Enterovirus 71 Uses Cell Surface Heparan Sulfate Glycosaminoglycan as an Attachment Receptor (original) (raw)
Abstract
Enterovirus 71 (EV-71) infections are usually associated with mild hand, foot, and mouth disease in young children but have been reported to cause severe neurological complications with high mortality rates. To date, four EV-71 receptors have been identified, but inhibition of these receptors by antagonists did not completely abolish EV-71 infection, implying that there is an as yet undiscovered receptor(s). Since EV-71 has a wide range of tissue tropisms, we hypothesize that EV-71 infections may be facilitated by using receptors that are widely expressed in all cell types, such as heparan sulfate. In this study, heparin, polysulfated dextran sulfate, and suramin were found to significantly prevent EV-71 infection. Heparin inhibited infection by all the EV-71 strains tested, including those with a single-passage history. Neutralization of the cell surface anionic charge by polycationic poly-d-lysine and blockage of heparan sulfate by an anti-heparan sulfate peptide also inhibited EV-71 infection. Interference with heparan sulfate biosynthesis either by sodium chlorate treatment or through transient knockdown of _N_-deacetylase/_N_-sulfotransferase-1 and exostosin-1 expression reduced EV-71 infection in RD cells. Enzymatic removal of cell surface heparan sulfate by heparinase I/II/III inhibited EV-71 infection. Furthermore, the level of EV-71 attachment to CHO cell lines that are variably deficient in cell surface glycosaminoglycans was significantly lower than that to wild-type CHO cells. Direct binding of EV-71 particles to heparin-Sepharose columns under physiological salt conditions was demonstrated. We conclude that EV-71 infection requires initial binding to heparan sulfate as an attachment receptor.
INTRODUCTION
Human enterovirus 71 (EV-71) belongs to the Enterovirus genus within the family Picornaviridae. The EV-71 genome is a positive-sense, single-stranded RNA enclosed in an icosahedral capsid assembled from 60 copies of each of the four structural proteins, VP1 to VP4 (1).
EV-71 infections usually cause mild hand, foot, and mouth disease (HFMD), characterized by the development of fever with rashes on the palms and feet and with oral ulcers (2). EV-71 also causes severe neurological manifestations, such as aseptic meningitis, brain stem encephalitis, and poliomyelitis-like acute flaccid paralysis (3). In recent years, EV-71 has emerged as an important cause of epidemic viral encephalitis in Asia, resulting in high fatalities among children below the age of 6 years (3, 4).
Receptor binding is an essential event during viral infection. The ability to recognize and interact with specific receptors is one of the factors that contribute to the host range and tissue tropism of a virus (5). To date, at least four EV-71 receptors have been reported. The first receptor to be discovered was the human P-selectin glycoprotein ligand-1 (PSGL-1). PSGL-1 is a sialomucin membrane protein expressed mainly in leukocytes and was hypothesized to facilitate the viremic phase of EV-71 infection (6). The second receptor reported was the human scavenger receptor class B-2 (SCARB2), a type III double-transmembrane protein located primarily in endosomes but also expressed on the cell surface (7–9). Sialylated glycan was also reported as one of the possible receptors for EV-71 infection. Removal of sialic acid residues from plasma membranes was able to protect DLD-1, RD, and SK-N-SH cells from EV-71 infection (10, 11). Annexin II has recently been identified as another possible receptor for EV-71 in RD cells. Pretreatment of EV-71 with soluble annexin II resulted in reduced viral attachment to the cell surface (12). However, inhibitors that blocked all of the receptors identified failed to inhibit EV-71 infection completely (13). This may imply the involvement of one or more EV-71 receptors that have yet to be discovered.
Our previous study identified a novel antiviral peptide, SP40, derived from the VP1 capsid protein (14). This peptide was found to contain a heparan sulfate-specific glycosaminoglycan (GAG) binding domain (-RRKV-), and the positively charged amino acids were found to be critical for antiviral properties. We postulated that SP40 interacted with heparan sulfate and blocked EV-71 attachment (14). Similarly, bovine and human lactoferrin exhibited antiviral activities against EV-71 when tested in vitro and in vivo, possibly due to direct interaction with heparan sulfate, resulting in interference with EV-71 binding (15, 16). In a recent study, heparin and heparan sulfate were reported to exhibit antiviral activities against EV-71 infection (17). These findings led us to hypothesize that surface heparan sulfate might play an important role in EV-71 attachment.
GAGs are negatively charged linear polysaccharides composed of hexosamine/hexuronic acid repeats. GAGs acquire negative charges through N- and O-sulfation of the carbohydrate moieties (18). The types of GAG include heparin, heparan sulfate, and chondroitin sulfate. GAG-ligand interactions are complex and are characterized either by merely electrostatic forces or by specific interactions (19). Many viral pathogens, such as herpes simplex virus (20, 21), HIV (22, 23), dengue virus (24), human papillomavirus (25), vaccinia virus (26), Theiler's murine encephalomyelitis virus (TMEV) (27), adeno-associated virus type 2 (28, 29), hepatitis B virus (30), hepatitis C virus (31), Sindbis virus (32), echovirus (33), coxsackievirus B3 and A9 (34, 35), and foot-and-mouth disease virus (36, 37), are known to utilize cell surface heparan sulfate as an attachment or entry receptor.
In the present study, we investigate the role of heparan sulfate in the binding of EV-71 to the surface of rhabdomyosarcoma (RD) cells. We provide evidence that the ability of EV-71 to bind to RD cells was reduced after the enzymatic removal of cell surface heparan sulfate and through the obstruction of heparan sulfate biosynthesis. Our results show that cell surface heparan sulfate, and not chondroitin sulfate, serves as an attachment receptor for EV-71.
MATERIALS AND METHODS
Reagents.
Soluble GAGs (heparin sodium salt from porcine intestinal mucosa, chondroitin sulfate sodium salt from shark cartilage, _N_-acetyl-de-O-sulfated heparin sodium salt, and de-N-sulfated heparin sodium salt), dextran sulfate sodium salt from Leuconostoc mesenteroides, suramin, heparinase I/II/III from Flavobacterium heparinum, chondroitinase ABC from Proteus vulgaris, and sodium chlorate were all purchased from Sigma. Heparinase I from recombinant Bacteroides thetaiotaomicron was supplied by R&D Systems. An anti-EV-71 monoclonal antibody and poly-d-lysine were purchased from Millipore, and Alexa Fluor 488-labeled anti-mouse IgG was purchased from Invitrogen. Anti-heparan sulfate peptides G1 (LRSRTKIIRIRH) and G2 (MPRRRRIRRRQK) (underlining indicates positively charged amino acids), described previously (38), were synthesized by Mimotopes Pty Ltd. (Australia) with >92% purity as determined by high-performance liquid chromatography (HPLC). All the primers and probes were synthesized by IDT.
Cell lines and viruses.
All the cell lines used in this experiment were obtained from the American Type Culture Collection (ATCC). Human rhabdomyosarcoma (RD; ATCC no. CCL-136) and Chinese hamster ovary (CHO-K1; ATCC no. CCL-61) cells were grown in Dulbecco's modified Eagle medium (DMEM) (HyClone) supplemented with 10% fetal bovine serum (FBS). Mutant CHO-pgsD677 (ATCC no. CRL-2244) and CHO-pgsA745 (ATCC no. CRL-2242) cells were grown in Kaighn's modification of Ham's F-12 (F-12K) medium (ATCC) supplemented with 10% FBS. All the experiments were carried out using 1.5 × 104 RD cells, unless otherwise stated. EV-71 laboratory strains BrCr (GenBank accession number AB204852), 41 (a gift from Tan Eng Lee, Singapore Polytechnic, Singapore) (GenBank accession number AF316321), UH1/PM/97 (GenBank accession number AM396587), and SHA66/97 (GenBank accession number AM396586) were propagated in RD cells in maintenance medium supplemented with 2% FBS. All the other EV-71 isolates (14716, 35017, 1687413, and 1657640) and the poliovirus (PV) vaccine strain were obtained from the Diagnostic Virology Laboratory, University Malaya Medical Center, Kuala Lumpur, Malaysia. Isolates 14716, 1687413, and 1657640 were all passaged once in tissue culture, while isolate 35017 had been passaged twice. Throughout the study, EV-71 strain 41 was used, unless otherwise stated.
TaqMan quantitative real-time PCR assay.
TaqMan quantitative real-time PCR was performed as described previously (14). Forward primer 5′-GAGCTCTATAGGAGATAGTGTGAGTAGGG-3′, reverse primer 5′-ATGACTGCTCACCTGCGTGTT-3′, and TaqMan probe 5′-6-carboxyfluorescein (FAM)-ACTTACCCA/ZEN/GGCCCTGCCAGCTCC-Iowa Black FQ-3′ were used. Viral RNA samples were extracted by using a QIAamp viral RNA minikit (Qiagen, Germany) according to the manufacturer's instructions. The TaqMan real-time reverse transcription (RT)-PCR assay was performed with the StepOnePlus real-time system (ABI) using the TaqMan Fast Virus 1-step master mix (ABI), with cDNA synthesis from RNA by reverse transcription for 5 min at 50°C and subsequent amplification for 40 cycles at 95°C for 3 s and 60°C for 30 s.
Evaluation of the role of GAGs and inhibitors in EV-71 infection of RD cells.
Viral inactivation experiments were performed by preincubation of EV-71 particles with various concentrations of GAGs (heparin, chondroitin sulfate, de-N-sulfated heparin, and _N_-acetyl-de-O-sulfated heparin) and inhibitors (dextran sulfate and suramin) for 60 min at 37°C before inoculation of the RD cells. For cell protection studies, RD cells were preincubated with GAGs, poly-d-lysine, or anti-heparan sulfate peptides at various concentrations for 60 min at 37°C. The cells were washed twice and were subsequently infected with various EV-71 isolates for 1 h at a multiplicity of infection (MOI) of 0.1. Viral infectivity was evaluated and quantified by TaqMan real-time PCR and a plaque assay after 24 h postinfection.
Inhibition of cellular GAG sulfation by sodium chlorate.
Sodium chlorate inhibition experiments were carried out in DMEM supplemented with 10% FBS. RD cells were cultured for 24 h in the presence of sodium chlorate at concentrations ranging from 0 mM to 50 mM, followed by infection using different EV-71 strains at an MOI of 0.1. The viral titers were quantified by TaqMan quantitative real-time PCR.
siRNA silencing of the gene involved in heparan sulfate biosynthesis.
Small interfering RNAs (siRNAs) targeting the heparan sulfate-modifying enzyme _N_-deacetylase/_N_-sulfotransferase 1 (NDST-1) and the heparan sulfate polymerase exostosin-1 (EXT-1) were purchased from Santa Cruz Biotechnology. Negative-control siRNA with no homology to any known mammalian gene was obtained from Bioneer. Different concentrations of the siRNAs (0 nM, 1 nM, 5 nM, 10 nM, and 20 nM in 50 μl of Opti-MEM) were incubated with Lipofectamine 2000 in Opti-MEM I (Invitrogen) for 20 min. The siRNA was then transfected into 1.0 × 104 RD cells for 24 h. Prior to EV-71 infection, the transfection medium was removed, and the cells were washed twice with growth medium (DMEM with 10% FBS) and were then incubated with the growth medium for at least 3 h. The cells transfected with the respective siRNAs were infected with EV-71 at an MOI of 0.1 for 1 h at room temperature. After 1 h of incubation, the inoculum was removed, washed twice, and replaced with maintenance medium (DMEM with 2% FBS). The viral titers were determined at 24 h postinfection by using TaqMan quantitative real-time PCR.
Enzymatic removal of GAGs from the surfaces of RD cells.
Chondroitinase ABC and heparinase I/II/III were reconstituted in digestion buffer I (phosphate-buffered saline [PBS] containing 0.05 M sodium acetate and 0.02% bovine serum albumin [BSA] [pH 7.5]) and digestion buffer II (PBS containing 0.5 mM MgCl2, 0.9 mM CaCl2, and 0.1% BSA [pH 7.5]), respectively, and various concentrations of the enzymes (50 μl/well) were added to RD cells, which were then incubated for 1 h at 37°C. Cells were then washed with the respective digestion buffers and were subsequently infected with EV-71 for 1 h at 4°C. After the incubation, the cells were washed twice with serum-free medium, and infectivity was determined 24 h postinfection by TaqMan real-time RT-PCR.
Binding of EV-71 to GAG-deficient cell lines.
RD, CHO-K1, CHO-pgsD677, and CHO-pgsA745 cells in chamber slides (Lab-Tek) and CellCarrier-96 plates (Perkin-Elmer) were infected with EV-71 (MOI, 100) at 4°C for an hour. After 1 h of incubation, the inocula were removed and the cells were fixed with 4% formaldehyde. The fixed cells were permeabilized using 0.25% Triton X-100 (Sigma) for 5 min and were subsequently blocked with Image-iT FX signal enhancer (Invitrogen) for 1 h. EV-71 particles were immunostained with mouse anti-EV-71 monoclonal antibodies (Millipore) as the primary antibodies and 1:200-diluted Alexa Fluor 488-labeled anti-mouse IgG (Invitrogen) as the secondary antibody for 1 h at 37°C. For nuclear visualization, cells were treated with 0.01% 4′,6-diamidino-2-phenylindole (DAPI; Sigma) for 7 min at room temperature. Immunofluorescence was detected with a Leica TCS SP5 confocal microscope (Leica Microsystems, Germany). The CellCarrier-96 plate was evaluated using a Cellomics ArrayScan VTI HCS reader with Spot Detector BioApplication software (Thermo Scientific). The data are presented as the total viral spot count per cell (calculated as the total number of fluorescent viral spots divided by the total number of cells in the same field) and were subsequently verified by TaqMan quantitative real-time PCR.
Binding of EV-71 particles to immobilized heparin-Sepharose beads.
Six milliliters of EV-71 or PV vaccine strain supernatant was applied to a 1-ml HiTrap Heparin HP column (GE Healthcare, Sweden) previously equilibrated with the binding buffer (0.02 M Tris-HCl, 0.14 M NaCl [pH 7.4]) at a flow rate of approximately 0.5 ml/min. After loading, the column was washed with at least 5 to 10 column volumes of binding buffer. The bound viral particles were eluted using the elution buffer (0.02 M Tris-HCl, 2 M NaCl [pH 7.4]). Fractions of 1 ml were collected and analyzed by TaqMan quantitative real-time PCR and plaque assays for EV-71 and the PV vaccine strain, respectively.
Cytotoxicity analysis.
The cytotoxicities of heparin, dextran sulfate, anti-heparan sulfate peptides, and sodium chlorate were determined using the CellTiter 96 AQueous One solution cell proliferation assay reagent (Promega). Briefly, various concentrations of these compounds were added to overnight-cultured RD cells, which were then incubated overnight. Then 20 μl of the CellTiter 96 AQueous One solution cell proliferation assay reagent was added to each well. The plate was then analyzed at an absorbance of 490 nm after 2 h of incubation at 37°C.
Three-dimensional crystal structure and sequence analysis.
The crystal structure of EV-71 (Protein Data Bank [PDB] identification code 4AED) (39) was obtained from the PDB (http://www.rcsb.org/pdb). The 3-dimensional structure of the EV-71 pentamer was built using the DeepView-Swiss PDB Viewer, version 4.0.4 (Swiss Institute of Bioinformatics) (40). For sequence analysis, 174 VP1 sequences from different genotypes (41) were downloaded from GenBank (http://www.ncbi.nlm.nih.gov/protein/) and were aligned using ClustalW2 software (http://www.ebi.ac.uk/tools/msa/clustalw2/).
Statistical analysis.
The data presented are the means ± standard deviations (SD) obtained from at least two independent biological replicates. Error bars represent the SD. Statistical significance was calculated using the Mann-Whitney test. A P value of <0.05 was considered statistically significant.
RESULTS
Inhibition of EV-71 infection by heparin, dextran sulfate, and suramin.
To determine whether surface GAGs play a significant role in virus-receptor interactions, various concentrations of different GAGs were preincubated with EV-71 at an MOI of 0.1 for 1 h at 37°C before infection of RD cells. The viral titers were quantified 24 h postinfection by using TaqMan quantitative real-time PCR and plaque assays. Of the two GAGs evaluated, significant inhibition of EV-71 by heparin and chondroitin sulfate was observed only at concentrations above 500 μg/ml (Fig. 1A). The inhibitory effect of heparin was more significant, at 79.4% ± 2.9%, than that of chondroitin sulfate (38.5% ± 6.4%) when they were tested at a concentration of 1,000 μg/ml (P < 0.05). A similar inhibitory trend was observed in the plaque assays (Table 1).
Fig 1.
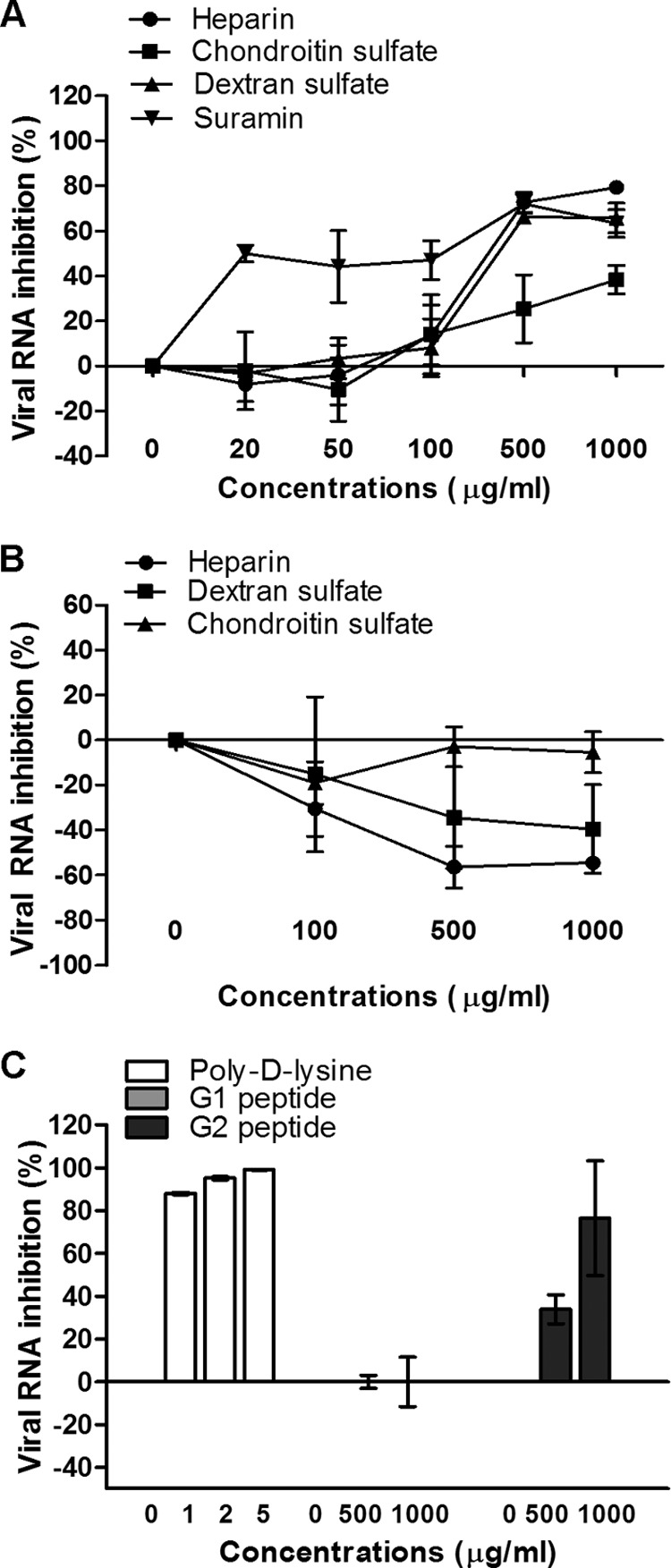
Inhibitory effects of GAGs and inhibitors. (A) For the viral inactivation assay, various concentrations of GAGs, polyanionic dextran sulfate, and suramin were preincubated with EV-71 particles for 1 h at 37°C before infection of RD cells at room temperature. (B and C) For the cell protection assay, various concentrations of GAGs (B) or of poly-d-lysine or anti-heparan sulfate peptides, designated peptides G1 and G2 (C), were preincubated with RD cells for 1 h at 37°C before EV-71 infection. The viral RNA was extracted and quantified by quantitative real-time PCR 24 h postinfection.
Table 1.
Effects of GAGs, GAG variants, and other inhibitors on EV-71 infection
| Inhibitor_a_ | Inhibition (%) | |
|---|---|---|
| Viral plaque | Viral RNA | |
| Heparin | 95.5 ± 0.7 | 79.4 ± 2.9 |
| Chondroitin sulfate | 52.4 ± 16.4 | 38.4 ± 6.4 |
| De-N-sulfated heparin | 70.0 ± 1.1 | 39.7 ± 15.6 |
| Dextran sulfate | 95.2 ± 0.7 | 65.8 ± 6.7 |
| Suramin | 76.3 ± 2.4 | 63.4 ± 6.1 |
To further investigate the significance of the carbohydrate backbone for the inhibition of EV-71, polyanionic dextran sulfate (2.3 sulfate groups/glucosyl group) and the polysulfonate pharmaceutical suramin (6.0 sulfate groups/molecule) were used. Previous studies have shown that suramin inhibits viruses such as dengue virus (24) and hepatitis B virus (30), which bind to cell surface heparan sulfate. Viral inhibition increased with the concentration of the inhibitors tested, and at 1,000 μg/ml, the levels of inhibition were 65.8% ± 6.7% for dextran sulfate and 63.4% ± 6.1% for suramin (Table 1). However, suramin was more effective, since it was able to inhibit EV-71 infection at a concentration as low as 20 μg/ml (Fig. 1A).
Preincubation of RD cells with heparin or dextran sulfate at concentrations from 100 to 1,000 μg/ml before viral infection had no inhibitory effect but enhanced virus infectivity. The results demonstrated that the inhibitory effect was due to direct interaction of these compounds with the virus and not with the target cells (Fig. 1A and B).
Neutralization of negative charges on cell surfaces reduces EV-71 infection.
To assess the important role of negative charges carried on the cell surface in the binding of EV-71, we preincubated RD cells with poly-d-lysine to neutralize negative charges on cell surfaces before infection. As shown in Fig. 1C, poly-d-lysine strongly decreased the level of EV-71 infection when applied at concentrations from 1 to 5 μg/ml. At 5 μg/ml, the inhibition was 99.1% ± 0.2%. To ascertain whether the negative charge carried by heparan sulfate present on the cell surface plays a significant role in viral attachment, two anti-heparan sulfate peptides, G1 (LRSRTKIIRIRH) and G2 (MPRRRRIRRRQK), were evaluated for their inhibitory effects. The inhibitory effect of G2 was significant, with viral RNA inhibition of 76.5% ± 26.8% at 1,000 μg/ml. The G1 peptide did not inhibit EV-71 infection (Fig. 1C).
Heparin reduces the infectivity of both laboratory strains and low-passage-number isolates.
Heparin at 2,500 μg/ml was observed to have inhibitory effects both on the laboratory strains and on the low-passage-number EV-71 isolates tested (Fig. 2). The inhibitory effects differed between strains. The level of inhibition of the laboratory strains ranged from 61.4% to 98.0%, while that of the low-passage-number isolates ranged from 30.4% to 78.4%. Interestingly, heparin failed to inhibit the PV vaccine strain even when tested at 2,500 μg/ml.
Fig 2.
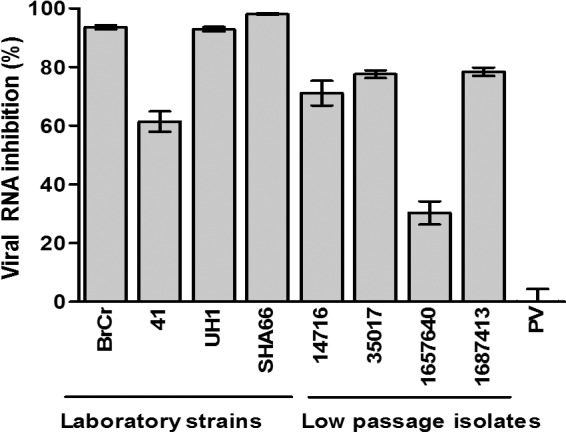
Inhibitory effects of heparin on EV-71 isolates and the PV vaccine strain. EV-71 and PV particles were pretreated with heparin at a final concentration of 2,500 μg/ml for 1 h at 37°C before infection of RD cells. The low-passage-number EV-71 isolates (14716, 35017, 1657640, and 1687413) and the PV vaccine strain were obtained from the Diagnostic Virology Laboratory, University Malaya Medical Center. The titers of EV-71 and the PV vaccine strain were quantified 24 h postinfection by TaqMan quantitative real-time PCR and plaque assays, respectively.
N- and O-sulfation on heparan sulfate are critical for EV-71 infection.
To establish whether the degree of sulfation of heparin is critical for viral inhibition, different sulfated heparin variants were investigated. Completely desulfated heparin failed to abolish EV-71 infection (Fig. 3A). In contrast, de-N-sulfated heparin showed moderate inhibition (39.7% ± 15.6%) at 1,000 μg/ml, indicating that the degree of sulfation within the GAG carbohydrate structure is functionally important. The inhibition of viral plaque formation by these GAGs and inhibitors is shown in Table 1.
Fig 3.
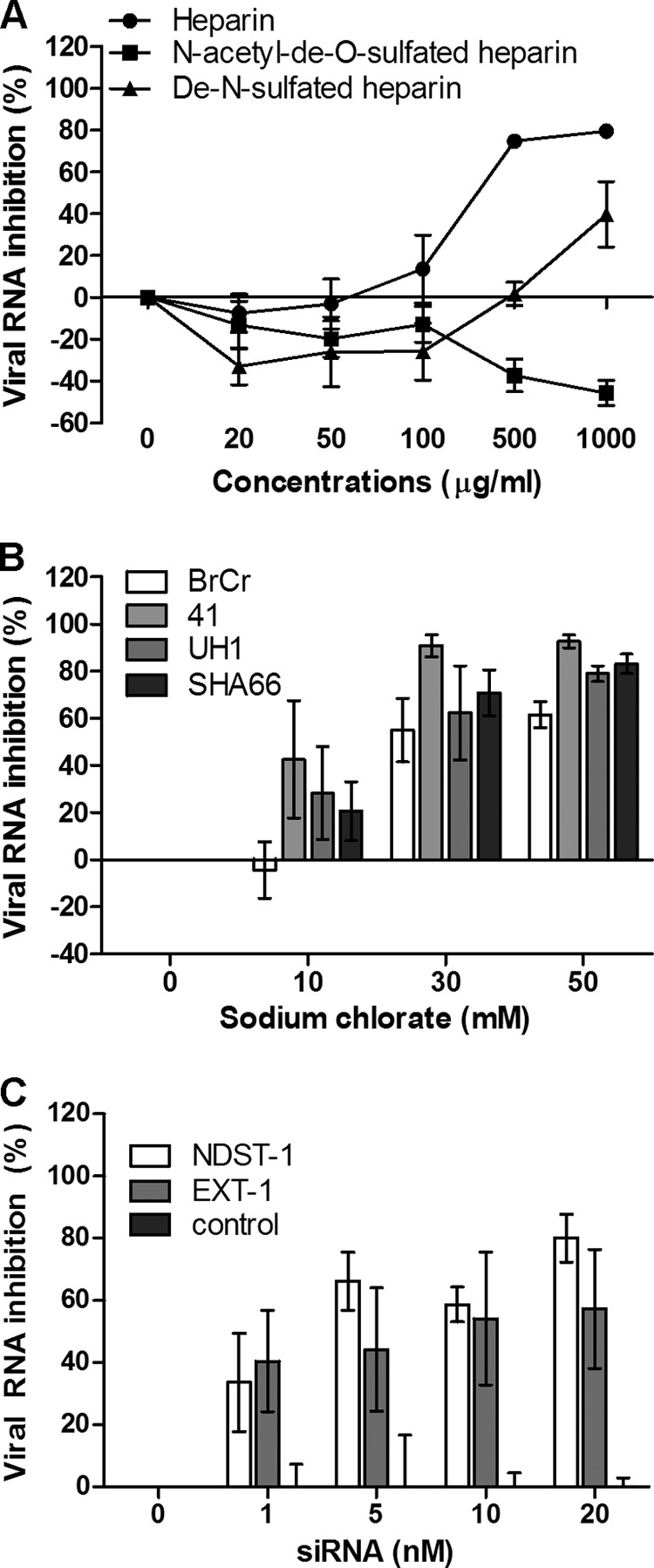
(A) EV-71 was preincubated with various concentrations of heparin and desulfated heparin variants for 1 h at 37°C before infection of RD cells. (B) Inhibitory effect of sodium chlorate on different EV-71 strains in RD cells. RD cells were pretreated with increasing concentrations (0 mM, 10 mM, 30 mM, and 50 mM) of sodium chlorate for 24 h before EV-71 infection at an MOI of 0.1. (C) Transient siRNA knockdown of NDST-1 and EXT-1 expression. NDST-1, EXT-1, and negative-control siRNAs in the Lipofectamine 2000 reagent were transfected into RD cells for 24 h before EV-71 infection. The viral load was determined 24 h postinfection by TaqMan quantitative real-time PCR.
To further confirm the role of the sulfation of heparan sulfate in EV-71 attachment, we carried out EV-71 infection of RD cells grown in a medium containing 0 to 50 mM sodium chlorate. The growth of cells in the presence of sodium chlorate, which inhibits cellular ATP-sulfurylase, has been shown previously to reduce the extent of sulfation of heparan sulfate by as much as 60%, and cells with such modified cell surfaces have been used to examine the role of GAGs in attachment by other viruses (25, 42). As shown in Fig. 3B, sodium chlorate was found to reduce the level of EV-71 infection of RD cells significantly, in a dose-dependent manner, with all strains tested. At 50 mM, sodium chlorate inhibited infection with EV-71 strains 41, UH1, SHA66, and BrCr; the percentages of viral RNA inhibition were 92.7% ± 2.7%, 79.0% ± 3.3%, 83.2% ± 4.2%, and 61.5% ± 5.5%, respectively. To rule out the possibility that the reduction in the level of EV-71 infection was due to cytotoxicity, the cytotoxicity of sodium chlorate was evaluated by the commercially available 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. The data revealed that sodium chlorate has minimal cytotoxicity at 50 mM.
To further confirm that heparan sulfate plays an important role in EV-71 infection, expression of the NDST-1 and EXT-1 genes was transiently knocked down using siRNA. NDST-1 is a heparan sulfate modification enzyme that removes _N_-acetyl groups from selected _N_-acetylglucosamine (GlcNAc) residues and replaces them with sulfate groups (43). EXT-1 is a heparan sulfate polymerase that adds alternating units of glucuronic acid (GlcA) and GlcNAc to the nonreducing end of the chain. As shown in Fig. 3C, transient knockdown of NDST-1 and EXT-1 expression in RD cells significantly reduced the level of EV-71 infection, with percentages of inhibition as high as 80.1% ± 7.7% and 57.2% ± 19.1%, respectively, at 20 nM siRNA. However, the negative-control siRNA was unable to reduce EV-71 infection.
Enzymatic removal of heparan sulfate from the cell surface reduces the levels of EV-71 binding and infection of RD cells.
To identify the type of GAG that is responsible for the binding of EV-71 to RD cells, we examined EV-71 binding to cells after enzymatic removal of cell surface GAGs by chondroitinase ABC and heparinase I/II/III digestion. Heparinase I degrades heparin and highly sulfated domains in heparan sulfate, while heparinase II cleaves both heparin and heparan sulfate, and heparinase III specifically degrades heparan sulfate (24). Treatment of RD cells with each of the heparinases at 2.5 mIU/ml and 5.0 mIU/ml for 1 h at 37°C was found to reduce viral RNA levels and plaque formation significantly (data not shown), in a dose-dependent manner, 24 h postinfection. Treatment of RD cells with heparinases I, II, and III at 5.0 mIU/ml significantly inhibited EV-71 RNA levels by 68.6% ± 6.2%, 91.7% ± 4.1%, and 82.2% ± 11.7%, respectively (Fig. 4A). Removal of cell surface chondroitin sulfate by chondroitinase ABC failed to inhibit EV-71 infection (Fig. 4A), even when chondroitinase ABC was tested at concentrations as high as 20 mIU/ml (data not shown). Removal of cell surface heparan sulfate significantly reduced the infectivities of the different EV-71 strains tested, but not that of the PV vaccine strain (Fig. 4B). Removal of surface heparan sulfate, but not chondroitin sulfate, was found to significantly reduce EV-71 attachment to the surfaces of RD cells (Fig. 4C).
Fig 4.

Treatment of RD cells with heparinase or chondroitinase ABC. (A) Inhibitory effects of heparinase I/II/III and chondroitinase ABC on EV-71 infection. RD cells were pretreated with heparinase or chondroitinase ABC for 1 h at 37°C before EV-71 infection at an MOI of 0.1. The viral RNA was extracted and evaluated by TaqMan quantitative real-time PCR. (B) Inhibitory effect of heparinase I on different EV-71 strains and the PV vaccine strain. (C) Confocal microscopy analysis (with a 40× objective) of EV-71 binding assays after heparinase I or chondroitinase ABC treatment. EV-71 particles were stained with a monoclonal anti-EV-71 antibody and were subsequently stained with Alexa Fluor 488-labeled anti-mouse IgG. Nuclei were stained with DAPI. EV-71 particles and nuclei are shown in green and blue, respectively.
Significant reduction in the level of EV-71 binding to GAG-deficient CHO cells.
CHO cells with defects in the biosynthesis of GAGs have been used extensively to demonstrate the involvement of heparan sulfate as the receptor for the binding of various viruses (29, 30, 33, 42, 44). Mutant CHO-pgsD677 cells are deficient in _N_-acetylglucosaminyltransferase and glucuronosyltransferase activities, which are required for heparan sulfate polymerization, and thus completely lack heparan sulfate; these cells also produce levels of chondroitin sulfate 3- to 4-fold higher than those in wild-type CHO-K1 cells (45). Mutant CHO-pgsA745 cells are deficient in the enzyme UDP-d-xylose:serine-1,3-d-xylosyltransferase, which catalyzes the first sugar transfer reaction in GAG formation, and thus completely lack GAGs (46). To investigate whether EV-71 binds differently to these cell lines, cells were seeded in CellCarrier-96 plates and chamber slides and were infected with EV-71 at an MOI of 100 for 1 h at 4°C. As shown in Fig. 5A and B, significantly fewer viral particles were attached to CHO-pgsD677 and CHO-pgsA745 cells than to wild-type CHO-K1 cells, which expressed heparan sulfate (P < 0.001). CHO-pgsD677 and CHO-pgsA745 cells showed reduced binding levels of 46.7% and 41.6%, respectively, compared to 100% for CHO-K1 cells. Interestingly, more viral particles were bound to RD cells than to CHO-K1 cells.
Fig 5.

Binding of EV-71 to CHO-K1 and CHO mutant cells. CHO-K1 cells and cells of CHO mutants defective in proteoglycan synthesis were assessed for their abilities to bind to EV-71. The CHO-pgsA745 cell line lacks heparan sulfate and chondroitin sulfate proteoglycans, while the CHO-pgsD677 cell line lacks heparan sulfate proteoglycan but produces 15% of normal proteoglycans. The binding of EV-71 to parental and mutant CHO cells was assayed by using confocal microscopic analysis (A) and a Cellomics ArrayScan VTI HCS reader with Spot Detector BioApplication software (B) and was verified by TaqMan quantitative real-time PCR.
Binding of EV-71 to immobilized heparin-Sepharose.
To characterize the interaction of EV-71 particles with GAGs, a virus-containing supernatant was applied to a heparin affinity chromatography column. EV-71 particles bound to immobilized heparin-Sepharose under physiological salt conditions (0.14 M NaCl) and were eluted by 2 M NaCl. The virus titers in each fraction collected were quantified by TaqMan quantitative real-time PCR. As shown in Fig. 6, most of the EV-71 particles were detected in all the eluates following the application of 2 M NaCl. The EV-71 particles present in eluate 1 were concentrated as much as 4.1-fold. In a control experiment, we used a column packed with Sepharose alone and found no binding of EV-71 particles to the column (data not shown). The results confirm that EV-71 particles bind to heparin and were eluted by high salt concentrations. In contrast to EV-71, the PV vaccine strain did not interact with heparin-Sepharose, and most of the PV particles were detected in the flowthrough fraction (Fig. 6).
Fig 6.
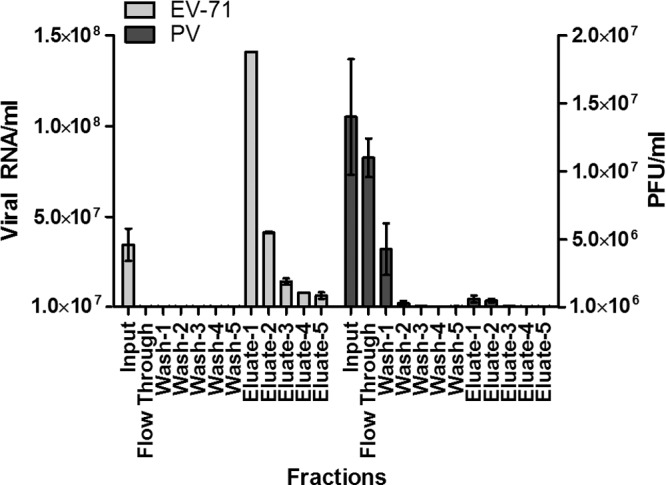
Binding of EV-71 and PV to an immobilized heparin-Sepharose column. EV-71 and PV supernatants were passed through a column of immobilized heparin-Sepharose and were eluted with 2 M NaCl. The titers of EV-71 and the PV vaccine strain from each fraction were quantified by TaqMan quantitative real-time PCR and plaque assays, respectively. EV-71 levels are presented as viral RNA copies per milliliter, and levels of the PV vaccine strain are presented as PFU per milliliter. Error bars represent means ± SD for each fraction.
Symmetry clustering of highly conserved amino acids Arg166, Lys242, and Lys244 of VP1 in the EV-71 pentamer structure.
To determine the possible heparan sulfate binding site(s) on EV-71 particles, the 3-dimensional crystal structure of EV-71 was built using the DeepView-Swiss PDB viewer (40). As shown in Fig. 7A, amino acids Arg166, Lys242, and Lys244 are arranged symmetrically in the 5-fold axis of the EV-71 pentamer structure. These amino acids are also located at positions that are highly exposed on the surface of the EV-71 particle (Fig. 7B). All these amino acids were highly conserved across all EV-71 genotypes except for Lys244, where lysine (K) was replaced by glutamic acid (E) in genotype A (Fig. 7C). This symmetrically arranged clustering of positively charged amino acids could serve as the binding site for heparan sulfate.
Fig 7.

Three-dimensional pentameric structure and sequence alignment of EV-71. The structure of the EV-71 pentamer was generated using the DeepView-Swiss PDB viewer. The molecular structures of EV-71 VP1, VP2, VP3, and VP4 are presented in blue, green, red, and purple, respectively. The amino acids Arg166, Lys242, and Lys244 are presented in yellow, white, and light blue, respectively. (A) Top view of the EV-71 pentamer. (B) Side view of the EV-71 pentamer. (C) Histogram showing sequence consensus in the VP1 region of EV-71. A total of 174 sequences were aligned and analyzed using ClustalW2. The arbitrary scale is shown at the left of the histogram; 1.0 denotes perfect consensus at a given amino acid site across all entries. The alignments of Arg166, Lys242, and Lys244 in representative EV-71 strains from genotypes A, B, and C are shown above the histogram.
DISCUSSION
Virus receptors may be one of the determinants of virus host range and tissue tropism (5). Receptors that are known to bind to EV-71 have been identified over the past few years. P-selectin glycoprotein ligand-1 (PSGL-1) was the first to be discovered as the EV-71 receptor. However, PSGL-1 is selectively expressed only in neutrophils, monocytes, and most lymphocytes (6). The second receptor, reported by Yamayoshi et al. (9), was human SCARB2, which is expressed on most types of cells (7). EV-71 binds to the SCARB2 receptor and is then internalized through clathrin-mediated endocytosis (47, 48). However, blocking of these receptors did not abolish EV-71 infection, and this finding led to the discovery of the third receptor, annexin II. Pretreatment of host cells with an anti-annexin II antibody was found to reduce the level of viral attachment. Preincubation of EV-71 with annexin II also reduced the level of EV-71 infection (12).
Besides these known receptors for EV-71, sialylated glycans were reported by Yang et al. (11) as receptors for EV-71. Treatment of RD, SK-N-SH, and DLD-1 cells with neuraminidase was found to reduce the ability of EV-71 to bind to the cell surface, confirming that EV-71 utilizes sialylated glycans as attachment factors (10). We have shown in this study that, in addition to the four receptors published to date, cell surface heparan sulfate is required for EV-71 attachment. The level of EV-71 infection of RD cells was significantly reduced when EV-71 particles were pretreated with soluble heparin, dextran sulfate, or suramin. However, preincubation of RD cells with heparin or dextran sulfate did not inhibit EV-71 infection but unexpectedly enhanced it. This may be caused by the creation of artificial binding sites through accumulation of heparin and dextran sulfate on the surfaces of cells (30, 49). These findings imply that heparin or dextran sulfate could bind to the positively charged surfaces of viral particles and prevent the interaction of viral particles with the cell surface, as demonstrated for foot-and-mouth disease virus (50). Removal of cell surface heparan sulfate using each of the heparinases (I, II, and III) also reduced the ability of EV-71 to bind to and infect RD cells, further confirming the important role of heparan sulfate in EV-71 infection. The possible involvement of chondroitin sulfate as a receptor for attachment was excluded based on the evidence that enzymatic removal of chondroitin sulfate did not impair EV-71 infection.
In the present study, we demonstrated that EV-71 can bind to CHO cells. However, CHO cells defective in the biosynthesis of heparan sulfate and chondroitin sulfate exhibited a reduced ability to bind to EV-71, further supporting the role of heparan sulfate in EV-71 infection. While EV-71 could bind to the CHO-K1 cells, no infection was observed, suggesting that EV-71 failed to internalize so as to initiate infection. TMEV GDVII bound to heparan sulfate has been postulated to use protein entry receptors for virus internalization (27). A similar mechanism could be utilized by EV-71. EV-71 particles bound to heparan sulfate may need to interact with known receptors, such as SCARB2 and sialylated glycan, or with an unknown protein entry receptor, to gain entry into cells. CHO cells, of hamster origin, may have a different SCARB2, since it has been reported that EV-71 binding domains in human SCARB2 and murine SCARB2 are different (51). In this study, we also showed that EV-71 was still able to bind to the CHO mutant completely lacking in heparan sulfate and chondroitin sulfate, which further suggests that multiple receptors are involved during EV-71 infection. This view is similar to what has been reported for several viruses that use heparan sulfate (27, 52, 53).
Heparan sulfate is expressed by all cell types. However, heparan sulfate expressed on different types of cells shows variations in structure, with differences in the degrees of sulfation, chain length, and the position of the sulfate group (54). Our data demonstrated that highly sulfated heparin could inhibit EV-71 infection, whereas decreased sulfation on heparin led to a loss of EV-71 inhibition. Completely desulfated heparin was found to have no inhibitory effect. These findings suggest that structural differences in the heparan sulfate present in different cell types could lead to differences in susceptibility to infection, which may contribute to the selective tropism of EV-71 (37).
Heparan sulfate binding could arise from adaptation to the cells in which isolates were propagated. Culture adaptations of heparan sulfate usage in picornaviruses (55, 56), flaviviruses (57), and alphaviruses (58) have been demonstrated. In vitro cultivation of viruses has been shown to rapidly select amino acid substitutions that increase the net positive charge of the envelope protein (59). However, the heparan sulfate binding phenotype has also been documented for clinical isolates of the porcine circoviruses (53) and echoviruses (33). In the present study, we showed that heparin was able to inhibit EV-71 isolates that had been passaged only once in tissue culture, suggesting that heparan sulfate binding phenotypes are not likely to be due to culture adaptation of EV-71. We also provide evidence that heparin failed to inhibit the infection of RD cells with the PV vaccine strain, further strengthening our view that EV-71 binds to heparan sulfate.
Furthermore, we also presented evidence that EV-71 binds to heparin-Sepharose under physiological salt concentrations and that the interaction could be disrupted by high salt concentrations. This finding suggests that EV-71–heparin interactions involve mainly electrostatic charge interactions. Replacement of a single positively charged amino acid with another amino acid is sufficient to retard the heparin binding phenotype in TMEV GDVII and coxsackievirus A9 (27, 34, 60). Thus, positively charged amino acids on the surfaces of viral particles are critical for the heparin binding phenotype. Sequence analysis of the VP1 capsid protein revealed two possible heparan sulfate mimic binding domains, from residues 120 to 123 (-RRKV-) and residues 241 to 244 (-SKSK-). Interestingly, our previous study has shown that peptides with these heparan sulfate binding domains exhibited significant antiviral activities against EV-71 infection in RD cells (14), suggesting that the inhibitory effect could have resulted from direct binding to heparan sulfate and blocking of viral attachment. Heparan sulfate-interacting regions could be linear heparan sulfate binding domains, as well as clusters of basic residues that arise as a result of the 3-dimensional structure, as shown for foot-and-mouth disease virus and coxsackievirus A9 (34, 50). With the recently available crystal structure of EV-71, the electrostatic surface of EV-71 was determined (61). A few conserved positively charged amino acids (Arg166, Lys242, and Lys244) were clustered symmetrically at the 5-fold axis of the EV-71 pentamer. The linear arrangement of these amino acids in the cluster could also be significant for the interactions. These amino acids were also located on the surfaces of EV-71 particles. These findings suggest that the interaction could be due to the electrostatic interactions between these clusters of basic amino acids arranged in a 3-dimensional array on the virions and concentrated negative charges on the sulfated heparan sulfate chain.
In this study, we have shown that EV-71 particles utilized cell surface heparan sulfate as an attachment receptor. Although the exact mechanism of interaction of EV-71 particles with negatively charged heparan sulfate on the cell surface remains unclear, determination of the molecular basis of the virus-receptor interaction in target cells will be useful for understanding the pathogenicity of EV-71 and for the development of antiviral agents against EV-71.
ACKNOWLEDGMENTS
We acknowledge Lim Fei Tieng (Hi-Tech Instruments Sdn Bhd, Malaysia) for assistance in the confocal microscopy analysis.
This work was supported by University of Malaya research grants (RG245/10HTM and RG298/11HTM), University of Malaya High Impact research grants (E000013-20001 and UM.C/625/1/HIR/014), a Fundamental Research Grant Scheme (FP015/2012A) from the Ministry of Education, Malaysia, a postgraduate research grant (PV013/2012A) from the University of Malaya, and a Sunway University research grant (SHNS-0111-01).
Footnotes
Published ahead of print 24 October 2012
REFERENCES
- 1.Brown BA, Pallansch MA. 1995. Complete nucleotide sequence of enterovirus 71 is distinct from poliovirus. Virus Res. 39:195–205 [DOI] [PubMed] [Google Scholar]
- 2.Chan YF, Sam IC, Wee KL, AbuBakar S. 2011. Enterovirus 71 in Malaysia: a decade later. Neurol. Asia 16:1–15 [Google Scholar]
- 3.Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. 2010. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 9:1097–1105 [DOI] [PubMed] [Google Scholar]
- 4.Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. 2010. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 10:778–790 [DOI] [PubMed] [Google Scholar]
- 5.Haywood AM. 1994. Virus receptors: binding, adhesion strengthening, and changes in viral structure. J. Virol. 68:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. 2009. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat. Med. 15:794–797 [DOI] [PubMed] [Google Scholar]
- 7.Yamayoshi S, Iizuka S, Yamashita T, Minagawa H, Mizuta K, Okamoto M, Nishimura H, Sanjoh K, Katsushima N, Itagaki T, Nagai Y, Fujii K, Koike S. 2012. Human SCARB2-dependent infection by coxsackievirus A7, A14, and A16 and enterovirus 71. J. Virol. 86:5686–5696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamayoshi S, Koike S. 2011. Identification of a human SCARB2 region that is important for enterovirus 71 binding and infection. J. Virol. 85:4937–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, Koike S. 2009. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 15:798–801 [DOI] [PubMed] [Google Scholar]
- 10.Su PY, Liu YT, Chang HY, Huang SW, Wang YF, Yu CK, Wang JR, Chang CF. 2012. Cell surface sialylation affects binding of enterovirus 71 to rhabdomyosarcoma and neuroblastoma cells. BMC Microbiol. 12:162 doi:10.1186/1471-2180-12-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang B, Chuang H, Yang KD. 2009. Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol. J. 6:141 doi:10.1186/1743-422X-6-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang SL, Chou YT, Wu CN, Ho MS. 2011. Annexin II binds to capsid protein VP1 of enterovirus 71 and enhances viral infectivity. J. Virol. 85:11809–11820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu KX, Ng MM, Chu JJ. 2010. Developments towards antiviral therapies against enterovirus 71. Drug Discov. Today 15:1041–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan CW, Chan YF, Sim KM, Tan EL, Poh CL. 2012. Inhibition of enterovirus 71 (EV-71) infections by a novel antiviral peptide derived from EV-71 capsid protein VP1. PLoS One 7:e34589 doi:10.1371/journal.pone.0034589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin TY, Chu C, Chiu CH. 2002. Lactoferrin inhibits enterovirus 71 infection of human embryonal rhabdomyosarcoma cells in vitro. J. Infect. Dis. 186:1161–1164 [DOI] [PubMed] [Google Scholar]
- 16.Weng TY, Chen LC, Shyu HW, Chen SH, Wang JR, Yu CK, Lei HY, Yeh TM. 2005. Lactoferrin inhibits enterovirus 71 infection by binding to VP1 protein and host cells. Antiviral Res. 67:31–37 [DOI] [PubMed] [Google Scholar]
- 17.Pourianfar HR, Poh CL, Fecondo J, Grollo L. 2012. In vitro evaluation of the antiviral activity of heparan sulfate mimetic compounds against enterovirus 71. Virus Res. 169:22–29 [DOI] [PubMed] [Google Scholar]
- 18.Honke K, Taniguchi N. 2002. Sulfotransferases and sulfated oligosaccharides. Med. Res. Rev. 22:637–654 [DOI] [PubMed] [Google Scholar]
- 19.Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. 1998. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 20:156–167 [DOI] [PubMed] [Google Scholar]
- 20.Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC. 1998. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 72:6119–6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.WuDunn D, Spear PG. 1989. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63:52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mondor I, Ugolini S, Sattentau QJ. 1998. Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J. Virol. 72:3623–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roderiquez G, Oravecz T, Yanagishita M, Bou-Habib DC, Mostowski H, Norcross MA. 1995. Mediation of human immunodeficiency virus type 1 binding by interaction of cell surface heparan sulfate proteoglycans with the V3 region of envelope gp120-gp41. J. Virol. 69:2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ, Marks RM. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3:866–871 [DOI] [PubMed] [Google Scholar]
- 25.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75:1565–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72:1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddi HV, Lipton HL. 2002. Heparan sulfate mediates infection of high-neurovirulence Theiler's viruses. J. Virol. 76:8400–8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dechecchi MC, Melotti P, Bonizzato A, Santacatterina M, Chilosi M, Cabrini G. 2001. Heparan sulfate glycosaminoglycans are receptors sufficient to mediate the initial binding of adenovirus types 2 and 5. J. Virol. 75:8772–8780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Summerford C, Samulski RJ. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72:1438–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulze A, Gripon P, Urban S. 2007. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 46:1759–1768 [DOI] [PubMed] [Google Scholar]
- 31.Barth H, Schafer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, Kinoshita-Toyoda A, Toida T, Van Kuppevelt TH, Depla E, Von Weizsacker F, Blum HE, Baumert TF. 2003. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 278:41003–41012 [DOI] [PubMed] [Google Scholar]
- 32.Byrnes AP, Griffin DE. 1998. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 72:7349–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodfellow IG, Sioofy AB, Powell RM, Evans DJ. 2001. Echoviruses bind heparan sulfate at the cell surface. J. Virol. 75:4918–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLeish NJ, Williams CH, Kaloudas D, Roivainen M, Stanway G. 2012. Symmetry-related clustering of positive charges is a common mechanism for heparan sulfate binding in enteroviruses. J. Virol. 86:11163–11170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zautner AE, Jahn B, Hammerschmidt E, Wutzler P, Schmidtke M. 2006. N- and 6-O-sulfated heparan sulfates mediate internalization of coxsackievirus B3 variant PD into CHO-K1 cells. J. Virol. 80:6629–6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harwood LJ, Gerber H, Sobrino F, Summerfield A, McCullough KC. 2008. Dendritic cell internalization of foot-and-mouth disease virus: influence of heparan sulfate binding on virus uptake and induction of the immune response. J. Virol. 82:6379–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson T, Ellard FM, Ghazaleh RA, Brookes SM, Blakemore WE, Corteyn AH, Stuart DI, Newman JW, King AM. 1996. Efficient infection of cells in culture by type O foot-and-mouth disease virus requires binding to cell surface heparan sulfate. J. Virol. 70:5282–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiwari V, Liu J, Valyi-Nagy T, Shukla D. 2011. Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J. Biol. Chem. 286:25406–25415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plevka P, Perera R, Cardosa J, Kuhn RJ, Rossmann MG. 2012. Crystal structure of human enterovirus 71. Science 336:1274 doi:10.1126/science.1218713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723 [DOI] [PubMed] [Google Scholar]
- 41.Chan YF, Sam IC, AbuBakar S. 2010. Phylogenetic designation of enterovirus 71 genotypes and subgenotypes using complete genome sequences. Infect. Genet. Evol. 10:404–412 [DOI] [PubMed] [Google Scholar]
- 42.Guibinga GH, Miyanohara A, Esko JD, Friedmann T. 2002. Cell surface heparan sulfate is a receptor for attachment of envelope protein-free retrovirus-like particles and VSV-G pseudotyped MLV-derived retrovirus vectors to target cells. Mol. Ther. 5:538–546 [DOI] [PubMed] [Google Scholar]
- 43.Presto J, Thuveson M, Carlsson P, Busse M, Wilen M, Eriksson I, Kusche-Gullberg M, Kjellen L. 2008. Heparan sulfate biosynthesis enzymes EXT1 and EXT2 affect NDST1 expression and heparan sulfate sulfation. Proc. Natl. Acad. Sci. U. S. A. 105:4751–4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vlasak M, Goesler I, Blaas D. 2005. Human rhinovirus type 89 variants use heparan sulfate proteoglycan for cell attachment. J. Virol. 79:5963–5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lidholt K, Weinke JL, Kiser CS, Lugemwa FN, Bame KJ, Cheifetz S, Massague J, Lindahl U, Esko JD. 1992. A single mutation affects both _N_-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 89:2267–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esko JD, Stewart TE, Taylor WH. 1985. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 82:3197–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hussain KM, Leong KL, Ng MM, Chu JJ. 2011. The essential role of clathrin-mediated endocytosis in the infectious entry of human enterovirus 71. J. Biol. Chem. 286:309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin YW, Lin HY, Tsou YL, Chitra E, Hsiao KN, Shao HY, Liu CC, Sia C, Chong P, Chow YH. 2012. Human SCARB2-mediated entry and endocytosis of EV71. PLoS One 7:e30507 doi:10.1371/journal.pone.0030507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hilgard P, Stockert R. 2000. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology 32:1069–1077 [DOI] [PubMed] [Google Scholar]
- 50.Fry EE, Lea SM, Jackson T, Newman JW, Ellard FM, Blakemore WE, Abu-Ghazaleh R, Samuel A, King AM, Stuart DI. 1999. The structure and function of a foot-and-mouth disease virus-oligosaccharide receptor complex. EMBO J. 18:543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen P, Song Z, Qi Y, Feng X, Xu N, Sun Y, Wu X, Yao X, Mao Q, Li X, Dong W, Wan X, Huang N, Shen X, Liang Z, Li W. 2012. Molecular determinants of enterovirus 71 viral entry: cleft around GLN-172 on VP1 protein interacts with variable region on scavenge receptor B2. J. Biol. Chem. 287:6406–6420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delputte PL, Vanderheijden N, Nauwynck HJ, Pensaert MB. 2002. Involvement of the matrix protein in attachment of porcine reproductive and respiratory syndrome virus to a heparin like receptor on porcine alveolar macrophages. J. Virol. 76:4312–4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Misinzo G, Delputte PL, Meerts P, Lefebvre DJ, Nauwynck HJ. 2006. Porcine circovirus 2 uses heparan sulfate and chondroitin sulfate B glycosaminoglycans as receptors for its attachment to host cells. J. Virol. 80:3487–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi X, Zaia J. 2009. Organ-specific heparan sulfate structural phenotypes. J. Biol. Chem. 284:11806–11814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Escarmis C, Carrillo EC, Ferrer M, Arriaza JF, Lopez N, Tami C, Verdaguer N, Domingo E, Franze-Fernandez MT. 1998. Rapid selection in modified BHK-21 cells of a foot-and-mouth disease virus variant showing alterations in cell tropism. J. Virol. 72:10171–10179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sa-Carvalho D, Rieder E, Baxt B, Rodarte R, Tanuri A, Mason PW. 1997. Tissue culture adaptation of foot-and-mouth disease virus selects viruses that bind to heparin and are attenuated in cattle. J. Virol. 71:5115–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee E, Hall RA, Lobigs M. 2004. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J. Virol. 78:8271–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klimstra WB, Ryman KD, Johnston RE. 1998. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 72:7357–7366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee E, Lobigs M. 2002. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray Valley encephalitis virus. J. Virol. 76:4901–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reddi HV, Kumar AS, Kung AY, Kallio PD, Schlitt BP, Lipton HL. 2004. Heparan sulfate-independent infection attenuates high-neurovirulence GDVII virus-induced encephalitis. J. Virol. 78:8909–8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X, Peng W, Ren J, Hu Z, Xu J, Lou Z, Li X, Yin W, Shen X, Porta C, Walter TS, Evans G, Axford D, Owen R, Rowlands DJ, Wang J, Stuart DI, Fry EE, Rao Z. 2012. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 19:424–429 [DOI] [PMC free article] [PubMed] [Google Scholar]