JAB1/CSN5 inhibits the activity of Luman/CREB3 by promoting its degradation (original) (raw)
. Author manuscript; available in PMC: 2016 Sep 14.
Published in final edited form as: Biochim Biophys Acta. 2013 Apr 11;1829(9):921–929. doi: 10.1016/j.bbagrm.2013.04.001
Abstract
Luman/CREB3 (also called LZIP) is an endoplasmic reticulum (ER)-bound transcription factor that has been implicated in the ER stress response. In this study, we used the region of Luman containing the basic DNA-binding domain as bait in a yeast two-hybrid screen and identified the Jun activation domain-binding protein 1 (JAB1) or the COP9 signalosome complex unit 5 (CSN5) as an interacting protein. We confirmed their direct binding by glutathione _S_-transferase pull-down assays, and verified the existence of such interaction in the cellular environment by mammalian two-hybrid and co-immunoprecipitation assays. Deletion mapping studies revealed that the MPN domain in JAB1 was essential and sufficient for the binding. JAB1 also colocalized with Luman in transfected cells. More interestingly, the nuclear form of Luman was shown to promote the translocation of JAB1 into the nucleus. We found that overexpression of JAB1 shortened the half-life of Luman by 67%, and repressed its transactivation function on GAL4 and unfolded protein response element (UPRE)-containing promoters. We therefore propose that JAB1 is a novel binding partner of Luman, which negatively regulates the activity of Luman by promoting its degradation.
Keywords: Luman/CREB3/LZIP, JAB1/CSN5, Unfolded protein response, Protein degradation, Transcription factor
1. Introduction
Luman/CREB3 [1], also called LZIP [2], is identified through its association with herpes simplex virus (HSV)-related host cell factor 1 (HCF-1) [3,4]. The mode of interaction between Luman and HCF-1 is mimicked by the HSV-1 protein VP16, which has led to the hypothesis that Luman may play a role in the viral reactivation from latency [2,5,6]. Current data suggest that the primary cellular function of Luman may be in the endoplasmic reticulum stress response [2]. Luman has been implicated in HIV replication [7], dendritic cell maturation [8], breast cancer metastasis [9] and monocyte cell migration [10].
Luman is a cAMP response element (CRE)-binding protein that has four other closely related CREB3 family members, including CREB-H or CREB3-like 1 (CREB3L1) [11,12], BBF2H7/CREB3L2 [13], OASIS/CREB3L3 [14] and CREB4/AIbZIP/Atce1/Tisp40/CREB3L4 [15–19]. As transcript factors, they share a unique structural feature — a hydrophobic transmembrane domain that tethers them to the endoplasmic reticulum (ER). All CREB3 family members appear to play a role in the ER stress response or the unfolded protein response (UPR) [reviewed in 20,21], although with different tissue specificities. In the event of the UPR, these CREB3 proteins are believed to be cleaved and released from the ER by regulated intramembrane proteolysis, translocating into the nucleus and activating downstream target genes [8,13,16,19,22–24].
In addition to CRE, these CREB3 proteins differentially bind to various enhancer elements commonly found in the promoter region of UPR-related genes. Luman can bind the CAAT enhancer binding protein (C/EBP) element [1] and the Tax responsive element (TxRE) [25]. Recently, the ER-associated degradation (ERAD)-related protein Herp (homocysteine-induced ER protein) [26] or Mif1 [27] has been found to be a direct downstream target of Luman [23]. Luman induces cellular Herp expression during the UPR via transactivation of an ERSE-II enhancer element in the promoter. Luman may also induce another ERAD protein EDEM, through a UPRE-like element [28]. On the basis of viral mimicry, we have thus proposed that Luman may play a unique role in the ERAD that is fundamental to HSV lytic/latent replication cycle [23].
Here we report the identification of JAB1 (Jun activation domain-binding protein-1) as a cellular ligand of Luman using a yeast two-hybrid strategy. We verified the interaction between Luman and JAB1 with in vitro and in vivo assays. We found that JAB1 could repress the activation potential of Luman and decrease its protein stability.
2. Material and methods
2.1. Yeast two-hybrid screen
The Matchmaker 3 yeast two-hybrid system and a pre-transformed adult human brain Matchmaker cDNA library (BD Biosciences Clontech) were used in the screen. All experimental procedures followed the manufacturer’s instruction. Briefly, the bait plasmid pGBKT7/LU(AD-BD) was constructed by cloning a PCR fragment of Luman cDNA (a.a. 55–181) into the pGBKT7 vector. The bait plasmid was then transformed into the Saccharomyces cerevisiae MATα reporter strain AH109 using the small-scale LiAc protocol. For library screens, the AH109[pGBKT7/LU(AD-BD)] reporter strain was mated with S. cerevisiae MATa strain Y187, pre-transformed with an adult human brain Matchmaker cDNA library, and plated on SD/-Leu/-Trp/-Ade/-His minimal media supplemented with 10 mM 3-amino-1,2,4-triazole (3-AT). Plates were incubated at 30 °C for up to 16 days. Approximately 5 × 105 colonies were screened. Forty-three colonies were picked up and re-streaked onto to new SD/-Ade/-His/-Leu/-Trp plates, out of which 24 grew rapidly. Plasmids were recovered from these yeast strains, and were cotransformed back into the yeast strain AH109 with the bait plasmid pGBKT7/LU(AD-BD), and selected on SD/-Ade/-His/-Leu/-Trp plates. Among them, 7 clones were found to be positive and were subsequently sequenced, one of which contained JAB1 cDNA.
2.2. Cell culture and transfection
Human embryonic kidney (HEK)-293, COS7 and Vero cells were grown in Dulbecco’s modified Eagle’s medium [high glucose (4500 mg/L), 4 mM L-glutamine; HyClone] supplemented with 10% fetal bovine serum (Sigma) and 1% v/v penicillin/streptomycin. Cells were maintained at 37 °C in a humidified 5% CO2 incubator and passaged every 2–3 days. Cells were transfected by the calcium phosphate precipitation method as previously described [6].
2.3. Plasmids
The full-length JAB1 cDNA was amplified using primers 5′-CTGAATTCCACACCCGGAAACCTAGC and 5′-GTACCTCGAGTATCAGATTTTGG GTAACT (attached _Eco_RI and _Xho_I restriction sites are underlined), and was cloned between _Eco_RI and _Xho_I sites of pcDNA3 with an N-terminal FLAG or HA epitope tag, yielding pFLAG-JAB1 and pHA-JAB1. The same strategy was used to clone the fragment into pGEX-KG (encoding a glutathione-S-transferase fusion protein at the N-terminus; gift from Gerry Weinmaster, University of California, Los Angeles) and pM1 (encoding an N-terminal GAL4 DNA binding domain fusion protein; gift from Ian Sadowski, University of British Columbia), which gave rise to pGEX-JAB1 and pM-JAB1.
JAB1 deletion mutant plasmids pHA-JAB1(1–195), pHA-JAB1(44–195), and pHA-JAB1(190–334) were constructed using the same cloning scheme as the full-length JAB1, and inserted into the _Eco_RI/_Xho_I sites of the pcDNA3 vector. The primer pairs for amplifying mutant JAB1 fragments a.a. 1–195, 44–195 and 190–334 were 5′-CTGAATTCAACGACAACTTCTCCGCTTCC/5′-GTGTCTCGAGTTAAGGTTTGTAGCCCTTTGG, 5′-GAGAATTCCCCTGGACTAAGGATCACCAT/5′-GTGTCTCGAGTTAAGGTTTGTAGCCCTTTGG and 5′-GTACCTCGAGTATCAGATTTTGGGTAACT/5′-CAGAATTCCCAAAGGGCTACAAACCT. The p5 × UPRE-luciferase plasmid [29] was a gift from Ron Prywes, Columbia University. Other plasmids used in this study have been described previously [23,28].
2.4. GST (glutathione S-transferase) pull-down
GST fusion proteins were produced in Escherichia coli strain BL21(DE3) (Novagen) and were purified using glutathione-sepharose beads (GE Healthcare) [1,5]. A rabbit reticulocyte in vitro transcription–translation system (TnT; Promega) was used to produce 35S-labeled Luman, JAB1 and its mutants, and the GAL4 activation domain (AD) fused to GFP (as a negative control) according to the manufacturer’s protocol. Protein concentrations of the bead slurry were calculated by comparison to BSA standards (Pierce). GST fusion proteins bound to glutathione-sepharose-4B beads were incubated for 1 h with in vitro 35S-labeled protein in the binding buffer [140 mM NaCl, 50 mM Tris, pH 8.0, 2 mM Na3VO4, 5 mg/mL BSA and 0.5% (vol/vol) Ipegal CA 630]. Beads were collected by centrifugation, washed and re-suspended in 2× SDS sample buffer. The eluted protein was separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), and visualized on a Typhoon 9400 PhosphorImager (GE Healthcare).
2.5. Dual luciferase reporter assays
Assays were performed according to the Dual Luciferase Reporting System manual (Promega). In each assay, 30 ng of pRL-SV40 and 1 μg of a reporter plasmid were used in the cell transfection. All reporter plasmids were based on the pGL3-promoter plasmid (Promega), encoding a firefly luciferase gene downstream of the response element. The pRL-SV40 plasmid encodes Renilla luciferase gene under the control of the SV40 promoter, which was used as an internal control for transfection efficiency.
At 16 h post-transfection, media were changed and the cells were allowed to recover for 8 h. Cells were harvested, lysed, and used in the dual luciferase assays. Luciferase activity was measured using a Turner TD-20e Luminometer and calculated as relative luciferase activity (firefly luciferase/Renilla luciferase). Assays were independently repeated at least 3 times. Data are shown with standard errors.
2.6. Co-immunoprecipitation
HEK-293 cells in 35-mm dishes were transiently transfected with a total of 5 μg of plasmid DNA (the blank vector pcDNA3 was used as filler DNA in single transfections). Cells were treated with a proteasome inhibitor MG132 at a final concentration of 5 μM for the last 6 h prior to cell harvest. At 24 h post-transfection cells were washed with cold PBS and lysed in RIPA buffer (2.5 M NaCl, 0.5 M Tris pH 7.4, 0.5 M EDTA, 10% v/v Triton X-100, 1 mM PMSF and 0.2 mM Na3VO4) at 4 °C. Cell lysates were pre-cleared with Protein G beads, and were incubated with 1 μg of either an anti-FLAG (Sigma) or anti-Luman antibody (M13) for 21 h. After incubation, the beads were collected by centrifugation, washed with cold RIPA buffer and resuspended in sample buffer. For Western blotting of immunoprecipitates, an anti-Luman (M13) [6] or anti-FLAG (Sigma) antibody was used as primary antibodies at 1:1000 dilution. HRP-conjugated anti-rabbit IgG or anti-mouse IgG antibodies (Promega) were used as secondary antibodies at 1: 30,000. Blots were visualized using ECL Plus (GE Healthcare) on a Typhoon 9400 PhosphorImager (GE Healthcare). The results are representative of at least two independent experiments.
2.7. Confocal immunofluorescence microscopy
Cells were fixed in 4% formaldehyde, and permeabilized in 0.1% Triton X-100. The cover slips were incubated with 1:100–200 dilution of primary antibodies anti-JAB1 B-17 and anti-HA HA.11 (Santa Cruz Biotechnologies), then with a 1:400 dilution of Alexa594- or Alexa488-conjugated anti-mouse IgG antibody (Molecular Probes Inc.) and were mounted with 50% glycerol containing 500 pM 4′,6-diamidino-2-phenylinole (DAPI). Images were captured with a Hamamatsu ORCA-ER Digital Camera under a Leica DMRE confocal microscope.
2.8. [35S]-methionine/cysteine pulse-chase
HEK-293 cells were seeded into 100-mm dishes one day prior to transfection. The cells were transfected with 20 μg of pFLAG-Luman only, or co-transfected with the pHA-JAB1 plasmid. Cells were split into 4 × 60-mm dishes at 16 h post-transfection. At 27 h post-transfection, cells were incubated in the pre-labeling media (DMEM without L-methionine and L-cysteine; Invitrogen) for 1 h. Cells were subsequently pulse-labeled for 1.5 h in pre-labeling media supplemented with a final concentration of 250 μCi/mL of [35S]-methionine/cysteine (MP Biomedicals). The cells were then chased with complete DMEM media for the desired time (0, 60, 120 or 240 min). Cells were lysed in the RIPA buffer described above. Equal amounts of total protein were subjected to immunoprecipitation by anti-FLAG antibody (M2, Sigma) and a control antibody (anti-XBP1, M-186, Santa Cruz Biotech.), resolved by SDS-PAGE, followed by autoradiography and visualization on a Typhoon 9400 PhosphorImager. Band intensities were analyzed using IMAGEQUANT TLv2003.01 (GE Healthcare) and expressed as relative units of density compared to that of time 0. Data is representative of two independent trials.
For Western blotting, a second gel was run in parallel and probed with anti-Luman (M13) [6]. Blots were visualized using ECL Plus (GE Healthcare) on a Typhoon 9400 PhosphorImager.
2.9. JAB1 siRNA knockdown and immunoprecipitation
Both the scrambled control siRNA (Silencer® Select Negative Control #1) and custom siRNA against JAB1 (5′-GCUCAGAGUAU CGAUGAAA-3′ with 3′ dTdT overhangs sense strand) were supplied by Ambion. HEK 293 cells were transfected with either the control siRNA or JAB1 siRNA at a final concentration of 10 nM using Lipofectamine™ RNAiMAX (Invitrogen) as per manufacturer’s instructions. Forty-eight hours of post transfection, cells were harvested in lysis buffer containing 150 mM NaCl, 1% NP40, 50 mM Tris pH-8, 5% glycerol, 1 mM EDTA and the Halt™ protease inhibitor cocktail (Thermo Scientific). The extracts were precleared with the Protein G Mag Sepharose Xtra beads (GE healthcare), and incubated with rabbit polyclonal anti-CREB3 antibody (Proteintech Group) for 1 h at 4 °C. The resulting antibody–protein complex was incubated with the protein G magnetic beads for 4 h at 4 °C, and the complex bound to the beads was washed three times with PBS and eluted with 100 mM glycine pH 2.5. Samples were neutralized with 1 M Tris buffer pH 9.5 prior to 10% SDS-PAGE and Western blotting using rabbit polyclonal anti-CREB3 and mouse monoclonal anti-ubiquitin (P4D1; Santa Cruz Biotechnology) antibodies.
2.10. Reverse transcription (RT)-PCR analysis
Cells were transfected with 10 nM siRNA directed against JAB1 for 48 h and then treated with 2 μg/mL tunicamycin. Cells transfected with 10 nM scrambled siRNA was used as a control. Total RNA was extracted using Trizol and the cDNA was synthesized using Superscript II RNaseH reverse transcriptase (Invitrogen) and oligo-dT primers. RT-PCR was carried out using the following primers: Herp, 5′-CTTGG AGCTGAGTGGCGAC and 5′-CAATGTCCAGGAGAGGCAATC; GAPDH, 5′-CCACCAACTGCTTAGCACC and 5′-GGGTGGCAGTGATGGCAT.
3. Results
3.1. Identification of JAB1 as a potential binding partner of Luman
To elucidate the cellular role of Luman, a yeast two-hybrid screen of a human fetal brain cDNA library was conducted to identify cellular ligands of Luman. The region of Luman used as bait in the screen, a.a. 55–182, is between the N-terminal activation domain and the leucine zipper region, containing an extended basic domain that is well conserved among CREB3 family members [1]. Among approximately 5 × 105 independent colonies screened, 7 positive clones were confirmed in repeated yeast two-hybrid assays. Among them, one contains JAB1/CSN5 cDNA, which is presented here.
3.2. Luman interacts with JAB1 both in vitro and in vivo
To confirm the direct interaction between Luman and JAB1, we first employed an in vitro biochemical method, the GST pull-down assay. Recombinant GST fusion proteins of N-terminal Luman a.a. 1–215 and JAB1 were made and purified using glutathione sepharose beads. The GST-fusion protein beads were incubated with [35S]-labeled Luman and JAB1 proteins, and precipitated to check the physical association of these proteins. As depicted in Fig. 1A, JAB1 was readily pulled down by GST-Luman(1–215), and the same was observed in the reciprocal assay. No interaction was seen in the controls, GST (lane 2) or GAL4 AD-GFP (Fig. 1A, row 3). These results confirmed that Luman and JAB1 can interact with each other directly. We also noticed that JAB1 also appeared to associate with itself (Fig. 1A, first row, lane 4). To our knowledge, this is the first report of JAB1 self-association.
Fig. 1.

Luman interacts with JAB1 both in vitro and in vivo. (A) Direct binding of Luman to JAB1 in GST pull-down assays. GST, GST-Luman(1–215) and GST-JAB1 proteins were coupled to glutathione-sepharose beads and incubated with [35S]-labeled GAL4 AD-GFP, FLAG-Luman and HA-JAB1. After extensive washing, proteins were eluted from the beads, separated on a 10% SDS-PAGE gel and visualized by autoradiography. The input lanes have 10% of the radio-labeled protein used in each pull-down assay. (B) Luman interacts with JAB1 in the cell as demonstrated by the mammalian two-hybrid assay. HEK-293 cells were transiently transfected with a combination of pM-JAB1 and a Luman construct, along with the reporter plasmids p5×GAL4 luciferase and pRL-SV40. Dual luciferase activities were measured 24 h post-transfection. Values are normalized to Renilla luciferase before being referenced to the pM-JAB1 background control. Data are based on 6 independent assays and shown with standard errors. (C) Luman interacts with JAB1 in mammalian cells as demonstrated by co-immunoprecipitation. HEK-293 cells were transiently transfected with pcDNA-Luman, pFLAG-JAB1 or both. Cells were treated with proteasome inhibitor MG132 for the last 6 h, prior to harvest of cell lysates at 24 h post-transfection. Cleared lysates were incubated with either a Luman- or FLAG-specific antibody, followed by precipitation with Protein G beads. Precipitated samples were subjected to SDS-PAGE and probed in Western blotting with antibodies against FLAG (top panel), Luman (middle panel) or HCF-1 (bottom panel). HCF-1 was included as a positive control for Luman since it is a known interacting protein. The input lanes have 2% of cleared cell lysates used in the immunoprecipitation assays. Abbreviations: IP, immunoprecipitation antibody; WB, Western blot antibody.
Next we sought to test whether the interaction exists in the mammalian cellular environment using a mammalian two-hybrid assay. Analogous to the yeast two-hybrid system, JAB1 was fused to GAL4 DBD (DNA-binding domain) and used as the bait. The induction of GAL UAS reporter is dependent upon recruiting Luman to the GAL promoter and a functional activation domain at the N-terminus (a.a. 1–52) of Luman for transcription activation [1] in the prey plasmid. Luman with the activation domain that removed Luman(53–272), was included as the negative control. As seen in Fig. 1B, when GAL DBD-JAB1 is co-expressed with Luman (column 5), approximately 30-fold activation of the GAL4 reporter was observed. This activity is higher than the positive control sample containing Luman and GAL-Luman(53–371) (22-fold, column 4), indicating that Luman and JAB1 interact with each other in mammalian cells. Lack of JAB1 in the bait (column 3) or the activation domain of Luman in the prey (column 2) did not produce a significant level of reporter activation. These data suggest that the observed induction of the reporter was dependent upon successful recruitment of Luman by JAB1 and the transactivation function of Luman.
To verify the interaction between Luman and JAB1 in the cell, co-immunoprecipitation assays were also carried out. HEK-293 cells were transfected with plasmids encoding Luman and FLAG-JAB1. We found that a Luman specific antibody precipitated JAB1 efficiently (Fig. 1C, top panel). In reciprocal, precipitation of FLAG-JAB1 with an anti-FLAG antibody also brought down Luman readily (Fig. 1C, middle panel). In addition, Luman was able to pull out endogenous HCF-1, which was used as a positive control (bottom panel). To ensure that the antibodies used in the assays were specifically targeting their antigen, we also included controls to show that each antibody was able to precipitate its own antigen (panels A and B, right column). Thus, both mammalian two-hybrid and co-immunoprecipitation assays demonstrated that Luman can interact with JAB1 in the mammalian cell environment.
3.3. JAB1 co-localization of Luman in mammalian cells
To further confirm the interaction in the mammalian cells, we sought to examine the subcellular localization of the two proteins by confocal immunofluorescence microscopy. Without co-transfection with Luman, JAB1 exhibited a diffuse staining pattern throughout the cell with a prominent nuclear staining (Fig. 2, row 1). As shown previously [6], the full-length Luman localized to the endoplasmic reticulum (ER); while Luman(1–215) was nuclear, which represents the proteolytically processed form of the protein (Fig. 2, row 2 and 3). Interestingly in co-transfected Vero cells, JAB1 was apparently tethered onto the ER by the full-length Luman in the cytosol, while the nuclear activated form of Luman protein, Luman(1–215), also colocalized with JAB1 in the nucleus. These results suggest a strong interaction between the two proteins.
Fig. 2.

Co-localization of JAB1 with Luman demonstrated by confocal fluorescence microscopy. Vero cells were transiently transfected with plasmids expressing JAB1 or GFP-Luman protein. JAB1 was visualized indirectly with a JAB1-specific antibody and a fluorophore Alexa488- or Alexa594-conjugated secondary antibody. DAPI was used to counter-stain the nuclei of the cells.
3.4. Luman interacts with JAB1 via the MPN domain
The MPN (Mpr1, Pad1 N-terminal) domain of JAB1 (a.a. 44–195) has been found to mediate protein–protein interactions [30]. We asked whether Luman also interacts with JAB1 through this region. Three JAB1 mutants were made, including regions encoding the MPN domain, and the segment N-terminal or C-terminal to the MPN domain (Fig. 3A). The ability of direct interaction with these JAB1 deletion mutants by Luman was examined by the GST pull-down assay. We found that GST-Luman(1–215) bound with all of the JAB1 mutants except JAB1 190–334 (Fig. 3B), which lacks the MPN domain. Notably, JAB1 (44–195) associated with Luman, indicating that the MPN domain itself is sufficient for the Luman–JAB1 interaction. This result is consistent with other reports that the MPN domain is necessary for protein–protein interactions [31,32].
Fig. 3.
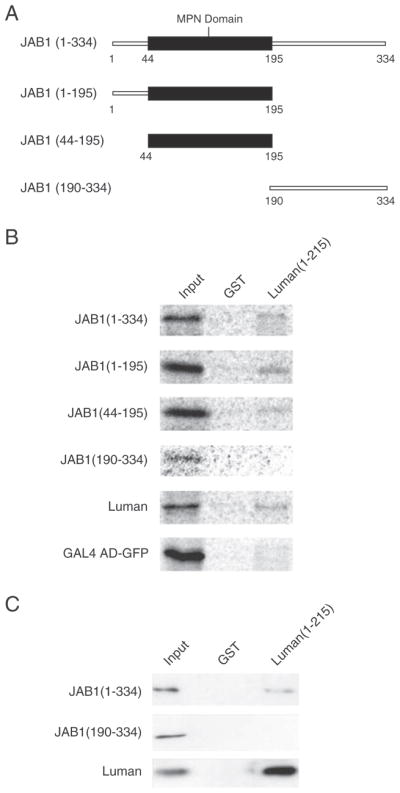
The MPN domain of JAB1 is necessary and sufficient for interaction with Luman. (A) Schematic diagram showing the JAB1 deletion mutants used in the study. (B) GST and GST-Luman(1–215) proteins were coupled to glutathione-sepharose beads and incubated with 35S-labeled HA-JAB1 proteins, as well as with the controls FLAG-Luman and GAL4 AD-GFP. After extensive washing, proteins were eluted from the beads, separated on a 10% SDS-PAGE gel and visualized by autoradiography. The input lanes have 10% of the radio-labeled protein used in each assay. (C) To confirm the pulldown results of JAB1(190–334), the experiment was repeated with the full-length JAB1 protein and Luman as positive controls.
3.5. Interaction of JAB1 represses the transactivation potential of Luman
To determine the functional relevance of JAB1–Luman interaction, with Luman being a transcription factor, we sought to investigate whether JAB1 could affect the transactivation ability of Luman. Previous studies have shown that Luman may play a role in the unfold protein response, and it can activate a key UPR enhancer element, the unfolded protein response element (UPRE) [1,23,28]. Dual luciferase assays were performed to determine the effect of JAB1 on the activation potential of Luman on a UPRE-containing reporter.
For the UPRE luciferase assay, HEK-293 cells were transfected with either pcDNA or pcHA-JAB and a Luman construct (pcFLAG-Luman or pcFLAG-Luman(1–215)) along with the 5× UPRE luciferase reporter and the control reporter pRL-SV40. Consistent with previous data [28], Luman showed approximately 12-fold activation while Luman1-215 had ~25-fold (Fig. 4A top, columns 3 and 5). Co-expression with JAB1 reduced this activation by 40% for Luman (compare columns 3 and 4) and 27% for Luman(1–215) (compare columns 5 and 6 in Fig. 4A top panel).
Fig. 4.

JAB1 represses transactivational activity of Luman and promotes degradation of the Luman protein. (A) Dual luciferase reporter assays. HEK-293 cells were transiently transfected with pHA-JAB1 and FLAG-Luman (top panel) or Gal DBD-Luman (bottom panel) constructs, along with the firefly luciferase reporter plasmids 5×UPRE or 5×GAL4 respectively. The control Renilla luciferase reporter plasmid pRL-SV40 was included in all samples. The firefly luciferase values are normalized to Renilla luciferase before being referenced to the pcDNA control. Data are based on 3 independent assays and shown with standard errors. (B) Pulse-chase assays. HEK-293 cells were transiently transfected with pcFLAG-Luman alone, or co-transfected with HA-JAB1. The cells were methionine/cysteine starved for 1 h, pulse-labeled with [35S]-methionine/cysteine for 1.5 h and chased for the time as indicated. The Luman protein was then immunoprecipitated with an anti-FLAG antibody, resolved by SDS-PAGE, and visualized by autoradiography. The negative control represents a pooled sample immunoprecipitated with a non-specific antibody. The half-life of the protein was determined by densitometric analysis using data from two independent trials collected on a Typhoon 9400 Phosphorimager. Values are plotted with standard errors.
To determine whether the JAB1 repression is specific to the UPRE enhancer, the GAL4 UAS reporter system was also included in the dual luciferase assay. The activation potential of Luman was tested when tethered to the GAL promoter through fusion of the GAL DBD. We found that the transcription potential of Luman was reduced by ~50% with overexpression of JAB1 (Fig. 4A bottom panel). The results indicate that JAB1 interaction can repress the general activation potential of Luman, and that the repression appears to be independent of the promoter sequences.
3.6. JAB1 promotes degradation of the Luman protein
Next we were interested to investigate that mechanism by which JAB1 represses the transactivation potential of Luman. The presence of an acidic domain, such as the one present at the amino-terminus of Luman, is thought to increase the probability of a polypeptide being targeted for rapid degradation by the proteasome [33]. We also noticed a rapid turnover of the Luman protein. Since JAB1 has previously been shown to affect the degradation rate of other proteins such as p27Kip1 through protein–protein interaction [31,34], we were interested to examine whether the interaction with JAB1 could affect the protein stability of Luman. The pulse-chase experiment was thus carried out. Two parallel cultures of HEK-293 cells expressing FLAG-Luman or co-expressing FLAG-Luman and HA-JAB1 were pulse-labeled with [35S]-methionine/cysteine for 1.5 h, and then chased with regular media for 0, 60, 120 or 240 min. The Luman protein was immunoprecipitated, resolved by SDS-PAGE and subjected to autoradiography. Identification of the Luman bands was made with the aid of comparative Western blotting. From the pulse-chase experiment (Fig. 4B), the half-life of Luman was estimated to be ~135 min. With the co-expression of JAB1, its half-life shortened to ~45 min, representing a change of 90 min or a reduction of 67%.
To confirm this observation and to examine if JAB1 regulation of the Luman protein stability involves ubiquitin and/or proteasomes, endogenous Luman protein from HEK 293 cells, with or without JAB1 siRNA treatment, was immunoprecipitated and examined for ubiquitination (Fig. 5). In agreement with the pulse-chase experiment findings (Fig. 4B), the JAB1 knockdown cells had significantly higher levels of Luman (Fig. 5A, left panel). In the immunoprecipitates using an anti-Luman antibody, there were markedly higher levels of Luman protein in the JAB1 knockdown samples; however, no ubiquitinated Luman was detected (Fig. 5A, right panel). To examine the effects of JAB1 and siRNA JAB1 on expression of Luman-induced UPR target genes, we have performed semi-quantitative PCR of Luman downstream target gene Herp, with or without tunicamycin treatment (Fig. 5B). Compared to the controls, both JAB1 knockdown and Luman overexpression samples showed higher levels of Herp. It may be worth mentioning that Herp, as a key ER stress protein, is under the control of many factors. Therefore, one may not expect a drastic impact of JAB1 knockdown via Luman on the expression levels of Herp.
Fig. 5.
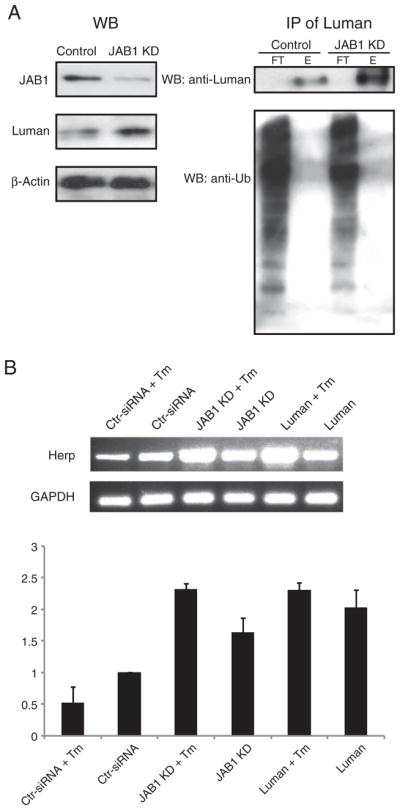
JAB1 siRNA knockdown increases the levels of Luman and the downstream target Herp. (A) HEK293 cells treated with either scrambled siRNA or JAB1 siRNA, were immunoprecipitated using an anti-Luman antibody. The flowthrough (FT) and eluted (E) samples were separated on a 10% SDS-PAGE, and analyzed by Western blotting using both Luman and ubiquitin antibodies. The left panel depicts the levels of Luman, JAB1 and β-actin in the input lysates prior to immunoprecipitation. The right panel shows the levels of total Luman and ubiquitinated Luman in the precipitates. (B) RT-PCR analysis of Herp and GAPDH in HEK293 cells which were transfected with either scrambled siRNA or JAB1 siRNA or Luman, with or without tunicamycin (Tm) treatment. Densitometric analysis of the HERP bands normalized against GAPDH was performed. The experiment was repeated three times, and fold changes over the control siRNA sample were shown, along with standard errors.
4. Discussion
In the present study we provided both in vitro and in vivo evidence that JAB1 is a novel cellular binding partner of Luman. Mapping studies revealed that the MPN domain of JAB1 (amino acids 44–195) was necessary and sufficient for the interaction with Luman. We found that overexpression of JAB1 significantly shortened the half-life of the Luman protein and repressed its transactivation function. We therefore propose that JAB1 is potentially a key regulator of the activity of Luman.
JAB1 is also known as CSN5 or unit 5 of the COP9 signalosome complex (CSN), which is structurally similar to the 26S proteasome lid [35]. Both JAB1 and Rpn11, the JAB1 paralogue in the proteasome, has a JAMM motif in the MPN domain that resembles the active site of metalloproteases [36,37]. Rpn11 is responsible for the cleavage of ubiquitin molecules at the proteasome from proteins targeted for degradation. In parallel, JAB1 is responsible for the cleavage of Nedd8, a ubiquitin-like molecule, from the cullin subunit of the E3 ubiquitin ligase [38]. It is thus not surprising that JAB1 interaction with Luman via the MPN domain promoted Luman degradation (Fig. 4B).
As a potential regulator of Luman, one possible mechanism by which JAB1 controls the transactivation function of Luman is through affecting the binding affinities of Luman to target DNA elements. For instance, JAB1 enhances c-Jun transactivation by enhancing binding to their cognate DNA sequences [39,40]. In the case of Luman, this regulatory mechanism does not seem plausible, since JAB1 repression of Luman was also observed in the GAL4 reporter system, in which Luman was recruited to the artificial GAL4 UAS promoter, independent of known target elements of Luman, such as UPRE or CRE.
Another common modus operandi of JAB1 is to inhibit protein function by destabilizing the protein and promoting its degradation [41]. JAB1 has been previously shown to destabilize proteins, such as the estrogen receptor [42], DNA topoisomerase IIα [32], and transcription factors Smad7 [43] and RUNX3 [44]. Overexpression of JAB1 is found to reduce the half-life of p27Kip1 from more than 5 h to 1.4 h [34], and increase the degradation rate of rLHR by 3 fold [45]. The pulse–chase experiment presented in this study suggests that JAB1 may also exert its repressive effect on Luman by decreasing the half-life of Luman. Being an unstable protein, the half-life of Luman was estimated to be approximately 135 min (Fig. 4B), similar to that of ATF6 (half-life of 120 min), which is another ER-stress responsive transcription factor that bears many structural and functional resemblance to Luman [46]. With overexpression of JAB1, the half-life of Luman was decreased by ~67%, to approximately 45 min. Thus, we believe that JAB1 may be a critical factor that regulates the duration of the action of Luman during the UPR.
The mechanism of JAB1-mediated degradation of Luman is unclear, but likely through the proteasome pathway. The low levels of ubiquinated Luman protein observed in Fig. 5B may indicate rapid degradation of the protein. We have observed previously that the proteasome inhibitor MG132 substantially increases the stability of Luman [23]. Other known JAB1-binding partners, such as c-Jun [47], p53 [48,49], and p27Kip1 [34], are altered in their susceptibility to proteasomal degradation by JAB1, which is believed to have resulted from changes in their phosphorylation status by the action of CSN-associated kinases. JAB1, as part of the CSN, has been shown to regulate the phosphorylation of transcriptional regulators, including c-Jun [50]; thus it is possible that interaction with JAB1 may also alter the phosphorylation status of Luman, thereby affecting its protein stability. As this being said, however, it is possible that the protein stability of Luman may also be regulated by an alternative non-proteasomal degradation mechanism induced by ER stress [51].
JAB1 can exist in two forms in the cell: the nuclear CSN and the cytoplasmic small-JAB1 containing complex [31,43,47,52–55]. No detectable level of endogenous JAB1 was observed in the cells examined in our studies, likely due to low expression level of the protein and its natural diffused distribution throughout the cell. In our transfected cells, JAB1 exhibited a staining pattern that agrees with previous findings and with its known cellular functions [31,43,53,55–57]. Most interestingly we have found that Luman could drastically change the subcellular localization of JAB1. In particular, co-expression of the proteolytically processed Luman, a.a. 1–215, apparently caused JAB1 translocation to the nucleus entirely. JAB1 has previously been shown to have an altered localization when co-expressed with interacting proteins [31,34,55,57]. This observation suggests an intricate regulatory relationship between JAB1 and Luman in terms of their cellular functions. JAB1 may play a critical role in regulation of the stability of Luman and represses its transactivation potential; however, once activated by proteolytic processing, Luman may also affect the function of JAB1 by controlling its subcellular localization, although the functional significance of JAB1 nuclear translocation is unclear.
As an additional support of their functional connections in the cell, JAB1 has also been linked to the UPR recently through its interaction with the ER-stress sensor IRE1 [58]. Under the conditions of no or low ER-stress, interaction with JAB1 downregulates IRE1-mediated activation of the UPR. In this capacity, JAB1 appears to function as a “brake” on the UPR, preventing premature or unnecessary activation of this cellular stress signaling pathway. Here we show that, in a similar manner, JAB1 might also downregulate the transactivation activity of Luman during the UPR. The interaction of JAB1 and Luman provides yet another example of complex crosstalks between different regulatory circuitries in this highly orchestrated signaling event.
Acknowledgments
We thank Ron Prywes for the p5×UPRE-Luc plasmid. We thank Alayne Skinner for her technical assistance with the yeast two-hybrid screens. This work was supported by an operating grant from Canadian Institutes of Health Research to R.L.
Abbreviations
ER
endoplasmic reticulum
ERAD
ER-associated degradation
HSV
herpes simplex virus
UPR
unfolded protein response
UPRE
unfolded protein response
References
- 1.Lu R, Yang P, O’Hare P, Misra V. Luman, a new member of the CREB/ATF family, binds to herpes simplex virus VP16-associated host cellular factor. Mol Cell Biol. 1997;17:5117–5126. doi: 10.1128/mcb.17.9.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freiman RN, Herr W. Viral mimicry: common mode of association with HCF by VP16 and the cellular protein LZIP. Genes Dev. 1997;11:3122–3127. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kristie TM, Sharp PA. Purification of the cellular C1 factor required for the stable recognition of the Oct-1 homeodomain by the herpes simplex virus alpha-trans-induction factor (VP16) J Biol Chem. 1993;268:6525–6534. [PubMed] [Google Scholar]
- 4.Wilson AC, LaMarco K, Peterson MG, Herr W. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell. 1993;74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 5.Lu R, Yang P, Padmakumar S, Misra V. The herpesvirus transactivator VP16 mimics a human basic domain leucine zipper protein, Luman, in its interaction with HCF. J Virol. 1998;72:6291–6297. doi: 10.1128/jvi.72.8.6291-6297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu R, Misra V. Potential role for Luman, the cellular homologue of herpes simplex virus VP16 (alpha gene trans-inducing factor), in herpesvirus latency. J Virol. 2000;74:934–943. doi: 10.1128/jvi.74.2.934-943.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blot G, Lopez-Verges S, Treand C, Kubat NJ, Delcroix-Genete D, Emiliani S, Benarous R, Berlioz-Torrent C. Luman, a new partner of HIV-1 TMgp41, interferes with Tat-mediated transcription of the HIV-1 LTR. J Mol Biol. 2006;364:1034–1047. doi: 10.1016/j.jmb.2006.09.080. [DOI] [PubMed] [Google Scholar]
- 8.Eleveld-Trancikova D, Sanecka A, van Hout-Kuijer MA, Looman MW, Hendriks IA, Jansen BJ, Adema GJ. DC-STAMP interacts with ER-resident transcription factor LUMAN which becomes activated during DC maturation. Mol Immunol. 2010;47:1963–1973. doi: 10.1016/j.molimm.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 9.Kim HC, Choi KC, Choi HK, Kang HB, Kim MJ, Lee YH, Lee OH, Lee J, Kim YJ, Jun W, Jeong JW, Yoon HG. HDAC3 selectively represses CREB3-mediated transcription and migration of metastatic breast cancer cells. Cell Mol Life Sci. 2010;67:3499–3510. doi: 10.1007/s00018-010-0388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang SW, Kim YS, Kim YR, Sung HJ, Ko J. Regulation of human LZIP expression by NF-kappaB and its involvement in monocyte cell migration induced by Lkn-1. J Biol Chem. 2007;282:11092–11100. doi: 10.1074/jbc.M607962200. [DOI] [PubMed] [Google Scholar]
- 11.Chin KT, Zhou HJ, Wong CM, Lee JM, Chan CP, Qiang BQ, Yuan JG, Ng IO, Jin DY. The liver-enriched transcription factor CREB-H is a growth suppressor protein underexpressed in hepatocellular carcinoma. Nucleic Acids Res. 2005;33:1859–1873. doi: 10.1093/nar/gki332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Omori Y, Imai J, Watanabe M, Komatsu T, Suzuki Y, Kataoka K, Watanabe S, Tanigami A, Sugano S. CREB-H: a novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the box-B element with a liver-specific expression. Nucleic Acids Res. 2001;29:2154–2162. doi: 10.1093/nar/29.10.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, Iseki K, Wanaka A, Imaizumi K. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat Cell Biol. 2005;7:186–194. doi: 10.1038/ncb1213. [DOI] [PubMed] [Google Scholar]
- 14.Honma Y, Kanazawa K, Mori T, Tanno Y, Tojo M, Kiyosawa H, Takeda J, Nikaido T, Tsukamoto T, Yokoya S, Wanaka A. Identification of a novel gene, OASIS, which encodes for a putative CREB/ATF family transcription factor in the long-term cultured astrocytes and gliotic tissue. Brain Res Mol Brain Res. 1999;69:93–103. doi: 10.1016/s0169-328x(99)00102-3. [DOI] [PubMed] [Google Scholar]
- 15.Cao G, Ni X, Jiang M, Ma Y, Cheng H, Guo L, Ji C, Gu S, Xie Y, Mao Y. Molecular cloning and characterization of a novel human cAMP response element-binding (CREB) gene (CREB4) J Hum Genet. 2002;47:373–376. doi: 10.1007/s100380200053. [DOI] [PubMed] [Google Scholar]
- 16.Stirling J, O’Hare P. CREB4, a transmembrane bZip transcription factor and potential new substrate for regulation and cleavage by S1P. Mol Biol Cell. 2006;17:413–426. doi: 10.1091/mbc.E05-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi H, Fillion C, Labrie Y, Grenier J, Fournier A, Berger L, El-Alfy M, Labrie C. AIbZIP, a novel bZIP gene located on chromosome 1q21.3 that is highly expressed in prostate tumors and of which the expression is up-regulated by androgens in LNCaP human prostate cancer cells. Cancer Res. 2002;62:721–733. [PubMed] [Google Scholar]
- 18.Stelzer G, Don J. Atce1: a novel mouse cyclic adenosine 3′,5′-monophosphate-responsive element-binding protein-like gene exclusively expressed in postmeiotic spermatids. Endocrinology. 2002;143:1578–1588. doi: 10.1210/endo.143.5.8822. [DOI] [PubMed] [Google Scholar]
- 19.Nagamori I, Yabuta N, Fujii T, Tanaka H, Yomogida K, Nishimune Y, Nojima H. Tisp40, a spermatid specific bZip transcription factor, functions by binding to the unfolded protein response element via the Rip pathway. Genes Cells. 2005;10:575–594. doi: 10.1111/j.1365-2443.2005.00860.x. [DOI] [PubMed] [Google Scholar]
- 20.Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- 21.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 22.Murakami T, Kondo S, Ogata M, Kanemoto S, Saito A, Wanaka A, Imaizumi K. Cleavage of the membrane-bound transcription factor OASIS in response to endoplasmic reticulum stress. J Neurochem. 2006;96:1090–1100. doi: 10.1111/j.1471-4159.2005.03596.x. [DOI] [PubMed] [Google Scholar]
- 23.Liang G, Audas TE, Li Y, Cockram GP, Dean JD, Martyn AC, Kokame K, Lu R. Luman/CREB3 induces transcription of the endoplasmic reticulum (ER) stress response protein Herp through an ER stress response element. Mol Cell Biol. 2006;26:7999–8010. doi: 10.1128/MCB.01046-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raggo C, Rapin N, Stirling J, Gobeil P, Smith-Windsor E, O’Hare P, Misra V. Luman, the cellular counterpart of herpes simplex virus VP16, is processed by regulated intramembrane proteolysis. Mol Cell Biol. 2002;22:5639–5649. doi: 10.1128/MCB.22.16.5639-5649.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogan MR, Cockram GP, Lu R. Cooperative interaction of Zhangfei and ATF4 in transactivation of the cyclic AMP response element. FEBS Lett. 2006;580:58–62. doi: 10.1016/j.febslet.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 26.Kokame K, Agarwala KL, Kato H, Miyata T. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J Biol Chem. 2000;275:32846–32853. doi: 10.1074/jbc.M002063200. [DOI] [PubMed] [Google Scholar]
- 27.van Laar T, Schouten T, Hoogervorst E, van Eck M, van der Eb AJ, Terleth C. The novel MMS-inducible gene Mif1/KIAA0025 is a target of the unfolded protein response pathway. FEBS Lett. 2000;469:123–131. doi: 10.1016/s0014-5793(00)01253-9. [DOI] [PubMed] [Google Scholar]
- 28.DenBoer LM, Hardy-Smith PW, Hogan MR, Cockram GP, Audas TE, Lu R. Luman is capable of binding and activating transcription from the unfolded protein response element. Biochem Biophys Res Commun. 2005;331:113–119. doi: 10.1016/j.bbrc.2005.03.141. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- 30.Burger-Kentischer A, Finkelmeier D, Thiele M, Schmucker J, Geiger G, Tovar GE, Bernhagen J. Binding of JAB1/CSN5 to MIF is mediated by the MPN domain but is independent of the JAMM motif. FEBS Lett. 2005;579:1693–1701. doi: 10.1016/j.febslet.2005.01.080. [DOI] [PubMed] [Google Scholar]
- 31.Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato JY. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem. 2002;277:2302–2310. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- 32.Yun J, Tomida A, Andoh T, Tsuruo T. Interaction between glucose-regulated destruction domain of DNA topoisomerase IIalpha and MPN domain of Jab1/CSN5. J Biol Chem. 2004;279:31296–31303. doi: 10.1074/jbc.M401411200. [DOI] [PubMed] [Google Scholar]
- 33.Salghetti SE, Muratani M, Wijnen H, Futcher B, Tansey WP. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci U S A. 2000;97:3118–3123. doi: 10.1073/pnas.050007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398:160–165. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- 35.Bech-Otschir D, Seeger M, Dubiel W. The COP9 signalosome: at the interface between signal transduction and ubiquitin-dependent proteolysis. J Cell Sci. 2002;115:467–473. doi: 10.1242/jcs.115.3.467. [DOI] [PubMed] [Google Scholar]
- 36.Cope GA, Suh GS, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–611. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 37.Verma R, Aravind L, Oania R, McDonald WH, Yates JR, III, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 38.Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Deshaies RJ. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–1385. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- 39.Claret FX, Hibi M, Dhut S, Toda T, Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature. 1996;383:453–457. doi: 10.1038/383453a0. [DOI] [PubMed] [Google Scholar]
- 40.Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000;408:211–216. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- 41.Lue H, Thiele M, Franz J, Dahl E, Speckgens S, Leng L, Fingerle-Rowson G, Bucala R, Luscher B, Bernhagen J. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene. 2007;26:5046–5059. doi: 10.1038/sj.onc.1210318. [DOI] [PubMed] [Google Scholar]
- 42.Callige M, Kieffer I, Richard-Foy H. CSN5/Jab1 is involved in ligand-dependent degradation of estrogen receptor alpha by the proteasome. Mol Cell Biol. 2005;25:4349–4358. doi: 10.1128/MCB.25.11.4349-4358.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim BC, Lee HJ, Park SH, Lee SR, Karpova TS, McNally JG, Felici A, Lee DK, Kim SJ. Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor beta signaling by binding to Smad7 and promoting its degradation. Mol Cell Biol. 2004;24:2251–2262. doi: 10.1128/MCB.24.6.2251-2262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JH, Choi JK, Cinghu S, Jang JW, Lee YS, Li YH, Goh YM, Chi XZ, Lee KS, Wee H, Bae SC. Jab1/CSN5 induces the cytoplasmic localization and degradation of RUNX3. J Cell Biochem. 2009;107:557–565. doi: 10.1002/jcb.22157. [DOI] [PubMed] [Google Scholar]
- 45.Li S, Liu X, Ascoli M. p38JAB1 binds to the intracellular precursor of the lutropin/choriogonadotropin receptor and promotes its degradation. J Biol Chem. 2000;275:13386–13393. doi: 10.1074/jbc.275.18.13386. [DOI] [PubMed] [Google Scholar]
- 46.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seeger M, Kraft R, Ferrell K, Bech-Otschir D, Dumdey R, Schade R, Gordon C, Naumann M, Dubiel W. A novel protein complex involved in signal transduction possessing similarities to 26S proteasome subunits. FASEB J. 1998;12:469–478. [PubMed] [Google Scholar]
- 48.Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, Dubiel W. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001;20:1630–1639. doi: 10.1093/emboj/20.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang XC, Chen J, Su CH, Yang HY, Lee MH. Roles for CSN5 in control of p53/MDM2 activities. J Cell Biochem. 2008;103:1219–1230. doi: 10.1002/jcb.21504. [DOI] [PubMed] [Google Scholar]
- 50.Naumann M, Bech-Otschir D, Huang X, Ferrell K, Dubiel W. COP9 signalosome-directed c-Jun activation/stabilization is independent of JNK. J Biol Chem. 1999;274:35297–35300. doi: 10.1074/jbc.274.50.35297. [DOI] [PubMed] [Google Scholar]
- 51.Shenkman M, Tolchinsky S, Lederkremer GZ. ER stress induces alternative nonproteasomal degradation of ER proteins but not of cytosolic ones. Cell Stress Chaperones. 2007;12:373–383. doi: 10.1379/CSC-281.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carrabino S, Carminati E, Talarico D, Pardi R, Bianchi E. Expression pattern of the JAB1/CSN5 gene during murine embryogenesis: colocalization with NEDD8. Gene Expr Patterns. 2004;4:423–431. doi: 10.1016/j.modgep.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Bounpheng MA, Melnikova IN, Dodds SG, Chen H, Copeland NG, Gilbert DJ, Jenkins NA, Christy BA. Characterization of the mouse JAB1 cDNA and protein. Gene. 2000;242:41–50. doi: 10.1016/s0378-1119(99)00525-9. [DOI] [PubMed] [Google Scholar]
- 54.Oh W, Lee EW, Sung YH, Yang MR, Ghim J, Lee HW, Song J. Jab1 induces the cytoplasmic localization and degradation of p53 in coordination with Hdm2. J Biol Chem. 2006;281:17457–17465. doi: 10.1074/jbc.M601857200. [DOI] [PubMed] [Google Scholar]
- 55.Bianchi E, Denti S, Granata A, Bossi G, Geginat J, Villa A, Rogge L, Pardi R. Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature. 2000;404:617–621. doi: 10.1038/35007098. [DOI] [PubMed] [Google Scholar]
- 56.Oh W, Yang MR, Lee EW, Park KM, Pyo S, Yang JS, Lee HW, Song J. Jab1 mediates cytoplasmic localization and degradation of West Nile virus capsid protein. J Biol Chem. 2006;281:30166–30174. doi: 10.1074/jbc.M602651200. [DOI] [PubMed] [Google Scholar]
- 57.Luo W, Wang Y, Hanck T, Stricker R, Reiser G. Jab1, a novel protease-activated receptor-2 (PAR-2)-interacting protein, is involved in PAR-2-induced activation of activator protein-1. J Biol Chem. 2006;281:7927–7936. doi: 10.1074/jbc.M510784200. [DOI] [PubMed] [Google Scholar]
- 58.Oono K, Yoneda T, Manabe T, Yamagishi S, Matsuda S, Hitomi J, Miyata S, Mizuno T, Imaizumi K, Katayama T, Tohyama M. JAB1 participates in unfolded protein responses by association and dissociation with IRE1. Neurochem Int. 2004;45:765–772. doi: 10.1016/j.neuint.2004.01.003. [DOI] [PubMed] [Google Scholar]