A Novel, Broad-Spectrum Inhibitor of Enterovirus Replication That Targets Host Cell Factor Phosphatidylinositol 4-Kinase IIIβ (original) (raw)
Abstract
Despite their high clinical and socioeconomic impacts, there is currently no approved antiviral therapy for the prophylaxis or treatment of enterovirus infections. Here we report on a novel inhibitor of enterovirus replication, compound 1, 2-fluoro-4-(2-methyl-8-(3-(methylsulfonyl)benzylamino)imidazo[1,2-_a_]pyrazin-3-yl)phenol. This compound exhibited a broad spectrum of antiviral activity, as it inhibited all tested species of enteroviruses and rhinoviruses, with 50% effective concentrations ranging between 4 and 71 nM. After a lengthy resistance selection process, coxsackievirus mutants resistant to compound 1 were isolated that carried substitutions in their 3A protein. Remarkably, the same substitutions were recently shown to provide resistance to inhibitors of phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ), a lipid kinase that is essential for enterovirus replication, suggesting that compound 1 may also target this host factor. Accordingly, compound 1 directly inhibited PI4KIIIβ in an in vitro kinase activity assay. Furthermore, the compound strongly reduced the PI 4-phosphate levels of the Golgi complex in cells. Rescue of coxsackievirus replication in the presence of compound 1 by a mutant PI4KIIIβ carrying a substitution in its ATP-binding pocket revealed that the compound directly binds the kinase at this site. Finally, we determined that an analogue of compound 1, 3-(3-fluoro-4-methoxyphenyl)-2-methyl-_N_-(pyridin-4-ylmethyl)imidazo[1,2-_a_]pyrazin-8-amine, is well tolerated in mice and has a dose-dependent protective activity in a coxsackievirus serotype B4-induced pancreatitis model.
INTRODUCTION
The genus Enterovirus belongs to the family of Picornaviridae, a group of positive-strand RNA viruses that includes many important human pathogens. Within the Enterovirus genus, there are four human enterovirus (HEV) species, called HEV-A to HEV-D, which in total comprise more than 100 virus (sero)types. Well-known members of the HEV-A species are the coxsackie A viruses and the emerging neurotropic enterovirus 71 (EV71) (1). These viruses are the major ethological agents of hand-foot-and-mouth disease, especially in young children. EV71 can also cause severe neurological diseases, including brain stem encephalitis and poliomyelitis-like paralysis (2). The HEV-B species comprise the coxsackie B viruses and echoviruses, which are the main causes of viral meningitis, myocarditis, and pancreatitis (3, 4). Coxsackieviruses are also associated with type 1 diabetes (5). The most extensively studied enterovirus is poliovirus (PV), which belongs to the HEV-C species and is the causative agent of paralytic poliomyelitis. The HEV-D species contains five viruses, including EV68 and EV70, which can cause clinical symptoms ranging from hand-foot-and-mouth disease to respiratory tract infections and acute hemorrhagic conjunctivitis (6, 7).
The three species of human rhinoviruses (HRV), HRV-A to HRV-C, are also classified within the Enterovirus genus and all together contain ∼150 serotypes. HRV is the main cause of the common cold, which poses a significant socioeconomic burden, with millions of days of absence from work or school, and often leads to improper use of antibiotics (8, 9). Furthermore, HRV infections can trigger severe asthma attacks and exacerbations of chronic obstructive pulmonary disease (COPD) in high-risk patients. The World Health Organization (WHO) has predicted that COPD will become the third leading cause of death worldwide by the year 2030 (http://www.who.int/mediacentre/events/annual/world_copd_day/en/).
There are two strategies to combat viral infections: the use of vaccines to prevent disease, or drugs to inhibit viral replication. For enteroviruses, a vaccine is only available for PV, while EV71 vaccine candidates are currently being evaluated in clinical trials (10, 11). The development of vaccines against other nonpolio enteroviruses seems essentially impossible, given the large variety of (sero)types. Hence, antiviral drugs are urgently needed for the prophylaxis and/or treatment of enterovirus infections. Antiviral compounds can affect different stages of the enterovirus replication cycle. This cycle starts with attachment and entry of a virus particle into the host cell. Subsequently, the virus uncoats to deliver its genome into the cytoplasm (12). Here, the positive-strand RNA genome is directly translated into a large polyprotein, which is subsequently processed into four capsid proteins (VP1 to VP4) and seven nonstructural proteins (2Apro, 2B, 2C, 3A, 3B, 3Cpro, and 3Dpol) that mediate viral RNA replication (13). Enteroviruses reorganize Golgi complex membranes into tubular and vesicular structures that serve as a platform for viral RNA replication (14–16), possibly via recruitment of essential membrane-modifying host factors, such as guanine nucleotide exchange factor GBF1 (17–19) and phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ) (20–22). Replication starts with the synthesis of negative-strand RNA, which in turn serves as a template for production of more positive-strand RNA (13). These new viral genomes are then packaged in capsid proteins to yield progeny virions, which are released into the external environment upon cell lysis.
To date, only two compounds (the capsid binders BTA798 and V-073) are under clinical development for the treatment of enterovirus infections (23). A major concern with these compounds that directly target a viral protein is the rapid emergence of drug-resistant viruses as a consequence of the high mutation rate of RNA viruses. One approach to overcome this problem is to pursue a combination therapy of several antiviral compounds that target different viral proteins (24). Another strategy is the development of compounds that interfere with or modulate the function of host factors that play a critical role in the virus replication cycle. Since host factors are unlikely to mutate and develop resistance in response to therapy, they are attractive targets for antiviral drugs. Importantly, this “host-targeting” approach may allow the development of broad-spectrum inhibitors, since all viruses within a single genus, or even within a whole family, usually exploit host factors of the same cellular pathway. A potential disadvantage of host-targeting compounds is that interference with a cellular target may be associated with toxic side effects. An example of a safe host-targeting antiviral agent that has successfully passed phase II clinical trials for treatment of infections by the Flavivirus hepatitis C virus (HCV) is Alisporivir, a compound that binds to the host factor cyclophillin (25, 26). Here, we present a novel inhibitor of enterovirus replication that specifically interacts with the host factor PI4KIIIβ.
MATERIALS AND METHODS
Cells and reagents.
Buffalo green monkey (BGM) kidney cells, HeLa R19 cells, and HeLa Rh cells were grown at 37°C, 5% CO2 in minimal essential medium (MEM; Gibco) supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Compound 1, 2-fluoro-4-(2-methyl-8-(3-(methylsulfonyl)benzylamino)imidazo[1,2-_a_]pyrazin-3-yl)phenol, and compound 2, 3-(3-fluoro-4-methoxyphenyl)-2-methyl-_N_-(pyridin-4-ylmethyl)imidazo[1,2-_a_]pyrazin-8-amine, were provided by Galapagos NV, had a purity of more than 95%, and were dissolved in dimethyl sulfoxide (DMSO). The synthesis of these compounds will be described elsewhere. Guanidine hydrochloride (GuaHCl) was purchased from Sigma-Aldrich and dissolved in water.
Plasmids.
The p53CB3/T7 wild-type (wt) and 3A mutant full-length infectious clones have been described previously (19, 28). The coxsackievirus serotype B3 (CVB3) replicon has also been described previously (18). The EV71 replicon (BrCr strain) was constructed with the same approach as the CVB3 replicon by replacing the P1 capsid coding region with the firefly luciferase gene by using standard techniques. The cDNA was placed behind a hammerhead ribozyme coding sequence to remove the extra nucleotides at the 5′ end. The plasmids carrying FAPP1-PH-GFP (FAPP1 is 4-phosphate adaptor protein 1; PH is pleckstrin homology; GFP is green fluorescent protein), PI4KIIIβ-HA (HA is hemagglutinin), PI4KIIIβ-Y583M-HA, and kinase-dead PI4KIIIβ-D656A-HA were kindly provided by T. Balla (NICHD, National Institutes of Health, Bethesda, MD).
Viruses.
Mutant CVB3 and CVB3-Rluc, which contains the Renilla luciferase gene upstream of the capsid coding region, were obtained by transfection of RNA transcripts derived from the full-length infectious clones into BGM cells as described before (29). A mengovirus strain of the cardiovirus encephalomyocarditis virus (EMCV) was obtained from cDNA clone pM16.1, generously provided by A. C. Palmenberg (University of Wisconsin, Madison, WI). Enterovirus 71 (BrCr) and coxsackievirus A21 (Coe) were received from the National Institute for Public Health and Environment (RIVM; The Netherlands). Equine rhinitis A virus (ERAV; NM11/67) was kindly provided by D. Rowlands and T. Tuthill (University of Leeds, Leeds, United Kingdom). Human rhinoviruses 2 and 14 were a kind gift of J. Seipelt (Medical University of Vienna, Vienna, Austria). Virus titers were determined by endpoint titration according to the method of Reed and Muench, and they are expressed as the 50% cell culture infective dose (CCID50).
Determination of antiviral activity and cytotoxicity.
The assays to determine the 50% effective concentration (EC50) and 50% cytotoxic concentration (CC50) of compound 1 were performed as described elsewhere (30). Briefly, cells were infected with 100 CCID50 for 2 h, after which the virus was removed and serial dilutions of the compound were added. For determination of the CC50, serial dilutions of the compound were added to the cells. Following 3 to 4 days of incubation, the medium was replaced with CellTiter 96 AQueous One solution reagent (Promega). Optical densities at 490 nm were corrected for background absorbance, which was determined from wells that lacked cells. The resulting values for untreated cells were set to 100%.
Virus infections and RNA transfection.
Subconfluent layers of cells were infected with virus at a multiplicity of infection (MOI) of 0.1 to 1. Alternatively, cells were transfected with RNA transcripts of either the full-length infectious clones or the replicon constructs. After 30 min, cells were washed, and fresh (compound-containing) medium was added to the cells. At 8 h postinfection (p.i.), cells were subjected to three cycles of freeze-thawing, after which virus titers were determined by endpoint titration. Alternatively, cells were lysed to determine the intracellular Renilla luciferase activity, by using the Renilla luciferase assay system (Promega).
PI4K in vitro activity assay.
The PI4K in vitro activity assay was performed as described previously (20). Briefly, recombinant PI4KIIIβ (SignalChem) or PI4KIIIα (Millipore) and their substrate, phosphatidylinositol (PI)-phosphatidylserine (PS), were diluted in buffer containing Triton X-100. The reaction was started by addition of a mixture of ATP and 0.25 μCi of [γ-33P]ATP. After 75 to 90 min of incubation at 30°C, the reaction was terminated by addition of phosphoric acid. The incorporated radioactivity was measured by using a TopCount NXT microplate scintillation counter (PerkinElmer). Data were converted to the percent inhibition relative to controls.
Immunofluorescence assay.
HeLa R19 cells were grown to subconfluency on coverslips in 24-well plates. Cells were transfected with 300 ng DNA for each construct by using FuGENE (Roche) according to the manufacturer's protocol. After 16 h, cells were treated with 1 μM compound 1 for 1 h at 37°C. Subsequently, cells were fixed and stained with a mouse monoclonal anti-HA antibody (Covance) and an Alexa 568-conjugated goat anti-mouse antibody (Molecular Probes) as described previously (20). Cells were analyzed with a Leica BMR microscope. Fluorescent images of typical examples of observations that we consistently obtained in three or more experiments are presented in the figures.
Replication rescue assay.
A replication rescue assay was conducted as described before (20). Briefly, BGM cells were transfected with plasmids carrying wt or mutant PI4KIIIβ, or EGFP (enhanced green fluorescent protein) as a negative control. Two days posttransfection, the cells were infected with CVB3-Rluc in the absence or presence of compounds. At 8 h p.i., the intracellular Renilla luciferase activity was determined by using the Renilla luciferase assay system (Promega). Similar expression levels of the proteins were confirmed in parallel in an immunofluorescence assay with an antibody directed against HA.
PK studies.
Pharmacokinetics (PK) studies were performed in male NMRI mice administered a single dose of 1 mg/kg intravenously (i.v.) or 5 mg/kg orally. The vehicle for both routes of administration was DMSO-polyethylene glycol 200 (PEG 200)-saline (5:45:50, vol/vol). Intravenous doses were prepared at a concentration of 0.2 mg/ml and administered in a dose volume of 5 ml/kg of body weight. Oral doses were prepared at a concentration of 0.5 mg/ml and administered in a dose volume of 10 ml/kg of body weight. No adverse effects were noted for either route of administration. Terminal blood samples were collected from three animals per time point up to 8 h postdose and using heparin as anticoagulant. Plasma proteins were precipitated, and compound was extracted by the addition of three volumes of acetonitrile containing an analytical internal standard (carbamazepine). Samples were centrifuged for 30 min at 4,000 rpm, and the supernatant fractions were subjected to mass spectrometry analysis. Quantification of the compound was performed by extrapolation from calibration lines prepared in control mouse plasma and analyzed concurrently with experimental samples. Pharmacokinetic parameters were determined by noncompartmental analysis using WinNonlin software (Pharsight, version 5.2). AUC (area under the time-concentration curve) values were calculated by the trapezoidal method.
CVB4-induced pancreatitis mouse model.
All experimental procedures were in accordance with the KULeuven ethical committee for vertebrate animal experiments. Five-week-old male SJL mice (Harlan, The Netherlands) were fed ad libitum and weighed every day as a measure of compound tolerability. Mice (n = 4 per group) were treated orally twice daily. Compound 2 was dissolved at a concentration of 2.5 mg/ml in a vehicle with 10% hydroxypropyl-β-cyclodextrin (HPBCD) acidified to pH 3 with citric acid. For lower dosages, this solution was diluted with the vehicle solution. Fresh compound solutions were prepared every day. An administration volume of 200 μl was used per dose. The mice received the first dose (t = 0) in the morning on day 0 and were infected intraperitoneally with 1 × 105 PFU of CVB4 strain Edwards (E2) in a final volume of 300 μl of phosphate-buffered saline (PBS) 3 h later. The range of doses were administered at t = 8, 24, 32, 48, 56, and 72 h. The mice were sacrificed at 75 h. Blood was collected by cardiac puncture after euthanasia. After 1 h at room temperature (RT), blood was centrifuged at 13,000 rpm for 10 min. Serum amylase and lipase were quantified by using an enzymatic colorimetric reaction (Cobas/Roche, Basel, Switzerland) on the Modular P analyzer (Roche Diagnostics, Basel, Switzerland). The mice were perfused transcardially with phosphate-buffered saline, and the pancreas was removed. One part was fixed in 4% formaldehyde and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (H&E) for routine histological examination. The severity of pancreatitis (corresponding to the degree of edema, inflammation, and necrosis) was assessed blindly by using a standardized scoring system and expressed quantitatively. The second half of the pancreas was flash-frozen in liquid nitrogen and stored for quantification of infectious virus content. To this end, the tissue parts were homogenized in cell culture medium by mechanical disruption in 2-ml tubes containing ceramic beads on the automated Precellys24 apparatus (Bertin, France). Homogenization was performed at 6,500 rpm for 3 cycles of 5 s, with intervals of 5 s. Next, the tubes were centrifuged for 10 min at 4°C, 13,000 rpm, and the cleared supernatant was collected. The infectious virus content was determined in cell cultures and expressed as the CCID50 according to the method of Reed and Muench.
RESULTS
Compound 1 is a broad-spectrum inhibitor of enterovirus replication.
The BioFocus SoftFocus kinase inhibitor library was screened for antiviral activity against different viruses, as recently reported (27). Compound 1 (Fig. 1) was identified as an inhibitor of CVB3. During the development of the series, the analogue compound 2 was identified, which showed improved bioavailability. We evaluated the effect of compound 1 on a large panel of representative members of the different HEV and HRV species in a multicycle cytopathic effect (CPE) assay. The antiviral effect was assessed at 3 to 4 days postinfection to allow maximal CPE. Table 1 shows that compound 1 potently inhibited all viruses tested, with EC50s ranging between 4 and 71 nM. The cytotoxicity of compound 1, determined in parallel with the EC50 and using the same culture conditions for 3 to 4 days, was low, with CC50 values ranging from 11 to 65 μM, resulting in high selectivity indices.
Fig 1.
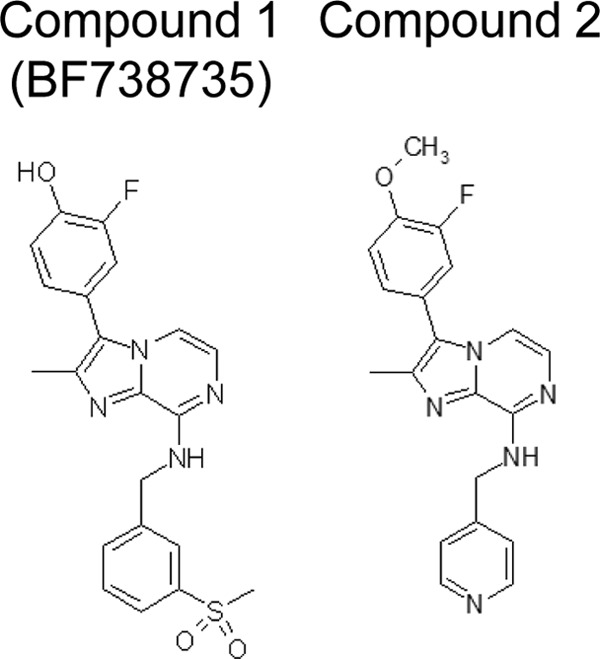
Molecular structures of compounds 1 and 2.
Table 1.
Broad-spectrum antienterovirus activity of compound 1
| Virus group and species or strain | Cells | EC50 (μM)a | SI_b_ |
|---|---|---|---|
| Human enterovirus A | |||
| EV71 | BGM | 0.011 ± 0.003 | 5,945 |
| Human enterovirus B | |||
| CVA9 | BGM | 0.021 ± 0.004 | 3,114 |
| CVB3 | BGM | 0.071 ± 0.018 | 921 |
| ECHO9 | BGM | 0.054 ± 0.006 | 1,211 |
| ECHO11 | BGM | 0.026 ± 0.007 | 2,515 |
| Human enterovirus C | |||
| PV1 | HeLa Rh | 0.019 ± 0.008 | 2,505 |
| PV2 | HeLa Rh | 0.013 ± 0.004 | 3,661 |
| PV3 | HeLa Rh | 0.005 ± 0.002 | 9,520 |
| Human enterovirus D | |||
| EV68 | HeLa R19 | 0.031 ± 0.001 | 371 |
| EV70 | HeLa R19 | 0.025 ± 0.020 | 460 |
| Human rhinovirus A | |||
| HRV2 | HeLa Rh | 0.009 ± 0.005 | 5,288 |
| HRV9 | HeLa Rh | 0.006 ± 0.005 | 7,933 |
| HRV15 | HeLa Rh | 0.021 ± 0.010 | 2,267 |
| HRV29 | HeLa Rh | 0.005 ± 0.005 | 9,520 |
| HRV39 | HeLa Rh | 0.004 ± 0.005 | 11,900 |
| HRV41 | HeLa Rh | 0.017 ± 0.006 | 2,800 |
| HRV59 | HeLa Rh | 0.006 ± 0.005 | 7,933 |
| HRV63 | HeLa Rh | 0.006 ± 0.003 | 7,933 |
| HRV85 | HeLa Rh | 0.024 ± 0.025 | 1,983 |
| HRV89 | HeLa Rh | 0.024 ± 0.019 | 1,983 |
| Human rhinovirus B | |||
| HRV14 | HeLa Rh | 0.031 ± 0.024 | 1,535 |
| HRV42 | HeLa Rh | 0.027 ± 0.032 | 1,762 |
| HRV70 | HeLa Rh | 0.024 ± 0.027 | 1,983 |
| HRV72 | HeLa Rh | 0.028 ± 0.026 | 1,700 |
| HRV86 | HeLa Rh | 0.024 ± 0.031 | 1,983 |
Inhibition of virus replication in a multicycle assay could be the result of a direct effect of the compound on virus replication or an indirect effect on virus spreading by stimulation of an antiviral pathway in the host cell. To investigate whether the compound directly or indirectly affected virus replication, we tested its effects in a single-cycle assay with a result determined at 8 h postinfection. Compounds 1 and 2 inhibited all enteroviruses used for this assay, while members of other picornavirus genera, namely, the cardiovirus EMCV and the aphthovirus ERAV, were not affected by the compound (Fig. 2). This result demonstrated that compounds 1 and 2 inhibit virus replication directly.
Fig 2.

Antiviral activities of compounds 1 and 2 on a single virus replication cycle. Cells were infected at low MOI with various HEV and HRV species, the cardiovirus EMCV, or the aphthovirus ERAV. After infection, 1 μM compound 1 or 3 μM compound 2 was added to the cells. After 8 h, cells were lysed by freeze-thawing to release intracellular virus particles, and the total virus titer was determined by endpoint titration. Bars represent means of three samples ± the SD.
The compounds act at the level of viral RNA replication.
To determine the step in the virus replication cycle at which the compounds inhibited replication, we used CVB3-Rluc, a genetically engineered virus that encodes Renilla luciferase upstream of the capsid coding region (18). Replication of CVB3-Rluc yields, besides viral proteins, large amounts of Renilla luciferase. Quantification of intracellular luciferase levels is a sensitive measure to delineate the steps of viral RNA translation and replication and eliminates possible effects of the compound on virion assembly and egress in the viral replication cycle. GuaHCl, a well-known inhibitor of viral RNA replication, was used to distinguish between translation and replication of the viral RNA. Treatment with GuaHCl resulted in a strong reduction in the amount of luciferase (Fig. 3A, dashed line). Low concentrations of compound 1 reduced the amount of luciferase to GuaHCl-treated levels, suggesting that the compound blocked viral RNA replication. The EC50 of compound 1 in this assay was 77 nM, which was comparable to the inhibition observed in the multicycle assay for CVB3 (Table 1). Compound 2 was found to be slightly less potent, with an EC50 of 238 nM.
Fig 3.

Compound 1 is a potent inhibitor of enterovirus RNA replication. (A) BGM cells were infected with CVB3-Rluc at an MOI of 1. Following infection, the virus was removed and compound 1 or compound 2 was added to the cells. The values obtained with the replication inhibitor GuaHCl, used at 2 mM, are shown as a dashed line. (B) BGM cells were transfected with RNA transcripts of EV71 or CVB3 replicons. Immediately after transfection, the medium was replaced by fresh (compound-containing) medium. For the experiments in both panels A and B, at 8 h postinfection or posttransfection, cells were lysed to quantify the intracellular amount of luciferase as a measure of viral RNA replication. Data points or bars represent means of three samples ± the SD.
To ensure that the compounds exerted their antiviral effects at the step of viral RNA replication in the enterovirus replication cycle, we employed subgenomic CVB3 and EV71 replicons in which the capsid coding region was replaced by the firefly luciferase gene (18). For the CVB3 as well as the EV71 replicon, 1 and 5 μM compound 1 strongly inhibited replication, as demonstrated by the reduction in luciferase amounts to levels observed after GuaHCl treatment (Fig. 3B). Taken together, these results indicate that the compounds act at the stage of viral RNA replication.
Substitutions in 3A rendered CVB3 resistant to compound 1.
In order to obtain more insight into the mechanism of action, we generated CVB3 mutants resistant to compound 1 by culturing the virus in the presence of the compound. After 32 passages (∼16 weeks), three independent cultures of CVB3 were obtained that replicated efficiently in the presence of compound 1 at concentrations exceeding the EC50 by more than 10-fold. The genomes of all virus isolates carried mutations in two or more different nonstructural proteins (Table 2). Only a single substitution was detected in the 3A protein, either H57Y (two virus isolates) or I54F (one isolate). Interestingly, we previously found that these 3A substitutions provide resistance against enviroxime, GW5074, and TTP-8307 (20, 28). Besides these substitutions, 3A-V45A was also shown to allow CVB3 to replicate in the presence of these compounds (20, 28). We therefore evaluated whether 3A-V45A also confers resistance against compound 1, by employing our previously established CVB3 3A-V45A and CVB3 3A-H57Y mutants (20, 28). Wild-type CVB3 replication was strongly inhibited (to virus input levels) at 1 and 5 μM compound 1 (Fig. 4A). In contrast, the substitutions 3A-V45A and 3A-H57Y greatly restored the ability of CVB3 to replicate in the presence of compound 1. During the production of the mutant viruses, comprising multiple replication cycles, the viruses may have acquired other substitutions in addition to 3A-V45A or 3A-H57Y that are essential for providing resistance. To rule out this possibility, we transfected into cells RNA transcripts of the infectious clones and determined the virus titers after one replication cycle. Comparable results were obtained with RNA transfection as with infection (Fig. 4B). Thus, the single substitutions V45A and H57Y in 3A allowed CVB3 to replicate in the presence of compound 1.
Table 2.
Substitutions identified in the genome of CVB3 mutants resistant to compound 1
| Serial passage no. | Amino acid substitution(s) within: | |||
|---|---|---|---|---|
| VP1 | 2A | 2C | 3A | 3D |
| 1 | N2D | H57Y | ||
| H44R | ||||
| 2 | D152E | N2D | H57Y | |
| S109I | ||||
| 3 | E77Q | N2D | I54F | P336S |
| V40A | ||||
| A95V |
Fig 4.

Resistance of CVB3 mutants to compound 1. (A and B) BGM cells were infected with CVB3 wild type, CVB3 3A-V45A, or CVB3 3A-H57Y (A) or transfected with the corresponding RNA transcripts of full-length infectious CVB3 clones (B). Immediately after infection or transfection, various concentrations of compound 1 were added to the cells. After 8 h, cells were lysed by freeze-thawing to release intracellular virus particles, and the total virus titer was determined by endpoint titration. (B) Bars represent means of three samples ± the SD. Significant differences compared to wild-type virus are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Compounds 1 and 2 inhibit PI4KIIIβ activity.
Recently, we showed that the 3A substitutions described above rendered CVB3 resistant to the inhibitory effects of the specific PI4KIIIβ inhibitor PIK93 (20). Furthermore, we and others demonstrated that enviroxime and GW5074 target PI4KIIIβ (20, 22). The finding that the same 3A substitutions provided resistance to compound 1 prompted us to examine whether compounds 1 and 2 also target PI4KIIIβ. To this end, we first investigated whether the compounds were able to inhibit PI4KIIIβ in an in vitro kinase activity assay. Wortmannin, used as a positive control, had a 50% inhibitory concentration (IC50) of 353 nM for PI4KIIIβ and 622 nM for PI4KIIIα, which were in line with previous reports (31, 32). Compound 1 strongly inhibited PI4KIIIβ activity in vitro, with an IC50 of 5.7 nM (Fig. 5). Compound 1 also impaired PI4KIIIα, but only at an ∼300-fold-higher concentration (IC50 of 1.7 μM). Compound 2 was less potent in blocking PI4KIIIβ (IC50 of 91 nM), but it had an equal specificity for the β-isoform as compound 1. In addition, the activity of compound 1 was analyzed on a set of 150 cellular kinases (Reaction Biology Corporation), including 13 lipid kinases at a concentration of 10 μM (data not shown). For all kinases, the inhibition was less than 10%, indicating that compound 1 specifically inhibits PI4KIIIβ in vitro.
Fig 5.

Compounds 1 and 2 specifically inhibit PI4KIIIβ in vitro. Recombinant PI4KIIIβ or PI4KIIIα was incubated in the presence of compound 1 (upper panel) or compound 2 (lower panel) with their substrate, phosphatidylinositol, in the form of Triton micelles and radioactively labeled ATP. After termination of the enzyme reaction with phosphoric acid, the amount of radioactive ATP that was incorporated into the micelles was quantified as a measure of PI4K activity. Data were converted to the percent inhibition relative to controls. Data points represent the means of three samples ± the SD.
We next investigated whether compound 1 also inhibits PI4KIIIβ in cells. To this end, we employed a PI4P sensor called FAPP1-PH-GFP, a GFP-tagged PH domain of FAPP1. This PH domain contains a PI4P-binding pocket as well as an Arf1-binding site, which together determine its localization to the Golgi complex (33–36). Previously, we showed that PI4P produced by PI4KIIIβ is the major determinant of the Golgi complex localization of the PI4P sensor; hence, FAPP1-PH-GFP can be used to quantify the synthesis of PI4P lipids catalyzed by PI4KIIIβ at the Golgi apparatus (20). Following a 1-h treatment with 1 μM compound 1, a dramatic loss of the Golgi complex localization of the PI4P sensor compared to that of untreated cells was noted (Fig. 6A). Compound 1 reduced the amount of Golgi complex-localized FAPP1-PH-GFP by 76% ± 4.8% (mean ± standard deviation [SD]; n = 10). Similar to our previous observations with the PI4KIIIβ inhibitors enviroxime, GW5074, and PIK93 (52), we detected an increase in the intensity of the PI4KIIIβ staining on Golgi complex membranes after compound 1 treatment, which may have been the result of an unknown compensatory mechanism of the cell. Together, these results suggest that compound 1 reduces the PI4P levels at the Golgi complex through a direct inhibition of PI4KIIIβ activity.
Fig 6.
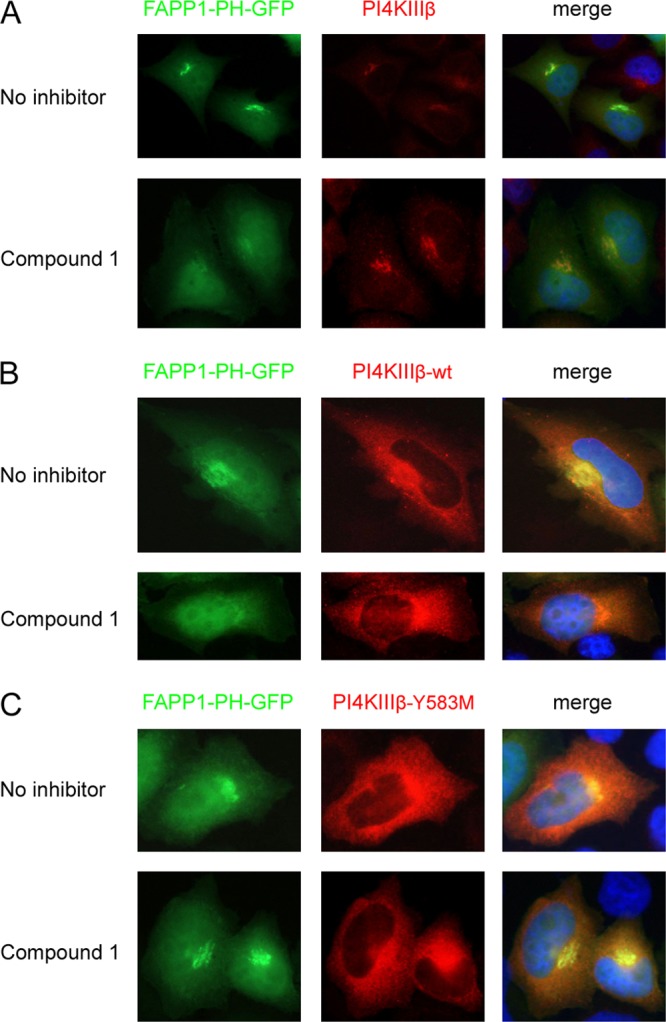
Compound 1 binds in the ATP-binding pocket of PI4KIIIβ. (A) HeLa R19 cells were transfected with FAPP1-PH-GFP and treated 1 day later with 1 μM compound 1 for 1 h, after which endogenous PI4KIIIβ was stained. (B and C) HeLa R19 cells were cotransfected with plasmids carrying FAPP1-PH-GFP and either HA-tagged PI4KIIIβ wt (B) or PI4KIIIβ-Y583M (C). The next day, cells were treated for 1 h with 1 μM compound 1, after which the overexpressed PI4KIIIβ was stained with an antibody against HA. Nuclei were stained with Hoechst stain.
Compounds 1 and 2 can inhibit kinases in two different manners (37). In the competitive manner, a compound acts as an ATP/GDP analogue and binds in the ATP-binding pocket. Alternatively, compounds may inhibit the kinase in a noncompetitive manner without interfering with ATP binding. To study the manner in which compound 1 inhibits PI4KIIIβ, we used the mutant PI4KIIIβ-Y583M, which carried the substitution Y583M in the ATP-binding pocket. This substitution weakens binding of the inhibitors wortmannin and PIK93 but does not affect the catalytic activity of the kinase (31). Plasmids carrying either PI4KIIIβ wt or PI4KIIIβ-Y583M were cotransfected with the FAPP1-PH-GFP construct into cells. One day later, cells were treated for 1 h with compound 1. Without treatment, the PI4P sensor was localized to the Golgi complex in cells expressing PI4KIIIβ wt or PI4KIIIβ-Y583M (Fig. 6B and C). Treatment with compound 1 resulted in diminished PI4P levels in cells that expressed PI4KIIIβ wt, as shown by the reduction of Golgi complex-localized FAPP1-PH-GFP (Fig. 6B). The PI4P sensor remained localized in the Golgi apparatus in cells expressing PI4KIIIβ-Y583M (Fig. 6C). These results suggested that compound 1 binds in the ATP-binding pocket of PI4KIIIβ and therefore inhibits PI4KIIIβ in a competitive manner.
PI4KIIIβ is the major target of the compounds accountable for the inhibition of CVB3 replication.
Competitive kinase inhibitors usually target more than one cellular kinase (38). Although compound 1 is a highly specific PI4KIIIβ inhibitor in vitro, we next verified whether PI4KIIIβ is the major target of compound 1 responsible for its detrimental effect on CVB3 replication. To this end, we performed a “replication rescue assay” with the PI4KIIIβ mutant. Cells were transfected with either PI4KIIIβ wt, PI4KIIIβ-Y583M, or as negative controls the kinase-dead PI4KIIIβ-D656A or EGFP. Two days later, the cells were infected with CVB3-Rluc in the presence of compound 1 and were lysed after 8 h to quantify the intracellular luciferase levels. Similar expression levels of all PI4KIIIβ proteins were confirmed in parallel in an immunofluorescence assay (data not shown). CVB3-Rluc was unable to replicate in cells transfected with EGFP, the kinase-dead PI4KIIIβ-D656A, or PI4KIIIβ wt in the presence of compound 1 (Fig. 7). In contrast, the expression of PI4KIIIβ-Y583M provided nearly complete protection against the inhibitory effect of compound 1 on CVB3 replication, since CVB3-Rluc replicated almost to untreated levels in these cells. Similar results were obtained with compound 2. These results corroborate PI4KIIIβ as the major target of compounds 1 and 2 accountable for the inhibition of CVB3 replication.
Fig 7.

CVB3 replication in the presence of compound 1 or 2 is rescued by expression of a PI4KIIIβ mutant. BGM cells were transfected with HA-tagged PI4KIIIβ wt, PI4KIIIβ-Y583M or, as negative controls, the kinase-dead PI4KIIIβ-D656A or EGFP. Two days posttransfection, cells were infected with CVB3-Rluc in the presence of 1 μM compound 1 or 3 μM compound 2. After lysis of the cells at 8 h p.i., the amount of luciferase activity was quantified in the samples. Bars represent means of three samples ± the SD.
Activity of compound 2 in a CVB4-induced pancreatitis mouse model.
Next, we investigated the in vivo antiviral activity of compound 2 in a CVB4-induced pancreatitis model (39). Since the pharmacokinetic properties of compound 1 were not suitable for analysis of its effects in vivo due to its rapid clearance (data not shown), we employed its analogue, compound 2, to explore protection against enterovirus infection in mice. Importantly, the in vitro characteristics of compound 2 were comparable to those of compound 1 (Fig. 2, 3, 5, and 7). The pharmacokinetics of compound 2 were assessed in male NMRI mice administered a dose of 5 mg/kg orally and 1 mg/kg i.v., using three mice per time point (Fig. 8). The compound was rapidly absorbed, with maximal plasma levels (Tmax) observed at 30 min postdosing, while the clearance (Cl) was low, approximately 25% of the liver blood flow, and the volume of distribution (_V_ss) was high (Table 3). Based on these results, we decided to perform a pilot experiment of the CVB4-induced pancreatitis model. Considering the PK profile of compound 2, we determined that application of an oral twice-a-day (BID) dosing regimen would give optimal coverage of the antiviral IC50 while keeping the maximum serum concentration (_C_max) in a lower range. Mice (n = 4 per group) were infected intraperitoneally with CVB4 and treated with compound 2 or left untreated. The compound was administered at three different doses (1, 5, and 25 mg/kg BID). No adverse effects or clinical signs were detected during the experiment, indicating that the mice tolerated compound 2.
Fig 8.
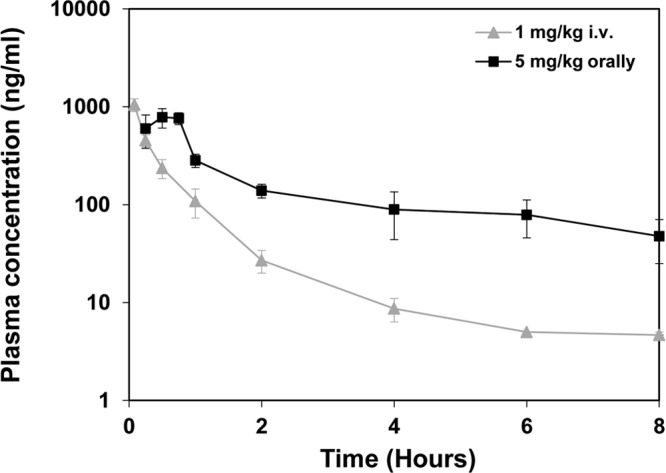
Plasma drug concentration levels of compound 2 in NMRI mice. Mice were treated with a single dose of compound 2 as either 1 mg/kg i.v. or 5 mg/kg orally. Terminal blood samples were collected from three animals per time point, up to 8 h postdosing. Samples were centrifuged, and the supernatant was subjected to mass spectrometry analysis. Data points represent the means of three samples ± the SD.
Table 3.
Pharmacokinetic parameters for compound 2 in male NMRI mice
| Parameter (units) | Result after treatment | |
|---|---|---|
| 1 mg/kg i.v. | 5 mg/kg orally | |
| _C_0 or _C_max (ng/ml) | 1,559 | 782 |
| Tmax (h) | 0.5 | |
| AUC(0–t) (ng · h/ml) | 503 | 1,281 |
| AUC(0–∞) (ng · h/ml) | 519 | 1,566 |
| Cl (liters/h/kg) | 1.93 | |
| _V_ss (liters/kg) | 2.23 | |
| _t_1/2 (h) | 2.38 | 4.15 |
| F (%) | 60 |
In H&E-stained sections of pancreas tissue collected 3 days after CVB4 infection, extensive exocrine tissue damage was obvious in infected, untreated animals (Fig. 9A). Histopathological analysis revealed diffuse interstitial inflammation, necrosis, and edema, along with a cellular infiltrate (Fig. 9B). On the other hand, pancreas sections obtained from mice treated with the highest compound 2 dose (25 mg/kg BID) showed no signs of inflammation, edema, or necrosis. Partial protection against CVB4-induced pancreatitis was noted at a lower dose (5 mg/kg BID), while no beneficial effect was observed at the lowest dose (1 mg/kg BID). Peak serum amylase and lipase levels were also measured on day 3 postinfection (39). In untreated CVB4-infected mice, serum lipase and amylase levels were increased more than 10-fold and 5-fold, respectively (Fig. 9C). A dose-dependent effect of compound 2 was observed with mice treated with the highest dose, as lipase and amylase levels were comparable to those in the uninfected control group. Moreover, infectious virus content was reduced by >2 log10 in mice treated with the highest dose (Fig. 9D). Taken together, these results indicated that compound 2 exhibited a clear dose-dependent antiviral effect in vivo, and a dose of 25 mg/kg BID fully protected against CVB4-induced pancreatitis.
Fig 9.

Antiviral activity of compound 2 in a CVB4-induced pancreatitis mouse model. (A) Pancreas histopathology (H&E stain) in CVB4-induced pancreatitis. Mice were infected with CVB4, treated with compound 2 (right panel) or left untreated (left panel), and sacrificed 3 days p.i. (B) Histopathological severity scoring for CVB4-induced pancreatitis. H&E-stained tissue sections were scored blindly for inflammation, necrosis, and edema by using a standardized scoring system. A score of 0 indicates complete absence of pathology, whereas a score of 3 refers to diffuse lesions throughout the tissue section. (C) Effect of compound 2 on serum markers for pancreatitis. At day 3 p.i., serum was collected and lipase/amylase were quantified in enzymatic colorimetric assays. (D) Effect of compound 2 on infectious virus production, which was quantified by titration of tissue homogenates from cell cultures.
DISCUSSION
We demonstrated here the specific and selective antiviral effects of a novel broad-spectrum inhibitor of enterovirus replication in vitro and in a CVB4-induced pancreatitis mouse model. To gain insight into the mechanism of action, compound-resistant CVB3 was generated, which revealed that single substitutions in 3A allowed the virus to replicate more efficiently in the presence of the compound. These same substitutions allowed us in our previous study to identify PI4KIIIβ as the target of the antiviral compounds enviroxime and GW5074. Similar to these earlier results, compounds 1 and 2 also specifically inhibited the activity of PI4KIIIβ in an in vitro kinase activity assay and strongly reduced the PI4P levels in Golgi complex membranes in intact cells. Finally, CVB3 replication in the presence of the compounds was nearly restored to untreated levels by the expression of a mutant PI4KIIIβ carrying a substitution in its ATP-binding pocket. Collectively, these results indicated that host factor PI4KIIIβ is the major target of the compounds responsible for their effects on enterovirus replication.
Enteroviruses are not the only viruses that exploit PI4KIIIβ for their replication. Within the Picornaviridae family, Aichi virus of the genus Kobuvirus was recently also reported to recruit PI4KIIIβ to its replication sites (40). Viruses belonging to the genera Cardiovirus and Apthovirus do not seem to rely on this host factor, which is in line with their insensitivity to enviroxime and GW5074 (data not shown). HCV also utilizes PI4Ks for its replication, predominantly PI4KIIIα (41–47), while PI4KIIIβ has also been reported to be involved in virus replication (21, 42, 45, 47). In addition, severe acute respiratory syndrome coronavirus also depends on PI4KIIIβ, but for cell entry instead of RNA replication (48).
Since PI4KIIIβ is an essential host factor for many viruses, it is an attractive target for the development of broad-spectrum antiviral drugs. Targeting a host factor is more likely to be associated with toxic side effects than targeting a viral protein. Indeed, a recent study in the context of HCV treatment showed that conditional transgenic mice that served as models for pharmacologic inhibition of PI4KIIIα displayed a lethal phenotype (49), which suggested that compounds targeting PI4KIIIα may have toxic side effects. The first specific inhibitor of PI4KIIIβ was PIK93, which had an ∼100-fold preference for the β-isoform over the α-isoform but also inhibited PI3Ks (50). GW5074 and enviroxime have long been known to inhibit enteroviruses, but they were only recently shown to exert their antiviral effect by inhibiting PI4KIIIβ (20, 22). GW5074 is a classic example of a promiscuous kinase inhibitor with an undesired wide range of other cellular kinases as targets, such as c-Raf, Pim-1 through Pim-3, HIPK2, and MST2 (38). Enviroxime is a far more potent inhibitor of enterovirus replication than GW5074 (28, 51), but the clinical development of enviroxime was discontinued at the point of phase II trials due to insufficient therapeutic effects and gastrointestinal side effects (52, 53). More recently, a number of investigators have reported novel antiviral PI4KIIIβ inhibitors, but they have also reported different adverse events in these studies. A compound from Novartis with antiviral activity against HCV was precluded from further development due to its antiproliferative effects on T and B cells in vitro, which were confirmed in vivo in a rat antibody formation assay (i.e., the sheep red blood cell assay) at one of the doses tested; however, no other clinical signs were noted in the 4 days of treatment (54). While this work was in progress, the antienterovirus compound T-00127-HEV1 (51), which is structurally similar to our compounds 1 and 2, was evaluated together with another novel PI4KIIIβ inhibitor from Boehringer-Ingelheim for toxicity in SJL mice (55). Oral doses of 50 mg/kg/day and higher of the BI compound were found to be lethal to the mice, with mortality starting at day 2 after the start of the treatment. However, in our study the mice treated orally twice a day with 25 mg/kg compound 2, which had a comparable potency against PI4KIIIβ, showed no signs of toxicity during 3 days of treatment. Therefore, further studies are needed to elucidate whether the toxicity effects observed in other studies were caused by the inhibition of PI4KIIIβ or, alternatively, by some as-yet-unknown other off-target effect.
Due to their high mutation rate, RNA viruses in general and picornaviruses in particular can rapidly acquire mutations that allow them to become resistant to the antiviral effects of compounds that directly target viral proteins, and this poses serious problems in a clinical context. For example, resistant HCV variants were already detected during the first days of treatment with the viral protease inhibitors telaprevir and boceprevir (56, 57). It is generally believed that compounds that target critical host factors pose a higher barrier for resistance development of RNA viruses. Cellular targets are unlikely to mutate in response to therapy and are therefore ideal targets for drug development. Indeed, PV was unable to acquire resistance against the Hsp90 inhibitor geldanamycin, which blocked the correct folding and maturation of enterovirus capsid proteins (58). Here, we showed that CVB3 can acquire resistance in cell culture to a novel PI4KIIIβ inhibitor by acquiring substitutions in its 3A protein. It is important to emphasize that this resistance developed only after a lengthy resistance selection process (∼16 weeks). As most enterovirus infections are acute and of limited duration, it is questionable whether resistance will develop in vivo upon (short-term) treatment with PI4KIIIβ inhibitors. If so, combination treatments with more than one drug might be needed for successful antiviral therapy.
ACKNOWLEDGMENTS
We acknowledge Stijn Delmotte, Mieke Flament, and Tom Bellon for their excellent assistance in generating the antiviral data and Carolien De Keyzer and Kim Donckers for performing the animal experiments and generating the in vivo data.
This work was supported by research grants from The Netherlands Organization for Scientific Research (NWO-ECHO-700.57.001 to F.V.K., NWO-VICI-820.02.018 to F.V.K., NWO-VENI-863.12.005 to H.V.D.S.), the “Covenant K.U. Leuven-Radboud University Nijmegen” framework to F.V.K. and J.N., the “Agency for Innovation by Science and Technology in Flanders (IWT)” to C.L., and from the KU Leuven Geconcerteerde Onderzoeksactie.
Footnotes
Published ahead of print 29 July 2013
REFERENCES
- 1.Palacios G, Oberste MS. 2005. Enteroviruses as agents of emerging infectious diseases. J. Neurovirol. 11:424–433 [DOI] [PubMed] [Google Scholar]
- 2.Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. 2010. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 9:1097–1105 [DOI] [PubMed] [Google Scholar]
- 3.Sawyer MH. 2002. Enterovirus infections: diagnosis and treatment. Semin. Pediatr. Infect. Dis. 13:40–47 [DOI] [PubMed] [Google Scholar]
- 4.Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3:765–776 [DOI] [PubMed] [Google Scholar]
- 5.Yeung W-CG, Rawlinson WD, Craig ME. 2011. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ 342:d35. 10.1136/bmj.d35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaida A, Kubo H, Sekiguchi J, Kohdera U, Togawa M, Shiomi M, Nishigaki T, Iritani N. 2011. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg. Infect. Dis. 17:1494–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lévêque N, Huguet P, Norder H, Chomel J-J. 2010. Enteroviruses responsible for acute hemorrhagic conjunctivitis. Med. Mal. Infect. 40:212–218 (In French) [DOI] [PubMed] [Google Scholar]
- 8.Rotbart HA. 2000. Antiviral therapy for enteroviruses and rhinoviruses. Antivir. Chem. Chemother. 11:261–271 [DOI] [PubMed] [Google Scholar]
- 9.Turner RB. 1998. The common cold. Pediatr. Ann. 27:790–795 [DOI] [PubMed] [Google Scholar]
- 10.Liang Z, Mao Q, Gao F, Wang J. 2013. Progress on the research and development of human enterovirus 71 (EV71) vaccines. Front. Med. 7:111–121 [DOI] [PubMed] [Google Scholar]
- 11.Zhu F-C, Liang Z-L, Li X-L, Ge H-M, Meng F-Y, Mao Q-Y, Zhang Y-T, Hu Y-M, Zhang Z-Y, Li J-X, Gao F, Chen Q-H, Zhu Q-Y, Chu K, Wu X, Yao X, Guo H-J, Chen X-Q, Liu P, Dong Y-Y, Li F-X, Shen X-L, Wang J-Z. 2013. Immunogenicity and safety of an enterovirus 71 vaccine in healthy Chinese children and infants: a randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet 381:1037–1045 [DOI] [PubMed] [Google Scholar]
- 12.Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. 2010. Picornaviruses. Curr. Top. Microbiol. Immunol. 343:43–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bedard KM, Semler BL. 2004. Regulation of picornavirus gene expression. Microbes Infect. 6:702–713 [DOI] [PubMed] [Google Scholar]
- 14.Bienz K, Egger D, Pasamontes L. 1987. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 160:220–226 [DOI] [PubMed] [Google Scholar]
- 15.Limpens RWAL, Kumar D, Transformation T, Structures ER, Study T, Compartments D, This S, Feeds RSS, Journal ASM. 2011. The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. mBio 2(5):e00166–11. 10.1128/mBio.00166-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. 2012. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 86:302–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E. 2008. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 4(11):e1000216. 10.1371/journal.ppat.1000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanke KHW, van der Schaar HM, Belov GA, Feng Q, Duijsings D, Jackson CL, Ehrenfeld E, van Kuppeveld FJM. 2009. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 83:11940–11949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wessels E, Duijsings D, Niu T-K, Neumann S, Oorschot VM, de Lange F, Lanke KHW, Klumperman J, Henke A, Jackson CL, Melchers WJG, van Kuppeveld FJM. 2006. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell 11:191–201 [DOI] [PubMed] [Google Scholar]
- 20.Van der Schaar HM, van der Linden L, Lanke KHW, Strating JRPM, Pürstinger G, de Vries E, de Haan C a M, Neyts J, van Kuppeveld FJM. 2012. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 22:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu N-Y, Ilnytska O, Belov G, Santiana M, Chen Y-H, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJM, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2011. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 85:2364–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thibaut HJ, De Palma AM, Neyts J. 2012. Combating enterovirus replication: state-of-the-art on antiviral research. Biochem. Pharmacol. 83:185–192 [DOI] [PubMed] [Google Scholar]
- 24.Norder H, De Palma AM, Selisko B, Costenaro L, Papageorgiou N, Arnan C, Coutard B, Lantez V, De Lamballerie X, Baronti C, Solà M, Tan J, Neyts J, Canard B, Coll M, Gorbalenya AE, Hilgenfeld R. 2011. Picornavirus non-structural proteins as targets for new anti-virals with broad activity. Antiviral Res. 89:204–218 [DOI] [PubMed] [Google Scholar]
- 25.Gallay PA, Lin K. 2013. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des. Dev. Ther. 7:105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coelmont L, Hanoulle X, Chatterji U, Berger C, Snoeck J, Bobardt M, Lim P, Vliegen I, Paeshuyse J, Vuagniaux G, Vandamme A-M, Bartenschlager R, Gallay P, Lippens G, Neyts J. 2010. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5(10):e13687. 10.1371/journal.pone.0013687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macleod AM, Mitchell DR, Palmer NJ, Van de Poel H, Conrath K, Andrews M, Leyssen P, Neyts J. 2013. Identification of a series of compounds with potent antiviral activity for the treatment of enterovirus infections. ACS Med. Chem. Lett. 4:585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Palma AM, Thibaut HJ, van der Linden L, Lanke K, Heggermont W, Ireland S, Andrews R, Arimilli M, Al-Tel TH, De Clercq E, van Kuppeveld F, Neyts J. 2009. Mutations in the nonstructural protein 3A confer resistance to the novel enterovirus replication inhibitor TTP-8307. Antimicrob. Agents Chemother. 53:1850–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wessels E, Duijsings D, Notebaart RA, Melchers WJG, van Kuppeveld FJM. 2005. A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-Golgi transport. J. Virol. 79:5163–5173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Palma AM, Heggermont W, Leyssen P, Pürstinger G, Wimmer E, De Clercq E, Rao A, Monforte A-M, Chimirri A, Neyts J. 2007. Anti-enterovirus activity and structure-activity relationship of a series of 2,6-dihalophenyl-substituted 1H,3H-thiazolo[3,4-a]benzimidazoles. Biochem. Biophys. Res. Commun. 353:628–632 [DOI] [PubMed] [Google Scholar]
- 31.Balla A, Tuymetova G, Toth B, Szentpetery Z, Zhao X, Knight ZA, Shokat K, Steinbach PJ, Balla T. 2008. Design of drug-resistant alleles of type-III phosphatidylinositol 4-kinases using mutagenesis and molecular modeling. Biochemistry 47:1599–1607 [DOI] [PubMed] [Google Scholar]
- 32.Downing GJ, Kim S, Nakanishi S, Catt KJ, Balla T. 1996. Characterization of a soluble adrenal phosphatidylinositol 4-kinase reveals wortmannin sensitivity of type III phosphatidylinositol kinases. Biochemistry 35:3587–3594 [DOI] [PubMed] [Google Scholar]
- 33.Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA. 2004. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat. Cell Biol. 6:393–404 [DOI] [PubMed] [Google Scholar]
- 34.He J, Scott JL, Heroux A, Roy S, Lenoir M, Overduin M, Stahelin RV, Kutateladze TG. 2011. Molecular basis of phosphatidylinositol 4-phosphate and ARF1 GTPase recognition by the FAPP1 pleckstrin homology (PH) domain. J. Biol. Chem. 286:18650–18657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tóth B, Balla A, Ma H, Knight ZA, Shokat KM, Balla T. 2006. Phosphatidylinositol 4-kinase IIIβ regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J. Biol. Chem. 281:36369–36377 [DOI] [PubMed] [Google Scholar]
- 36.Balla A, Tuymetova G, Tsiomenko A, Balla T. 2005. A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol. Biol. Cell 16:1282–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garuti L, Roberti M, Bottegoni G. 2010. Non-ATP competitive protein kinase inhibitors. Curr. Med. Chem. 17:2804–2821 [DOI] [PubMed] [Google Scholar]
- 38.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408:297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Palma AM, Verbeken E, Van Aelst I, Van den Steen PE, Opdenakker G, Neyts J. 2008. Increased gelatinase B/matrix metalloproteinase 9 (MMP-9) activity in a murine model of acute coxsackievirus B4-induced pancreatitis. Virology 382:20–27 [DOI] [PubMed] [Google Scholar]
- 40.Sasaki J, Ishikawa K, Arita M, Taniguchi K. 2012. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 31:754–76622124328 [Google Scholar]
- 41.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Nat. Acad. Sci. U. S. A. 106:7577–7582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borawski J, Troke P, Puyang X, Gibaja V, Zhao S, Mickanin C, Leighton-Davies J, Wilson CJ, Myer V, Cornellataracido I, Baryza J, Tallarico J, Joberty G, Bantscheff M, Schirle M, Bouwmeester T, Mathy JE, Lin K, Compton T, Labow M, Wiedmann B, Gaither LA. 2009. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 83:10058–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet M-S, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Bühler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tai AW, Benita Y, Peng LF, Kim S-S, Sakamoto N, Xavier RJ, Chung RT. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trotard M, Lepère-Douard C, Régeard M, Piquet-Pellorce C, Lavillette D, Cosset F-L, Gripon P, Le Seyec J. 2009. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 23:3780–3789 [DOI] [PubMed] [Google Scholar]
- 46.Vaillancourt FH, Pilote L, Cartier M, Lippens J, Liuzzi M, Bethell RC, Cordingley MG, Kukolj G. 2009. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387:5–10 [DOI] [PubMed] [Google Scholar]
- 47.Tai AW, Salloum S. 2011. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS One 6(10):e26300. 10.1371/journal.pone.0026300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang N, Ma P, Lang J, Zhang Y, Deng J, Ju X, Zhang G, Jiang C. 2012. Phosphatidylinositol 4-kinase IIIβ is required for severe acute respiratory syndrome coronavirus spike-mediated cell entry. J. Biol. Chem. 287:8457–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaillancourt FH, Brault M, Pilote L, Uyttersprot N, Gaillard ET, Stoltz JH, Knight BL, Pantages L, McFarland M, Breitfelder S, Chiu TT, Mahrouche L, Faucher A-M, Cartier M, Cordingley MG, Bethell RC, Jiang H, White PW, Kukolj G. 2012. Evaluation of phosphatidylinositol-4-kinase IIIα as a hepatitis C virus drug target. J. Virol. 86:11595–11607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. 2006. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 125:733–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arita M, Wakita T, Shimizu H. 2008. Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J. Gen. Virol. 89:2518–2530 [DOI] [PubMed] [Google Scholar]
- 52.Phillpotts RJ, Jones RW, Delong DC, Reed SE, Wallace J, Tyrrell DA. 1981. The activity of enviroxime against rhinovirus infection in man. Lancet i:1342–1344 [DOI] [PubMed] [Google Scholar]
- 53.Phillpotts RJ, Wallace J, Tyrrell DA, Tagart VB. 1983. Therapeutic activity of enviroxime against rhinovirus infection in volunteers. Antimicrob. Agents Chemother. 23:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamarche MJ, Borawski J, Bose A, Capacci-Daniel C, Colvin R, Dennehy M, Ding J, Dobler M, Drumm J, Gaither LA, Gao J, Jiang X, Lin K, McKeever U, Puyang X, Raman P, Thohan S, Tommasi R, Wagner K, Xiong X, Zabawa T, Zhu S, Wiedmann B. 2012. Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob. Agents Chemother. 56:5149–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spickler C, Lippens J, Laberge M-K, Desmeules S, Bellavance E, Garneau M, Guo T, Hucke O, Leyssen P, Neyts J, Vaillancourt FH, Décor A, O'Meara J, Franti M, Gauthier A. 2013. Phosphatidylinositol 4-kinase III beta is essential for the replication of human rhinovirus and its inhibition causes a lethal phenotype in vivo. Antimicrob. Agents Chemother. 57:3358–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Susser S, Vermehren J, Forestier N, Welker MW, Grigorian N, Füller C, Perner D, Zeuzem S, Sarrazin C. 2011. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J. Clin. Virol. 52:321–327 [DOI] [PubMed] [Google Scholar]
- 57.Susser S, Welsch C, Wang Y, Zettler M, Domingues FS, Karey U, Hughes E, Ralston R, Tong X, Herrmann E, Zeuzem S, Sarrazin C. 2009. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 50:1709–1718 [DOI] [PubMed] [Google Scholar]
- 58.Geller R, Vignuzzi M, Andino R, Frydman J. 2007. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 21:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]