MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis (original) (raw)
Significance
The bacterial envelope is composed of a diverse set of polysaccharides. Virtually all of these polymers are synthesized from lipid-linked precursors that are flipped across the cytoplasmic membrane by ATP-binding cassette transporters or multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily members. Transport of the cell wall precursor lipid II requires the MOP family member MurJ in Escherichia coli. Here, we provide evidence for a novel lipid II flippase in Bacillus subtilis called alternate to MurJ (Amj), which bears no similarity to either family of transporters. amj is up-regulated in the absence of B. subtilis MurJ (MurJBs) via an envelope stress-response pathway, suggesting this novel flippase may serve as a defense mechanism against naturally occurring MurJ antagonists.
Keywords: peptidoglycan, flippase, MurJ, sigM, lipid II
Abstract
Bacterial surface polysaccharides are synthesized from lipid-linked precursors at the inner surface of the cytoplasmic membrane before being translocated across the bilayer for envelope assembly. Transport of the cell wall precursor lipid II in Escherichia coli requires the broadly conserved and essential multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily member MurJ. Here, we show that Bacillus subtilis cells lacking all 10 MOP superfamily members are viable with only minor morphological defects, arguing for the existence of an alternate lipid II flippase. To identify this factor, we screened for synthetic lethal partners of MOP family members using transposon sequencing. We discovered that an uncharacterized gene amj (alternate to MurJ; ydaH) and B. subtilis MurJ (murJ Bs; formerly ytgP) are a synthetic lethal pair. Cells defective for both Amj and MurJBs exhibit cell shape defects and lyse. Furthermore, expression of Amj or MurJBs in E. coli supports lipid II flipping and viability in the absence of E. coli MurJ. Amj is present in a subset of gram-negative and gram-positive bacteria and is the founding member of a novel family of flippases. Finally, we show that Amj is expressed under the control of the cell envelope stress-response transcription factor σM and cells lacking MurJBs increase amj transcription. These findings raise the possibility that antagonists of the canonical MurJ flippase trigger expression of an alternate translocase that can resist inhibition.
The bacterial cell wall or peptidoglycan (PG) is composed of glycan strands cross-linked together by short peptides. This 3D meshwork protects the cell from osmotic lysis and determines shape, and its assembly is the target of some of our most successful antibiotics. Cell wall synthesis begins in the cytoplasm, where a set of highly conserved enzymes catalyze the formation of the lipid-linked precursor lipid II, which is composed of undecaprenyl-pyrophosphate (UndPP) linked to N-acetylglucosamine-N-acetylmuramic acid pentapeptide. Lipid II is synthesized on the inner face of the cytoplasmic membrane (1). The molecule is then translocated to the outer face of the membrane, where the disaccharide-peptide monomer is incorporated into the existing PG by cell wall synthetic machineries composed of penicillin-binding proteins and additional factors (2). The enzymes that transport lipid II across the membrane have been the subject of extensive research and speculation (3–6). Recent work in Escherichia coli has provided strong evidence that the polytopic membrane protein MurJ is required for lipid II transport across the membrane, and is likely to be a lipid II flippase (6). MurJ is a member of the multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily (7). It is broadly conserved among Eubacteria and essential for viability in many organisms. Intriguingly, work in the model gram-positive bacterium Bacillus subtilis indicates that cells lacking four MOP superfamily members similar to MurJ are viable and have no detectable defect in cell wall synthesis (8, 9). These findings have been interpreted as evidence that MurJ may not be a lipid II flippase or that an additional transporter exists in this bacterium (5, 10).
Here, we show that deletion of all 10 MOP superfamily members in B. subtilis has no significant impact on growth or cell morphology. We identify a previously uncharacterized protein Amj (alternate to MurJ; formerly YdaH) that is a synthetic lethal partner with one of the B. subtilis MurJ homologs, YtgP (renamed MurJ). For the sake of clarity, we refer to B. subtilis MurJ as MurJBs and to E. coli MurJ as MurJEc throughout this paper. Depletion of Amj in the absence of MurJBs causes cell shape defects and, ultimately, lysis, which are phenotypes also exhibited upon depletion of the enzyme (MurG) required for the last step in the synthesis of lipid II. Importantly, expression of either Amj or MurJBs in E. coli supports lipid II flipping and viability in the absence of MurJEc. Amj bears no sequence similarity to MOP family exporters or ATP-binding cassette (ABC) transporters, and therefore represents the founding member of a new family of flippases. Interestingly, amj is transcribed under the control of the cell envelope stress-response sigma factor σΜ, and we show that its expression increases in the absence of MurJBs. These results raise the possibility that B. subtilis responds to antagonists of its canonical flippase by inducing the expression of an alternate lipid II transporter.
Results
B. subtilis Cells Lacking All 10 Members of the MOP Superfamily Are Viable.
The recent discovery that MurJEc is essential for lipid II transport across the inner membrane in E. coli (6) prompted us to revisit the role of MurJ family members in B. subtilis. Previous studies revealed that B. subtilis cells lacking four putative MurJ paralogs are viable and have no morphological defects (8, 9). We wondered whether other MOP family members might function in their absence. Homology searches using HHpred (11) identified six additional B. subtilis proteins in the MOP exporter superfamily (TuaB, DinF, YoeA, YisQ, NorM, and EpsK). Some of these factors have been implicated in the transport of other polysaccharide precursors (12). Because some flippases in this superfamily have relaxed substrate specificity (13), we investigated whether one or more of these proteins can support lipid II transport.
Using the B. subtilis knockout collection, we constructed in-frame deletions of the 10 genes encoding MOP family members by sequential excision of the antibiotic resistance cassette using a temperature-sensitive plasmid encoding Cre recombinase (14) (Fig. S1 A_–_C). In the course of constructing the ∆10 strain, we discovered that the deletion of tuaB exhibited slow growth and a severe cell shape defect (Fig. S2), which are phenotypes consistent with impaired cell wall synthesis. tuaB is the second gene in the tua operon, which is required for the biogenesis of the extracellular polymer teichuronic acid under phosphate-limiting conditions (12). TuaB has been hypothesized to flip the lipid-linked precursor of this uronate-containing polysaccharide (12). Interestingly, mutations in the putative transporter of the phosphate-containing polymer wall teichoic acid (WTA) are lethal in B. subtilis and Staphylococcus aureus but can be suppressed by mutations in the gene encoding the first enzyme in the biosynthetic pathway (15, 16). Accordingly, we tested whether the phenotypes of the tuaB deletion could be similarly suppressed. Here, we deleted the entire tua operon. Cells lacking the full operon, including tuaB, grew at rates similar to WT and had normal rod-shaped morphology (Fig. S2). These results indicate that the ∆tuaB phenotypes are unrelated to lipid II transport and are likely caused by the toxic accumulation of intracellular teichuronic acid precursors or the depletion of the lipid carrier undecaprenyl-phosphate (16). Thus, to generate the ∆10 MOP strain, we used the tua operon deletion.
Construction of the decuple mutant lacking all 10 MOP exporter family members was successful. Strikingly, the cells were not only viable but grew at rates indistinguishable from WT and had only modest cell shape defects (Fig. 1). The absence of all 10 genes was confirmed by PCR (Fig. S1_D_). Furthermore, PCR analysis demonstrated that this strain, containing nine identical loxP (lox72) scars and harboring the Cre recombinase nine separate times during its construction, had not undergone rearrangements and was chromosomally stable (Fig. S1_E_). Collectively, these findings confirm and extend previous studies (8, 9) that the putative MurJ paralogs are not essential for growth in B. subtilis.
Fig. 1.

B. subtilis cells lacking all 10 MOP superfamily members are viable. (A) Growth curve of a strain lacking all 10 (∆10) MOP exporter superfamily members in B. subtilis. WT (BDR11) and the ∆10 mutant (BAM485) were grown to exponential phase in casein hydrolysate (CH) medium and back-diluted to an OD600 of 0.05, and growth was monitored over time. (B) Cytological analysis of the ∆10 mutant. Exponentially growing WT (BDR2648) and the ∆10 mutant (BAM486) were stained with the lipophilic dye 1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene _p_-toluenesulfonate and examined by fluorescence microscopy. Both strains express mCherry to label the cytoplasm. (Scale bar: 1 μm.)
A Genetic Screen Identifies an Alternative Flippase.
We reasoned that an additional lipid II flippase unrelated to MurJ must exist in B. subtilis. If our reasoning is correct, then this alternative flippase should become essential in the absence of the MurJ paralogs. To identify this factor, we performed a synthetic lethal screen using transposon-sequencing (Tn-seq). Genomic DNA from a Mariner transposon library was transformed into WT B. subtilis and into a strain (referred to as ∆4) lacking the four MOP family members (YtgP, YabM, SpoVB, and YkvU) most closely related to MurJEc. The ∆4 strain was used instead of the ∆10 mutant in hopes of focusing the screen on the identification of factors with lipid II flippase activity. Approximately 500,000 colonies from each transformation were separately pooled, and the transposon–chromosome junctions were mapped by massively parallel DNA sequencing (Fig. 2). For each library, we detected insertions in 30% of the Mariner insertion sites (TA dinucleotides) in the genome, and each insertion site had an average of 55 reads. On average, each nonessential gene was disrupted at 15 unique sites. Transposon insertions in four genes (divIVA, minJ, sigM, and ydaH) were statistically (Mann–Whitney U test) underrepresented in the ∆4 library compared with the WT library.
Fig. 2.
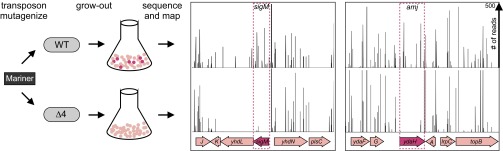
Synthetic lethal screen using transposon sequencing identifies amj. Mariner transposon libraries were generated in WT (BDR2413) and the ∆4 mutant (BAM490) lacking ytgP, yabM, spoVB, and ykvU. The sites of transposon insertion were identified by deep sequencing and mapped onto the B. subtilis 168 reference genome (National Center for Biotechnology Information RefSeq NC_000964.3). Two regions of the genome are depicted. The height of each line reflects the number of sequencing reads at this position. Transposon insertions in sigM and amj (ydaH) were significantly reduced in the ∆4 mutant compared with WT. No insertions were mapped to the essential gene plsC in either strain. Despite the essentiality of yhdL, which encodes an anti-σM factor, some insertions were detected in the WT background.
The conditionally essential genes identified by Tn-seq were confirmed by transforming a marked deletion of each into the ∆4 mutant and a WT control. Mutations in divIVA and minJ were synthetically sick in combination with the ∆4 mutant, forming smaller colonies compared with the transformants in the WT background, and were not characterized further. By contrast, transformations with ∆sigM::erm or ∆ydaH::erm chromosomal DNA into the ∆4 mutant yielded no transformants, whereas control transformations into WT produced >500 transformants. Thus, both sigM and ydaH appear to be conditionally essential in the absence of the four putative MurJ paralogs. SigM (σM) is an extracytoplasmic function sigma factor that activates the expression of many genes involved in cell wall synthesis in response to cell envelope stress (17–19). Intriguingly, the ydaH gene is among the most strongly induced genes expressed under σM control (18, 19), and it is predicted to encode a polytopic membrane protein with six transmembrane segments. YdaH bears no sequence similarity to MurJ or any other known flippase. Although not broadly conserved, ydaH is present in the genomes of a subset of gram-positive and gram-negative bacteria (Fig. S3), including Bordetella pertussis, Burkholdera mallei, and Clostridium botulinum. The phylum with the largest number of sequenced genomes containing YdaH orthologs is the Firmicutes. Based on experiments described below we have named this factor Amj.
amj and ytgP Are a Synthetic Lethal Pair.
To investigate which of the four putative MurJ paralogs were synthetically lethal with ∆amj and ∆sigM, we attempted to construct double mutants with the individual MOP mutants and either ∆amj or ∆sigM. The only double mutants that could not be generated were those double mutants that contained ∆ytgP. To more rigorously test the synthetic lethality of ∆ytgP and ∆amj, we constructed an isopropyl-β-d-thiogalactopyranoside (IPTG)–inducible allele of amj. Depletion of Amj in the ∆ytgP ∆amj double mutant led to loss of viability (Fig. 3_A_). Among all 10 MOP family members in B. subtilis, YtgP is the most similar to MurJEc. Furthermore, previous work (8) demonstrated that expression of ytgP could support growth in an E. coli strain depleted of MurJEc (see below). Based on these findings and the following findings, we propose renaming this gene _murJ_Bs.
Fig. 3.

MurJBs and Amj are synthetically lethal partners. (A) Spot dilutions of the indicated strains in the presence and absence of inducer. The three strains were grown in the presence of IPTG (500 μM) to an optical density of ∼1. The cultures were washed twice without inducer, resuspended at an OD600 of 1.25, and serially diluted. Five microliters of each dilution was spotted onto plates with and without inducer. (B) Cytological analysis of the terminal phenotype following Amj depletion in the absence of MurJBs. Depletion of the lipid II synthase MurG is shown for comparison. The indicated strains (BAM585 and BAM587) were grown to exponential phase in the presence of IPTG (500 μM), washed twice with media lacking inducer, back-diluted to an OD600 of 0.05 in CH medium, and grown to midexponential phase in the presence or absence of inducer. Cells were examined by fluorescence microscopy. Images of Amj depletion in the absence of MurJBs and MurG depletion are from 5 h and 4 h after removal of IPTG, respectively. Both strains contain mCherry, and an overlay of cytoplasmic fluorescence and phase contrast (merge) is shown. Lysis (yellow carets) and asymmetrical bulging (white carets) are highlighted. (Scale bar: 1 μm.)
Because amj is in the σM regulon, we suspected that σM-dependent expression of amj underlies the synthetic lethality of the ∆sigM ∆murJ_Bs double mutant. In support of this idea, expression of amj under IPTG control restored viability to the ∆_sigM, ∆_murJ_Bs mutant (Fig. S4).
Depletion of MurJBs and Amj Phenocopies Depletion of MurG.
As a first step toward investigating whether MurJBs and Amj constitute a pair of flippases, we sought to assess the terminal phenotype of cells depleted of Amj in the absence of MurJBs. For comparison, we constructed a strain in which we could deplete MurG, the glycosyltransferase required for conversion of lipid I into lipid II (20). Both depletion strains were inducer-dependent for growth and exhibited a reduction in optical density upon removal of IPTG (Fig. S5_A_). Examination of these strains during depletion by fluorescence microscopy revealed similar cell shape defects: The cells became asymmetrically swollen, generating aberrant morphologies (Fig. 3_B_). Consistent with the reduction in optical density, extensive cell lysis was observed upon further depletion (Fig. 3_B_ and Fig. S5 B and C). These results are consistent with the idea that Amj and MurJBs function in translocation of lipid II across the membrane.
Amj Can Substitute for E. coli MurJ.
As a direct test of the idea that Amj can support lipid II transport, we investigated whether expression of Amj in E. coli could suppress the essentiality of MurJEc. As a positive control for these experiments, we used MurJBs and SpoVB, which have previously been shown to complement a MurJEc depletion strain (8). In addition, we included the two other MOP family members (YabM and YkvU) used in our synthetic lethal screen. All five genes and gfp as a negative control were separately fused to the P_lac_ promoter on a low-copy plasmid and transformed into an E. coli strain harboring an arabinose-inducible promoter fusion to murJ Ec as the sole source of the gene. Strikingly, expression of Amj in the absence of arabinose supported growth in a manner similar to expression of MurJBs or SpoVB (Fig. 4_A_). By contrast, cells expressing YabM, YkvU, or GFP were inviable in the absence of arabinose, suggesting that these proteins are not involved in lipid II transport. Analysis of growth in liquid culture lacking arabinose revealed that E. coli cells expressing Amj or MurJBs grow at rates indistinguishable from WT (Fig. S6_A_) and support normal cell shape (Fig. 4_B_).
Fig. 4.

Amj can substitute for MurJEc in E. coli. (A) Spot dilutions of E. coli strains in which expression of murJ_Ec was under the control of the arabinose-inducible (PBAD) promoter. The indicated genes were present on a medium copy plasmid and expressed from the P_lac promoter. Strains were grown to stationary phase in the presence of 0.2% arabinose, washed twice without inducer, resuspended at an OD600 of 1.25, and serially diluted. Five microliters of each dilution was spotted in the presence of 0.2% arabinose (+ARA) or 25 μM IPTG (−ARA). (B) Amj expression suppresses the loss of viability following depletion of MurJEc. Strains CAM252 (MurJEc depletion + pGFP) and CAM249 (MurJEc depletion + pAmj) were grown to stationary phase in the presence of arabinose, washed twice in LB lacking inducer, and back-diluted to an OD600 of 0.01 in LB containing arabinose (+ARA) or IPTG (−ARA). The cultures were grown for seven generations and back-diluted to an OD600 of 0.1, maintaining arabinose or IPTG. Cells were examined by differential interference contrast microscopy at hour 1.5 after the second back-dilution. Examples of bulging (white carets) and lysis (yellow carets) are highlighted. (Scale bar: 1 μm.) (C) Cytological analysis of E. coli strains lacking MurJEc complemented with P_lac_-amj or P_lac_-_murJ_Bs compared with WT. Strains were grown in LB containing 1 mM IPTG. (Scale bar: 1 μm.)
To investigate whether Amj could complement a _murJ_Ec-null mutant, we constructed a ∆_murJ_Ec::kan insertion-deletion that contained a complementing allele at an ectopic locus. The marked deletion was then transduced into E. coli strains harboring plasmids with IPTG-inducible promoter fusions to amj, _murJ_Bs, or gfp. As expected for an essential gene, transduction of the null mutant into the GFP-expressing strain yielded no colonies. By contrast, transduction of _murJ_Ec::kan into the strains expressing Amj or MurJBs yielded hundreds of transductants (Fig. S6_B_). The presence of the _murJ_Ec deletion and the absence of the complementing _murJ_Ec allele were both confirmed by PCR (Fig. S6_C_). The complemented strains grew at rates similar to WT. Microscopic examination revealed that ∆_murJ_Ec::kan cells expressing Amj (but not MurJBs or MurJEc) were larger than WT and somewhat misshapen, although they retained a rod-like morphology (Fig. 4_C_). These phenotypes were more pronounced at lower IPTG concentrations in cells expressing either amj or _murJ_Bs (Fig. S6_D_), suggesting that expression levels were limiting.
Amj Supports Lipid II Flipping in E. coli.
To assess Amj-dependent lipid II flipping, we used a recently developed assay that monitors whether the lipid II precursor is present on the cytoplasmic face (unflipped) or the periplasmic face (flipped) of the inner membrane in E. coli (6). This assay takes advantage of the toxin colicin M, which cleaves periplasmic lipid II between the diphosphate and undecaprenyl moieties, generating a soluble product that can be detected by HPLC. Because colicin M cannot cross the inner membrane, unflipped lipid II is protected from cleavage. Previous studies established that in cells harboring MurJ, lipid II is accessible to colicin M and cleavage products are readily detected. However, upon inactivation of MurJ, the soluble colicin M products decrease and there is a concomitant accumulation of unflipped precursors. These data are consistent with the idea that MurJ translocates lipid II across the membrane. Using this assay, we analyzed the disposition of lipid II in E. coli cells expressing MurJBs and, separately, Amj (Fig. S7_A_). Cells expressing MurJEc and GFP were used as positive and negative controls, respectively. Similar to the previous study, all four strains harbored a functional MurJEc point mutant (A29C) that can be inactivated by the addition of the sulfhydryl-reactive compound (2-sulfonatoethyl)methanethiosulfonate (MTSES) (6). In the strain expressing GFP, addition of MTSES caused an immediate cessation of growth. However, expression of MurJEc, MurJBs, or Amj conferred resistance to MTSES (Fig. 5_A_). Cells were pulse-labeled with [3H]-_m-_DAP (American Radiolabeled Chemicals) to label PG precursors, and colicin M and/or MTSES was added 15 min later. The water-soluble colicin M products were identified and quantified by HPLC. Precursor levels in the lipid fraction were separately determined by organic extraction and scintillation counting. As expected based on previous results, in cells expressing GFP, the colicin M-dependent products were reduced upon inactivation of MurJEc(A29C) with MTSES (Fig. 5_B_ and Fig. S7_B_). By contrast, these cleavage products were largely unchanged in cells expressing Amj, MurJBs, or MurJEc. Furthermore, lipid-linked precursors only accumulated in the GFP-expressing strain upon MurJEc(A29C) inactivation (Fig. 5_C_). Taken together, these data indicate that both MurJBs and Amj can support lipid II translocation activity in E. coli.
Fig. 5.

Amj can support lipid II flippase activity in E. coli. (A) Growth curves of E. coli murJ(A29C) ∆lysA strains producing GFP, MurJBs, or Amj treated with MTSES. Cultures were grown in minimal media with 1 mM IPTG until midexponential phase, followed by addition of MTSES as indicated (arrows). Growth was monitored over time. (B) In vivo flippase assay. Cells were labeled with [3H]-_m_-DAP. After 15 min, colicin M and/or MTSES was added as indicated, and growth was continued for 10 min. Samples were then withdrawn and extracted sequentially with hot water and then butanol. Hot-water extracts were subjected to HPLC and radiodetection to quantify the radiolabeled colicin M product. Experimental details and peak identification are provided in Fig. S7. (C) Scintillation counting was used to quantify label in the lipid (butanol) fraction. Error bars represent SEM from two experiments. The P value determined with Student’s t test. *P < 0.05. ns, not significant.
Amj Expression Increases in the Absence of MurJBs.
amj is regulated by the stress-response sigma factor σM. σM is held inactive by the two integral membrane proteins YhdL and YhdK, and inhibition is relieved in response to an unknown signal that is thought to be generated by cell wall stress (21–23). Expression profiling experiments indicate that σM-controlled genes are transcribed at low levels under normal growth conditions and are induced two- to fivefold in response to acute exposure to cell wall synthesis inhibitors (19, 24). We wondered whether cells lacking MurJBs experience cell wall stress that is alleviated by σM-dependent expression of Amj. To investigate this possibility, we separately fused two σM-dependent promoters (P_sigM_ and P_amj_) to lacZ and yfp and monitored σM activity during growth. As reported previously (17), σM-dependent expression was detectable in WT cells (Fig. 6_A_) and an approximately threefold increase in expression was observed after 30 min of exposure to the cell wall synthesis inhibitor vancomycin (Fig. S8_A_). Consistent with the idea that cells lacking MurJBs experience cell wall stress, we found that transcription from both σM-dependent promoters increased approximately twofold in the absence of MurJBs (Fig. 6_A_ and Fig. S8_B_). We suspect that the increased level of Amj restores proper cell wall synthesis, which leads to negative feedback on σM activity. As Amj levels drop, σM is induced again. Thus, the twofold increase likely represents a new baseline activity for σM when cells lack MurJBs. Consistent with this idea, if we break the feedback loop by placing amj under inducible control and express it at low levels, we observed >10-fold higher expression from a σM-dependent promoter in cells lacking MurJBs (Fig. 6_B_). Although it is possible that this increase is a nonspecific effect of cell wall stress, we did not observe an increase in activity of the other six extracytoplasmic function sigma factors (25) under these conditions (Fig. S8_C_). Taken together, we conclude that cells respond to inhibition of MurJBs by inducing expression of amj.
Fig. 6.

Amj expression increases in the absence of MurJBs. (A) Bar graph showing β-gal activity of indicated strains harboring the σM-responsive promoter (P_amj_) fused to lacZ. Activity was assayed in exponentially growing cultures in LB. Indicated strains harboring an amj promoter fusion to yfp were analyzed by fluorescence microscopy. (Scale bar: 1 μm.) (B) β-Gal activity of P_amj_-lacZ in a strain lacking _murJ_Bs with amj under an IPTG-inducible control. Activity was assayed in exponentially growing cultures in LB containing either 500 μM IPTG (Amj High) or 50 μM IPTG (Amj Low). Error bars represent SEM from two experiments.
Discussion
Here, we have shown that B. subtilis cells lacking both MurJBs and Amj have a lethal defect in cell wall biogenesis. Moreover, both proteins can support lipid II transport and viability in E. coli cells lacking MurJEc. The simplest interpretation of the data, and one that we favor, is that both proteins are lipid II flippases. We cannot, however, rule out the possibility that both proteins are required to stimulate the activity of an unknown transporter. That Amj bears no similarity to MurJEc or any other protein encoded in the E. coli genome suggests it functions directly in lipid II transport. Additional support for the idea that MurJ homologs are flippases comes from their membership in the MOP exporter superfamily and mutagenesis studies in E. coli suggesting that MurJEc contains a solvent-exposed transmembrane channel (26). Collectively, our data lead us to propose that Amj is the founding member of a new family of lipid II flippases.
An additional conclusion that emerges from our study and previous work (6, 8) is that the MOP family member SpoVB required for synthesis of spore cortex PG (27) is a lipid II flippase. It is unclear why B. subtilis possesses a sporulation-specific flippase, but one possibility is that the spatial and temporal requirements for PG synthesis during development cannot be met by the vegetative enzymes.
Virtually all flippases implicated in translocating lipid-linked oligosaccharides across the cytoplasmic membrane fall into one of two families: ABC transporters or MOP exporters. For example, UndPP-linked homopolymeric O-antigen polysaccharides are flipped by the Wzt/Wzm ABC transporter, UndPP-linked WTA polymers require the putative ABC transporter TagGH, and UndPP-linked oligosaccharides involved in N- and O-linked protein glycosylation require distinct ABC transporters (28, 29). In a separate pathway, UndPP-linked oligosaccharide precursors are flipped by the MOP exporter Wzx and polymerized on the periplasmic face of the inner membrane to form heteropolymeric O-antigen polysaccharides (30, 31). Similarly, UndPP-linked oligosaccharide precursors required for synthesis of capsular polysaccharide, enterobacterial common antigen, and colanic acid all require distinct Wzx paralogs (32). In gram-positive bacteria, teichuronic acid precursors (UndPP-linked trisaccharides) require the MOP exporter family member TuaB (12). Finally, as discussed here, the PG precursor lipid II (UndPP-disaccharide pentapeptide) is likely flipped by the MOP family member MurJ. Amj shares no sequence similarity with either the ABC or MOP superfamilies, and therefore likely represents a new family of lipid-linked oligosaccharide transporters. Insofar as Amj does not appear to contain an ATPase domain, it more closely resembles MOP exporters. Amj is approximately half the size of MOP family members and is predicted to contain six transmembrane segments, whereas MOP family members contain 12–15 transmembrane segments. Thus, we anticipate that Amj oligomerizes to assemble a conduit for lipid II transport. It has been proposed that MurJ forms a C-shaped portal that allows transport of the hydrophilic diphosphate-disaccharide-pentapeptide portion of lipid II while accommodating its hydrophobic polyisoprenoid moiety within the lipid bilayer (26). Whether MurJ uses energy-coupled transport or facilitated diffusion in translocating lipid II remains an open question. Structural analysis and in vitro reconstitution of Amj and MurJ will be required to assess whether or not they use similar mechanisms to transport PG precursors.
Recent in vitro reconstitution experiments have led to the suggestion that FtsW and other shape, elongation, division, and sporulation (SEDS) family proteins could function as lipid II flippases (5). These polytopic membrane proteins are often found in complex with a cognate penicillin-binding protein (33) involved in cell wall synthesis, providing a mechanism to couple transport of precursors with their incorporation into the cell wall. That MurJ paralogs were dispensable for rod shape and viability in B. subtilis left open the possibility that SEDS proteins could function as alternate flippases (8–10). However, our discovery that MurJ and Amj are a synthetic lethal pair argues against this idea. Based on the data reported here and in previous work (6), we favor the idea that MurJ and Amj are the lipid II flippases in vivo. Because E. coli lacks an Amj homolog, it is unlikely that Amj interacts with the endogenous E. coli cell wall synthetic machineries. Accordingly, if Amj is indeed a lipid II flippase, its ability to suppress the lethality of an E. coli strain lacking MurJEc argues that coupling transport of precursors to their incorporation into PG is not required for cell wall synthesis. Moreover, MurJBs (which bears only 19% identity to MurJEc) was able to restore proper cell shape fully to the ∆_murJ_Ec mutant. Thus, our data suggest that the proposed “hand-off” mechanism in which lipid II transport is coupled to cell wall synthesis plays only a minor role or may not exist at all.
Why would B. subtilis encode two distinct enzymes to catalyze the same reaction? One possibility is that, like LPS and capsular polysaccharide biosynthesis, Amj and MurJBs flip different lipid-linked precursors. We find this possibility unlikely because both function in E. coli and generate similar colicin M cleavage profiles. An alternative possibility suggested by the expression of Amj under σM control and the increase in σM activity in the absence of MurJBs is that antagonists of the canonical and constitutive (24) lipid II flippase (MurJ) are produced by other soil-dwelling organisms. In this model, an encounter with these inhibitors triggers σM activation and production of an alternate flippase that is immune to these antibiotics.
The signal that triggers σM activation is currently unknown. However, recent work has suggested that available undecaprenyl-phosphate might serve as the signal that is sensed by the anti-σM factors YhdK/L to maintain inhibition of σM (22, 23). Our results are consistent with this idea. However, an alternative possibility that emerges from our work is that flipped lipid II is the species monitored in the σM pathway. This idea is also supported by the observation that the sensing domain of the anti-sigma factor YhdL resides on the extracytoplasmic face of the membrane. In this model, lipid II interacts with the sensing domain of YhdL and maintains the anti-σM factor complex in an inhibitory conformation or protected from proteolysis. Depletion of flipped lipid II would trigger relief of this inhibition, activating the σM envelope stress pathway. Future experiments will be required to establish whether or not the availability of flipped lipid II serves as a signal for this pathway.
Experimental Procedures
B. subtilis strains were derived from B. subtilis 168 (34) or PY79 (35). Unless otherwise indicated, cells were grown in Luria-Bertani (LB) or casein hydrolysate (CH) medium at 37 °C. E. coli strains were derived from MG1655. Plasmids were propagated in DH5α or TG1 backgrounds. β-Gal assays were performed as described previously (36). Insertion-deletion mutants come from the B. subtilis knockout collection or were generated by isothermal assembly (37) of PCR products, followed by direct transformation into B. subtilis. Antibiotic cassette removal was performed using a temperature-sensitive plasmid encoding Cre recombinase and is described in Fig. S1. Tn-seq was performed as described previously (38, 39). Lipid II flippase activity in E. coli was measured as described previously (6). Detailed protocols; tables of strains, plasmids, and oligonucleotide primers; and a description of strain and plasmid construction are provided in SI Experimental Procedures and Table S1.
Supplementary Material
Supplementary File
Acknowledgments
We thank members of the D.Z.R. and T.G.B. laboratories for advice and encouragement; Alan Grossman and Chris Johnson for sharing their transposon library; Craig Ellermeier and Andrew Camilli for plasmids; Max Schubert for plasmid constructions; Andrew Fenton, Yannick Brunet, and Xindan Wang for advice with Tn-seq; Nick Peters for help with imaging; and Andrew Kruse for stimulating discussion. Support for this work comes from NIH Grants GM086466 (to D.Z.R.), RC2 GM092616 (to D.Z.R. and C.A.G.), AI083365-01 and AI099144 (to T.G.B.), GM102790 (to C.A.G.), and the Center of Excellence for Translational Research (U19 AI109764) (to T.G.B. and D.Z.R.). L.-T.S. was funded, in part, by the American Heart Association (Grant 14POST18480014).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Bupp K, van Heijenoort J. The final step of peptidoglycan subunit assembly in Escherichia coli occurs in the cytoplasm. J Bacteriol. 1993;175(6):1841–1843. doi: 10.1128/jb.175.6.1841-1843.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Heijenoort J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol Mol Biol Rev. 2007;71(4):620–635. doi: 10.1128/MMBR.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehlert K, Höltje JV. Role of precursor translocation in coordination of murein and phospholipid synthesis in Escherichia coli. J Bacteriol. 1996;178(23):6766–6771. doi: 10.1128/jb.178.23.6766-6771.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc Natl Acad Sci USA. 2008;105(40):15553–15557. doi: 10.1073/pnas.0808352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohammadi T, et al. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011;30(8):1425–1432. doi: 10.1038/emboj.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sham LT, et al. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345(6193):220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hvorup RN, et al. The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur J Biochem. 2003;270(5):799–813. doi: 10.1046/j.1432-1033.2003.03418.x. [DOI] [PubMed] [Google Scholar]
- 8.Fay A, Dworkin J. Bacillus subtilis homologs of MviN (MurJ), the putative Escherichia coli lipid II flippase, are not essential for growth. J Bacteriol. 2009;191(19):6020–6028. doi: 10.1128/JB.00605-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasudevan P, McElligott J, Attkisson C, Betteken M, Popham DL. Homologues of the Bacillus subtilis SpoVB protein are involved in cell wall metabolism. J Bacteriol. 2009;191(19):6012–6019. doi: 10.1128/JB.00604-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young KD. Microbiology. A flipping cell wall ferry. Science. 2014;345(6193):139–140. doi: 10.1126/science.1256585. [DOI] [PubMed] [Google Scholar]
- 11.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21(7):951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 12.Soldo B, Lazarevic V, Pagni M, Karamata D. Teichuronic acid operon of Bacillus subtilis 168. Mol Microbiol. 1999;31(3):795–805. doi: 10.1046/j.1365-2958.1999.01218.x. [DOI] [PubMed] [Google Scholar]
- 13.Alaimo C, et al. Two distinct but interchangeable mechanisms for flipping of lipid-linked oligosaccharides. EMBO J. 2006;25(5):967–976. doi: 10.1038/sj.emboj.7601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan X, Yu HJ, Hong Q, Li S-PP. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl Environ Microbiol. 2008;74(17):5556–5562. doi: 10.1128/AEM.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Elia MA, Millar KE, Beveridge TJ, Brown ED. Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J Bacteriol. 2006;188(23):8313–8316. doi: 10.1128/JB.01336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Elia MA, et al. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J Bacteriol. 2006;188(12):4183–4189. doi: 10.1128/JB.00197-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thackray PD, Moir A. SigM, an extracytoplasmic function sigma factor of Bacillus subtilis, is activated in response to cell wall antibiotics, ethanol, heat, acid, and superoxide stress. J Bacteriol. 2003;185(12):3491–3498. doi: 10.1128/JB.185.12.3491-3498.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eiamphungporn W, Helmann JD. The Bacillus subtilis sigma(M) regulon and its contribution to cell envelope stress responses. Mol Microbiol. 2008;67(4):830–848. doi: 10.1111/j.1365-2958.2007.06090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jervis AJ, Thackray PD, Houston CW, Horsburgh MJ, Moir A. SigM-responsive genes of Bacillus subtilis and their promoters. J Bacteriol. 2007;189(12):4534–4538. doi: 10.1128/JB.00130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mengin-Lecreulx D, Texier L, Rousseau M, van Heijenoort J. The murG gene of Escherichia coli codes for the UDP-N-acetylglucosamine: N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J Bacteriol. 1991;173(15):4625–4636. doi: 10.1128/jb.173.15.4625-4636.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshimura M, Asai K, Sadaie Y, Yoshikawa H. Interaction of Bacillus subtilis extracytoplasmic function (ECF) sigma factors with the N-terminal regions of their potential anti-sigma factors. Microbiology. 2004;150(Pt 3):591–599. doi: 10.1099/mic.0.26712-0. [DOI] [PubMed] [Google Scholar]
- 22.Inoue H, Suzuki D, Asai K. A putative bactoprenol glycosyltransferase, CsbB, in Bacillus subtilis activates SigM in the absence of co-transcribed YfhO. Biochem Biophys Res Commun. 2013;436(1):6–11. doi: 10.1016/j.bbrc.2013.04.064. [DOI] [PubMed] [Google Scholar]
- 23.Lee YH, Helmann JD. 2013. Reducing the Level of Undecaprenyl Pyrophosphate Synthase Has Complex Effects on Susceptibility to Cell Wall Antibiotics. Antimicrob Agents Chemother 57(9):4267–4275.
- 24.Nicolas P, et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335(6072):1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- 25.Luo Y, Asai K, Sadaie Y, Helmann JD. Transcriptomic and phenotypic characterization of a Bacillus subtilis strain without extracytoplasmic function σ factors. J Bacteriol. 2010;192(21):5736–5745. doi: 10.1128/JB.00826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler EK, Davis RM, Bari V, Nicholson PA, Ruiz N. Structure-function analysis of MurJ reveals a solvent-exposed cavity containing residues essential for peptidoglycan biogenesis in Escherichia coli. J Bacteriol. 2013;195(20):4639–4649. doi: 10.1128/JB.00731-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popham DL, Stragier P. Cloning, characterization, and expression of the spoVB gene of Bacillus subtilis. J Bacteriol. 1991;173(24):7942–7949. doi: 10.1128/jb.173.24.7942-7949.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hug I, Feldman MF. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology. 2011;21(2):138–151. doi: 10.1093/glycob/cwq148. [DOI] [PubMed] [Google Scholar]
- 29.Lazarevic V, Karamata D. The tagGH operon of Bacillus subtilis 168 encodes a two-component ABC transporter involved in the metabolism of two wall teichoic acids. Mol Microbiol. 1995;16(2):345–355. doi: 10.1111/j.1365-2958.1995.tb02306.x. [DOI] [PubMed] [Google Scholar]
- 30.Liu D, Cole RA, Reeves PR. An O-antigen processing function for Wzx (RfbX): A promising candidate for O-unit flippase. J Bacteriol. 1996;178(7):2102–2107. doi: 10.1128/jb.178.7.2102-2107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldman MF, et al. The activity of a putative polyisoprenol-linked sugar translocase (Wzx) involved in Escherichia coli O antigen assembly is independent of the chemical structure of the O repeat. J Biol Chem. 1999;274(49):35129–35138. doi: 10.1074/jbc.274.49.35129. [DOI] [PubMed] [Google Scholar]
- 32.Whitfield C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem. 2006;75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 33.Mercer KL, Weiss DS. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J Bacteriol. 2002;184(4):904–912. doi: 10.1128/jb.184.4.904-912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeigler DR, et al. The origins of 168, W23, and other Bacillus subtilis legacy strains. J Bacteriol. 2008;190(21):6983–6995. doi: 10.1128/JB.00722-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Youngman PJ, Perkins JB, Losick R. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc Natl Acad Sci USA. 1983;80(8):2305–2309. doi: 10.1073/pnas.80.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudner DZ, Fawcett P, Losick R. A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc Natl Acad Sci USA. 1999;96(26):14765–14770. doi: 10.1073/pnas.96.26.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 38.van Opijnen T, Bodi KL, Camilli A. Tn-seq: High-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6(10):767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson CM, Grossman AD. Identification of host genes that affect acquisition of an integrative and conjugative element in Bacillus subtilis. Mol Microbiol. 2014;93(6):1284–1301. doi: 10.1111/mmi.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File