R263Q mutation, indicating that it is rare. This mutation, predicted to be damaging by various in silico algorithms (SIFT, Polyphen-2, Mutation Taster), affects a conserved residue in the Fe–S domain, which is also conserved in the other related SF2 helicases (FANCJ, ERCC2 [XPD], and RTEL) [Rudolf et al., 2006; Wu et al., 2009] (Fig. 1E). Interestingly, the Fe–S domain was shown to be essential for the helicase activity of FANCJ and XPD [Rudolf et al., 2006; Wu et al., 2010]. Moreover, a mutation of the homologous arginine residue in ERCC2 (XPD) (c.335G>A, p.R112H; NM_000400.3), which causes trichothiodystrophy (TTD; MIM# 601675), results in a loss of the helicase activity, a concomitant defect in nucleotide excision repair, and a reduction in the repair/transcription factor TFIIH (GTF2H2; MIM# 601748), all indicating that this arginine residue (R263 in DDX11; R112 in XPD) is functionally important [Botta et al., 2002; Dubaele et al., 2003].
Figure 1.
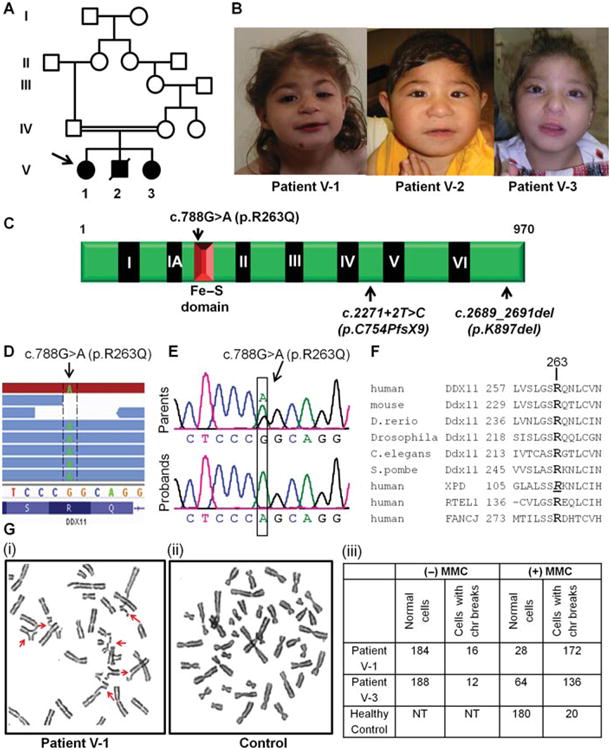
A pathogenic mutation identified in DDX11 in a family affected with WABS. A, B: Pedigree of the consanguineous Lebanese family studied here and their facial features. The proband is indicated by an arrow. Patient V-2 was deceased hence certain analyses could not be extended to biological sampling of this child. C: Schematic representation of DDX11 showing the helicase motifs (I–VI) as well as the Fe–S motif of DDX11. p.R263Q identified herein as well as the two previously published DDX11 mutations (c.2271+2T>C and p.K897del) are shown. D: Integrated Genomics Viewer (IGV) tracks of exome sequencing reads showing the presence of the homozygous DDX11 c.788G>A (p.R263Q) mutation in patient V-1. E: Sanger sequencing validation of c.788G>A (p.R263Q) DDX11 mutation. Representative chromatograms of the parents (both heterozygous for c.788G>A) and the three probands (all homozygous for c.788G>A) are shown. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon. F: Amino acid alignment of the beginning of Fe–S domain region of DDX11 in different species as well as the corresponding regions in the human-related DNA helicases: XPD (ERCC2), FANCJ, and RTEL1. R263 in human DDX11 is well conserved (in bold). R112 residue in human XPD whose mutation (c.335G>A, p.R112H) causes trichothiodystrophy is italicized and underlined. G: Mitomycin C (MMC)-induced chromosomal breakage. Representative metaphase from blood cultures treated with MMC showed increased chromosomal defects (arrows), including breakage, radial formations, centromeric heterochromatin repulsion (“railroads”) from patient V-1 (i), in contrast to the metaphases prepared from a similarly treated blood cultures prepared from a healthy control individual (ii). Table showing the representation of cells with chromosomal (chr) breaks in presence or absence of MMC. (NT, not tested.) A total of 200 cells (metaphases) were screened for each individual (iii).
Metaphase cells from lymphocytes of the two affected siblings (patients V-1 and V-3) showed an increase in chromosomal breakage (86% and 68%, respectively, compared to 10% for the healthy control) after treatment with the DNA cross-linking agent mitomycin C (MMC; see Supporting Information for methods) (Fig. 1G). Of these cells with MMC-induced chromosomal breaks, 46.5% and 44% in patients V-1 and V-3, respectively, showed centromeric heterochromatin repulsion (“railroads”) and premature chromatid separation. These data are consistent with observations in similarly treated cells from the original WABS patient [van der Lelij et al., 2010a]. Patient V-2 could not be studied at the cytogenetic level.
We next investigated whether the p.R263Q mutation affects DDX11 activity. For this, we purified recombinant human hexahistidine-tagged wild-type (WT) and mutant DDX11 (DDX11-R263Q) from transfected HEK293T and assayed the proteins activities (see Supporting Information for detailed methods) (Fig. 2). We first examined DNA unwinding activity catalyzed by DDX11-WT and DDX11-R263Q mutant. Using a forked 19-bp duplex DNA substrate, DDX11-WT unwound the substrate in a protein concentration dependent manner, as was previously shown [Wu et al., 2012] (Fig. 2B). In comparison, DDX11-R263Q unwound the forked duplex substrate much less efficiently (Fig. 2C). Quantitative analysis of the helicase data showed that 0.06 nM DDX11-WT unwound approximately 50% of the substrate whereas DDX11-R263Q displayed no detectable helicase activity. At 0.47 nM protein concentration, DDX1-R263Q unwound only approximately 10% of the substrate, whereas DDX11-WT unwound the substrate to near completion (Fig. 2D). We also tested the ability of the mutant DDX11 protein to unwind a two-stranded antiparallel G-quadruplex (G4) DNA substrate (OX-1-G2′), as we have previously performed for DDX11-WT [Wu et al., 2012]. As shown in Supp. Figure S1, DDX11-R263Q unwound the G4 substrate much less efficiently than DDX11-WT. On the basis of these results, we conclude that the R263Q mutation negatively affects DDX11 helicase activity on both duplex and G4 DNA substrates.
Figure 2.
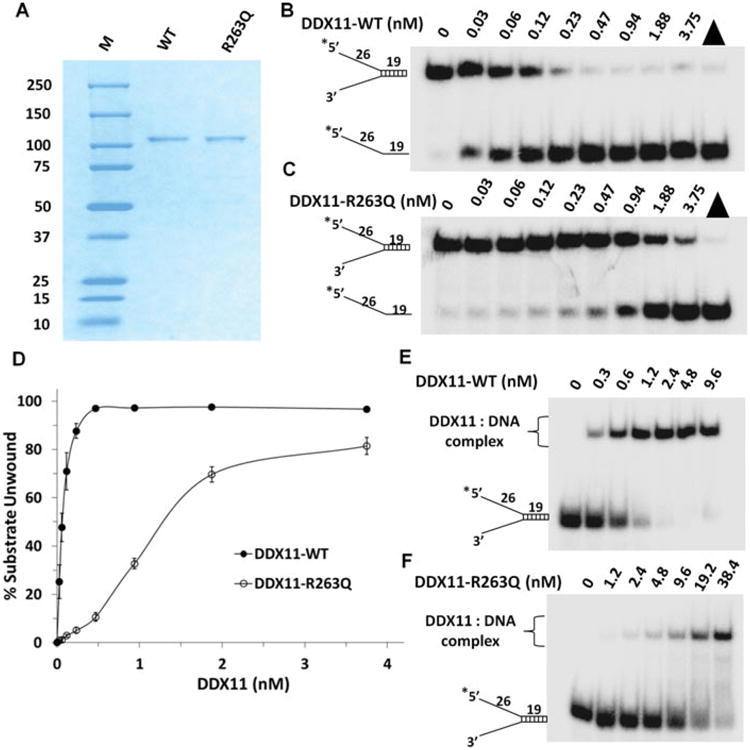
Purification and biochemical analyses of recombinant DDX11-WT and DDX11-R263Q for helicase activity and DNA binding. A: The purity of DDX11-WT and DDX11-R263Q was evaluated by Coomassie stained SDS PAGE (predicted MW approximately 110 kD). (M, molecular weight protein standards.) B,C: Helicase activity of DDX11-WT(B) and DDX11-R263Q (C) on forked duplex DNA substrate. Helicase reactions (20 _μ_l) were performed by incubating the indicated DDX11 protein with 0.5 nM forked duplex DNA substrate at 37°C for 15 min under standard helicase assay conditions as described in the methods section in the Supporting Information. Products were resolved on native 10% polyacrylamide gels and representative phosphorimages are shown. Triangle denotes heat-denatured DNA substrate control, and asterisk denotes 5′-32P end label. D: Quantitative analysis of data from helicase activity of DDX11-R263Q and DDX11-WT on forked duplex DNA substrate is shown. Data represent the mean of at least three independent experiments with standard deviations (SD) indicated by error bars. E, F: DNA binding by DDX11-WT and DDX11-R263Q as detected by gel mobility shift assays. The indicated concentrations of DDX11-WT (E) and DDX11-R263Q (F) were incubated with 0.5 nM forked duplex DNA substrate on ice for 30 min under standard gel shift assay conditions as described in the Supporting Information. The DNA-protein complexes were resolved on native 5% polyacrylamide gels. Representative phosphorimages for DNA-binding assays are shown. Asterisk denotes 5′-32P end label.
The reduced helicase activity of DDX11-R263Q might reflect an impaired ability to bind DNA. To address this issue, we performed electrophoresis mobility shift assays (EMSA) with the mutant and WT proteins in the absence of ATP using the same radiolabeled forked duplex DNA used for the helicase assays. DDX11-WT bound the forked duplex in a protein concentration dependent manner with approximately 75% of the substrate bound at a protein concentration of 1.2 nM (Fig. 2E). In contrast, little or no detectable DNA binding was observed at 1.2 nM DDX11-R263Q (Fig. 2F). Increasing the protein concentration to 2.4 nM resulted in nearly complete binding of the forked duplex by the WT DDX11, whereas only approximately 3% of the substrate was bound by DDX11-R263Q at the 2.4 nM protein concentration. From these results, we conclude that the p.R263Q mutation negatively affects the ability of DDX11 to bind DNA.
We next examined the DNA-dependent ATPase activity of DDX11-R263Q compared with DDX11-WT in the presence of M13 ssDNA circle as the DNA effector (Supp. Table S3). The _K_m for ATP binding by DDX11-R263Q was approximately threefold lower than that of DDX11-WT (0.19 ± 0.05 mM vs. 0.66 ± 0.04 mM, respectively), suggesting that the mutant protein bound ATP more favorably. From the Lineweaver-Burke analysis, however, it was evident that the DDX11-R263Q mutant showed a _V_max that was approximately 8.5-fold lower than that of DDX11-WT (0.066 ± 0.008 nmol min−1 vs. 0.56 ± 0.05 nmol min−1, respectively). A kinetic analysis of ATPase activity demonstrated that the _k_cat value for DDX11-R263Q was 18-fold lower than that of DDX11-WT (12 ± 4 min−1 vs. 220 ± 40 min−1, respectively), confirming that the mutant protein was defective in its ability to hydrolyze ATP. These results suggest that the R263Q mutation negatively affected the ability of DDX11 to perform DNA-dependent ATP hydrolysis, a result that is consistent with its DNA-binding defect and poor ability to unwind DNA substrates efficiently unwound by WT DDX11. The modestly reduced _K_m value for DDX11-R263Q compared to WT DDX11 may reflect a reduced ability of the mutant enzyme to turnover ATP. Collectively, these biochemical analyses of mutant DDX11 indicate that the p.R263Q mutation impairs the helicase activity of DDX11 by perturbing its DNA binding and DNA-dependent ATP hydrolysis. The reduced ability of the DDX11-R263Q mutant protein to bind DNA may be consistent with structural and biochemical data that indicate a role of the Fe-S cluster in the sequence-related XPD helicase in the interaction of the protein with DNA [Kuper et al., 2012; Pugh et al., 2012].
Members of the consanguineous family described here originate from North Lebanon. There was no history of birth defects, or increased miscarriage rate reported by this family and known relatives. All sibs were born at 32 weeks of gestation. No known toxic, medical exposure, or unusual events were reported during the gestations. Patient V-1 was first seen when she was 20 months old. At birth, her weight was 1,500_g_, her length was 35 cm, and her head circumference was 27 cm (all values far below the third percentile). Fontanelles were closed at the age of 3 months, and she walked unhelped at the age of 14 months. Upon examination, her weight was 7.5 kg, height was 74 cm, and head circumference was 36.5 cm (all values below the third percentile). She showed severe ID. She had a small and receding forehead, short nose, small nares, short neck, clinodactyly of the fifth fingers, and a single palmar crease on both hands. Neurological examination revealed a hypotonic girl. Abdominal ultrasonography and echocardiography did not show any abnormalities. CT imaging of the brain revealed rather small and rounded cochlea without visible cochlear turns or spirals. Bilateral sensorineural deafness was diagnosed by auditory evoked potential.
Patient V-2 was first evaluated by us at the age of 1 year for severe delayed milestones. At birth, his weight was 1,500_g_, length was 38 cm, and head circumference was 25 cm (all values below the third percentile). An echocardiogram evidenced a tetralogy of Fallot. Bilateral sensorineural deafness was diagnosed by auditory evoked potential at 6 months. His fontanelles were closed at the age of 7 months. At examination, his weight was 6.2 kg, height was 64 cm, and head circumference was 31 cm (all values below the third percentile). Physical examination showed the same dysmorphic features as those found in his sister with, in addition, prominent cheeks, flat philtrum, microretrognathism, and strabismus. He also showed severe ID. Abdominal ultrasonography did not show any abnormalities. He died at the age of 4 years because of heart failure. Patient V-3 showed the same dysmorphic features as her siblings, as well as sensorineural deafness and severe ID but no cardiac malformations.
The phenotypic profiles of our patients with the mutation p.R263Q in DDX11 overlap in presentation and severity with that of the previously described WABS patient who carried compound heterozygous mutations in DDX11 (Supp. Table S4) [van der Lelij et al., 2010a]. They showed ID growth retardation, and severe congenital abnormalities including microcephaly, facial dysmorphism, deafness due to cochlear abnormalities (in two of the sibs), and cardiac malformations (in one of the sibs). Unlike the previously reported WABS patient who had mild ID and skin pigmentation abnormality, the affected sibs reported herein had severe ID and no skin pigmentation abnormality, differences that may be attributed to the type of DDX11 mutations, and differences in genetic background. It is of interest that the DDX11 protein with the p.R263Q substitution in its Fe–S domain retained partial (albeit severely compromised) helicase activity at higher protein concentrations, whereas the previously characterized DDX11-K897del protein lacking a single lysine in the C-terminal end was completely inactive in DNA unwinding [Wu et al., 2012]. Our current findings demonstrate that the integrity of the Fe–S domain is critical for ATPase-driven DNA unwinding by DDX11. In this regard, a clinically relevant missense mutation in the conserved Fe–S domain of FANCJ interfered with its DNA helicase activity as well (Wu et al., 2010]. Future studies of clinically relevant mutations in Fe–S helicases should help elucidate their molecular and cellular defects.
In conclusion, we describe here the second WABS family with mutations in DDX11, and the first with multiple affected individuals who all carry a homozygous missense mutation in the Fe–S domain of DDX11 that impairs the protein's activity. Our data confirm that DDX11 mutations cause WABS and provide additional insights into the clinical phenotypes and functional consequences associated with pathogenic DDX11 mutations.
Supplementary Material
Supp Data
Acknowledgments
J.L.M. is a National Scientist of the Fonds de Recherche en Santé du Québec. J.M.C. holds a salary award from the Réseau de Médecine Génétique Appliquée du Québec (RMGA). We thank the patients and their family for participating in this study. We also thank the members of the RMGA bioinformatic team (Alexandre Dionne-Laporte, Dan Spiegelman, Edouard Henrion, and Ousmane Diallo) for the primary analysis of the exome sequencing data.
Contract grant sponsors: March of Dimes (grant no. 12-FY10-236 to Z.K., M.S., and J.L.M.); Lebanese CNRS (A.M.); Intramural Research program of the NIH, National Institute on Aging (to R.B. Jr).
Footnotes
Additional Supporting Information may be found in the online version of this article.
Disclosure Statement: The authors confirm that they have no conflicts of interest.
References
- Botta E, Nardo T, Lehmann AR, Egly JM, Pedrini AM, Stefanini M. Reduced level of the repair/transcription factor TFIIH in trichothiodystrophy. Hum Mol Genet. 2002;11:2919–2928. doi: 10.1093/hmg/11.23.2919. [DOI] [PubMed] [Google Scholar]
- Daoud H, Suhail H, Szuto A, Camu W, Salachas F, Meininger V, Bouchard JP, Dupre N, Dion PA, Rouleau GA. UBQLN2 mutations are rare in French and French–Canadian amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:2230e1–2230e5. doi: 10.1016/j.neurobiolaging.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E, et al. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell. 2004;117:873–886. doi: 10.1016/j.cell.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Dubaele S, Proietti De Santis L, Bienstock RJ, Keriel A, Stefanini M, Van Houten B, Egly JM. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–1646. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- Hirota Y, Lahti JM. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000;28:917–924. doi: 10.1093/nar/28.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuper J, Wolski SC, Michels G, Kisker C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2012;31:494–502. doi: 10.1038/emboj.2011.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy EJ. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–4865. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- Pugh RA, Wu CG, Spies M. Regulation of translocation polarity by helicase domain 1 in SF2B helicases. EMBO J. 2012;31:503–514. doi: 10.1038/emboj.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. The DNA repair helicases XPD and FancJ have essential iron–sulfur domains. Mol Cell. 2006;23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, Zdzienicka MZ, Joenje H, de Winter JP. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010a;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lelij P, Oostra AB, Rooimans MA, Joenje H, de Winter JP. Diagnostic overlap between Fanconi anemia and the cohesinopathies: Roberts syndrome and Warsaw breakage syndrome. Anemia. 2010b;2010:1–7. doi: 10.1155/2010/565268. 565268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sommers JA, Khan I, de Winter JP, Brosh RM., Jr Biochemical characterization of warsaw breakage syndrome helicase. J Biol Chem. 2012;287:1007–1021. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, Shin-Ya K, Kitao H, Brosh RM., Jr Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein–DNA complexes. Blood. 2010;116:3780–3791. doi: 10.1182/blood-2009-11-256016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Suhasini AN, Brosh RM., Jr Welcome the family of FANCJ-like helicases to the block of genome stability maintenance proteins. Cell Mol Life Sci. 2009;66:1209–1222. doi: 10.1007/s00018-008-8580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Data