Subcellular localization and RNAs determine FUS architecture in different cellular compartments (original) (raw)
Abstract
Mutations in Fused in sarcoma (FUS) gene cause a subset of familial amyotrophic lateral sclerosis (ALS), a fatal motor neuron degenerative disease. Wild-type FUS is largely localized in the nucleus, but mutant FUS accumulates in the cytoplasm and forms inclusions. It is unclear whether FUS depletion from the nucleus or FUS inclusions in the cytoplasm triggers motor neuron degeneration. In this study, we revealed that the nuclear and cytoplasmic FUS proteins form distinct local distribution patterns. The nuclear FUS forms oligomers and appears granular under confocal microscope. In contrast, the cytoplasmic FUS forms inclusions with no oligomers detected. These patterns are determined by the subcellular localization of FUS, regardless of wild-type or mutant protein. Moreover, mutant FUS remained or re-directed in the nucleus can oligomerize and behave similarly to the wild-type FUS protein. We further found that nuclear RNAs are critical to its oligomerization. Interestingly, the formation of cytoplasmic FUS inclusions is also dependent on RNA binding. Since the ALS mutations disrupt the nuclear localization sequence, mutant FUS is likely retained in the cytoplasm after translation and interacts with cytoplasmic RNAs. We therefore propose that local RNA molecules interacting with the FUS protein in different subcellular compartments play a fundamental role in determining FUS protein architecture and function.
Introduction
Fused in sarcoma (FUS) is a DNA/RNA-binding protein. Mutations in FUS cause a subset of familial amyotrophic lateral sclerosis (ALS) (1,2). ALS is a progressive neurodegenerative disease characterized by the loss of motor neurons (3).The ALS mutations result in the mislocalization of FUS from the nucleus to the cytoplasm and the formation of FUS-containing inclusions in the cytoplasm (1,2). It is unknown how the mislocalization of mutant FUS causes motor neuron dysfunction and degeneration. Cytoplasmic FUS inclusions are immune-positive of stress granule markers (4,5), a temporary cellular structure important for cell survival under a variety of stresses (6). The endogenous FUS has also been reported to participate in stress granule formation under various stresses (7,8). Mutant FUS has been shown to alter stress response dynamics, which is hypothesized to cause motor neuron degeneration (9–12). While proteinaceous inclusions involved in neurodegenerative diseases are often composed of amyloidal-aggregated misfolded proteins, FUS inclusions are structurally different (13,14). Specifically, FUS inclusions in the brain of frontal temporal dementia (FTD) patients were negative of thioflavin-S staining that specially stains cross-β-sheet structures in amyloidal aggregates (13). In addition, SDS-resistant oligomers were not detected in cells expressing various FUS mutants (14). The architecture of FUS protein in the cytoplasmic inclusions remains to be determined.
We previously reported that a sub-population of nuclear FUS is associated with active chromatin and participates in gene transcription regulation (15–17). The ALS mutations reduce FUS chromatin binding, disrupting FUS function in regulating gene transcription. We found that the chromatin-bound (CB) FUS was oligomerized and showed a granular pattern inside the nucleus. FUS oligomerization is mediated by the N-terminal QGSY-rich region (glutamine, glycine, serine and tyrosine-rich region) and requires RNA binding (15). The QGSY-rich region is part of a predicted prion-like domain (18) that is required for FUS aggregation in vitro (19). Therefore, we examined whether the QGSY-rich region-mediated oligomerization is also the structural basis of FUS cytoplasmic inclusions. The results will help us understand the nature of FUS cytoplamic inclusions. In addition, better understanding of the architectures of FUS proteins in the nuclear and cytoplasmic compartments will help determine whether redirecting cytoplasmic FUS mutants into the nucleus would be a potential therapeutic intervention for ALS as proposed in recent studies (20–24). Moreover, the requirement of RNA binding in FUS binding to chromatin raised the question whether RNAs directly mediate FUS protein organization in the nuclear granules and cytoplasmic inclusions.
In this study, we revealed that, in contrast to nuclear granules of FUS, the formation of FUS cytoplasmic inclusions does not require the QGSY-rich region-mediated oligomerization. By examining wild-type and mutant FUS proteins in different cellular compartments, we demonstrated that the subcellular localization plays an important role in determining the FUS protein architecture. We also discovered that chromatin-associated RNAs are critical to initiate the oligomerization of nuclear FUS. Moreover, the RNA binding ability was also required for FUS cytoplasmic inclusion formation. These results collectively suggest that subcellular localization and local RNA species are the determining factors for FUS distinct architectures and distribution patterns in different cellular compartments.
Results
FUS proteins are organized differently in nuclear granules and cytoplasmic inclusions
We have recently reported that the QGSY-rich region-mediated FUS oligomerization is essential for the formation of nuclear granules of FUS (15). When the QGSY-rich region (amino acids 1–164, Fig. 1A) was deleted, the granular distribution pattern disappeared and FUS molecules diffused in the entire nucleus (Fig. 1B). We examined whether the QGSY-rich region-mediated oligomerization was also the structural basis of cytoplasmic inclusions of mutant FUS. FUS R495X [arginine 495 to stop codon, a deletion mutant causing aggressive ALS (25,26)] formed cytoplasmic inclusions that were co-localized with the stress granule maker G3BP1 (Fig. 1C). We deleted the QGSY-rich region from FUS R495X and found that the truncation protein still formed cytoplasmic inclusions co-localized with G3BP1 (Fig. 1C). The results suggest that the QGSY-rich region is not required for the inclusion formation of ALS mutant FUS in the cytoplasm. The above results provide the initial evidence that FUS proteins in the nuclear granules and cytoplasmic inclusions are organized differently.
Figure 1.
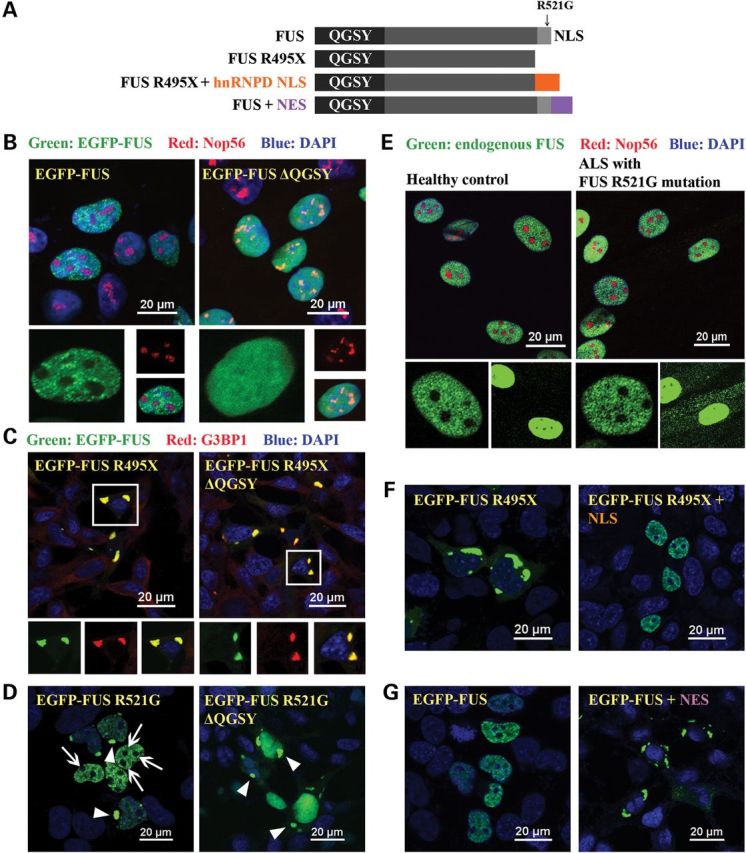
Subcellular localization is the determining factor of FUS distribution patterns. (A) Domain structure of FUS and various FUS constructs used in this study. (B) Full-length wild-type FUS was expressed in HEK cells and formed nuclear granules and was excluded from nucleoli in the nucleus (the left panel). These two features were lost in the truncated FUS ΔQGSY that lacks the QGSY-rich region (the right panel). Nop56 (red) is a marker for nucleoli. The green channel is shown in larger thumbnails and the red and merged images of the same area are shown in smaller thumbnails. (C) The R495X mutant FUS formed cytoplasmic inclusions that are co-localized with the stress granule marker G3BP1 (the left panel). Deleting the QGSY-rich region from R495X did not change the formation of G3BP1-positive cytoplasmic inclusions (the right panel). (D) R521G mutant FUS was expressed in HEK cells and formed both nuclear granules excluded from nucleoli and cytoplasmic inclusions (the left panel). Arrows: nucleoli. Arrow heads: cytoplasmic EGFP-FUS R521G inclusions. Deleting the QGSY-rich region did not change the cytoplasmic inclusions, but changed the distribution of R521G FUS inside the nucleus. The granular pattern disappeared and FUS was evenly distributed in the entire nucleus. (E) Immunostaining of FUS in fibroblast cells derived from familial ALS patients carrying the R521G FUS mutation and healthy controls. Thumbnails are zoomed-in images of two different areas. The solid frame area is zoomed in to show that both wild-type FUS in healthy control cells and R521G mutant FUS in familial ALS patient cells displayed a granular pattern excluded from nucleoli in the nucleus of respective cells (left thumbnails). The dashed frame area is zoomed in with enhanced brightness to show the cytoplasmic retention of R521G mutant FUS in the patient fibroblast cells compared with wild-type FUS in healthy control cells (right thumbnails). Green: FUS; Red: Nop56. (F) FUS R495X formed inclusions in the cytoplasm (the left panel). Tagging FUS R495X with a NLS targeted the chimeric protein in the nucleus (the right panel). The granular pattern of the chimeric protein in the nucleus was similar to wild-type FUS. (G) Tagging the wild-type FUS (the left panel) with a NES caused the cytoplasmic localization of the chimeric protein (the right panel). The wild-type FUS targeted to the cytoplasm also formed inclusions in a similar fashion to mutant FUS in (C) and (D). All images were taken with a Nikon A1 confocal microscope. Green: EGFP-tagged wild-type or mutant FUS. Blue: DAPI staining of DNA.
Subcellular compartment determines distinct local distribution patterns of FUS
The above results also raised an interesting question: what causes different local distribution patterns of FUS protein in different cellular compartments? Two factors can potentially contribute to it: subcellular compartment (nucleus versus cytoplasm) and protein mutation (wild-type versus ALS mutations). We rationalized that simultaneous examination of the local distribution pattern of nuclear and cytoplasmic sub-populations of mutant FUS protein can help delineating which factor plays a more important role. We took advantage of ALS point mutations (e.g. R521G) that cause partial retention of FUS protein in the cytoplasm while maintaining a significant portion in the nucleus. FUS R521G exhibited granular distribution and was excluded from nucleoli (arrows in Fig. 1D), which is very similar to wild-type FUS in the nucleus (compare with Fig. 1B). This pattern was also observed in the fibroblast cells derived from familial ALS patient carrying the R521G mutant FUS (Fig. 1E). The cytoplasmic retained FUS R521G mutant formed inclusions (Fig. 1D, left image, arrow heads). We deleted the QGSY-rich region of FUS R521G and examined how it changed the distribution of FUS protein in the nucleus and cytoplasm. The granular pattern of FUS R521G in the nucleus disappeared and FUS R521G was evenly distributed in the entire nucleus after the QGSY-rich region was deleted (Fig. 1D, right). The changes of FUS R521G in the nucleus were similar to the effect of QGSY-rich region deletion on wild-type FUS (Fig. 1B, right). In contrast, cytoplasmic inclusions of FUS R521G were still observed upon the deletion of the QGSY-rich region (Fig. 1D, right, arrow heads), which is similar to the effect of QGSY-rich region deletion on FUS R495X (Fig. 1C, right). These observations suggest that the subcellular compartment (i.e. nuclear or cytoplasmic) plays an important role in determining the local distribution pattern of FUS.
To further test this notion, we generated two chimeric proteins: FUS R495X fused with a nuclear localization sequence (NLS) from hnRNPD and wild-type FUS tagged with a typical nuclear export sequence (NES) (Fig. 1A). The chimeric protein R495X + hnRNPD NLS was largely localized inside the nucleus and showed a punctate pattern with nucleolar exclusion (Fig. 1F), resembling wild-type FUS (compare with Fig. 1B). Tagging the NES to wild-type FUS brought the protein outside the nucleus and the chimeric protein formed inclusions in the cytoplasm (Fig. 1G), which is similar to R495X (compare with Fig. 1C). These results solidify the notion that the subcellular compartment is the critical factor determining the local distribution pattern of FUS.
The ALS mutant FUS protein in the nucleus responds to transcription inhibition in a similar fashion to wild-type FUS
The above results suggest that ALS mutations have little effect on the local distribution pattern of the FUS protein localized in the nucleus. To further test this notion, we treated cells with transcription inhibitor 5,6-dichloro-1-(β-d-ribofuranosyl)-1H-benzimidazole (DRB) and examined how wild-type and mutant FUS proteins responded to this stress. The wild-type FUS protein accumulated into areas close to nucleoli upon DRB treatment (Fig. 2A), consistent with previous reports (27,28). Similarly, FUS R521G and FUS R495X (Fig. 2B) mutant proteins also accumulated next to nucleoli upon DRB treatment. The result again suggests that the nuclear localized mutant FUS responds to transcription inhibition similarly to wild-type FUS.
Figure 2.
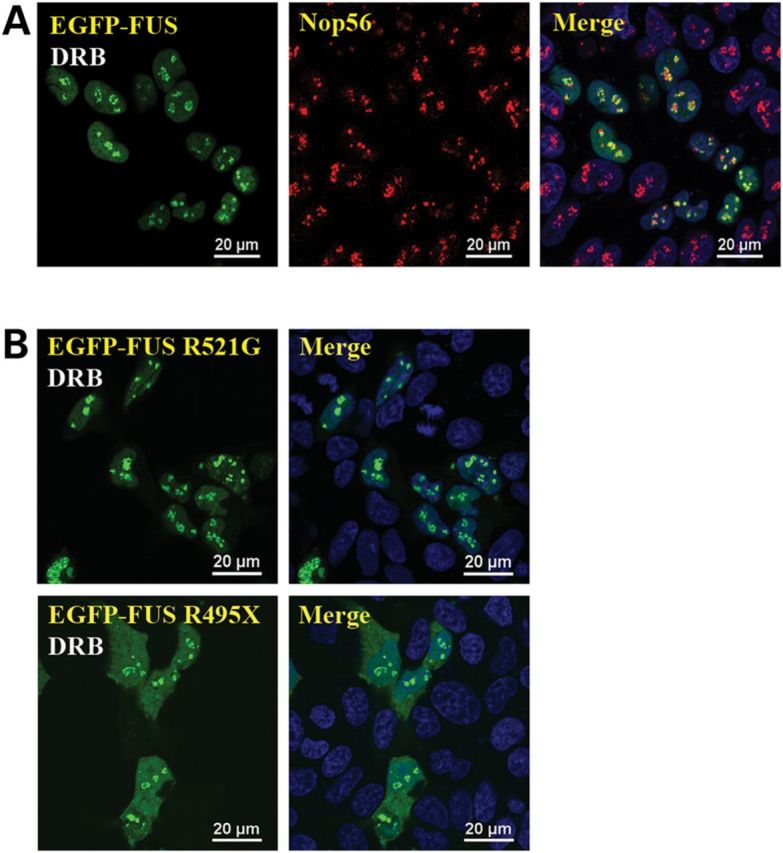
FUS mutants responded to transcription inhibition similarly to wild-type FUS. (A) Wild-type FUS accumulated in areas close to nucleoli in the presence of transcription inhibitor DRB. Cells expressing FUS were treated with 25 μm DRB for 2 h followed by formaldehyde fixation and Nop56 immunostaining. (B) FUS mutants R521G and R495X also accumulated in the peri-nucleolus areas in the presence of DRB. Cells expressing R521G or R495X FUS were treated with 25 μm DRB for 2 h before formaldehyde fixation. All images were taken with a Nikon A1 confocal microscope. Green: EGFP-tagged FUS. Red: Nop56. Blue: DAPI staining of DNA. The distribution of FUS in the absence of DRB was the same as those shown in Figure 1.
Chromatin-associated mutant FUS oligomerizes similarly to wild-type FUS
Our recently published results suggest that there are two pools of FUS inside the nucleus (CB and nuclear soluble) and the granular distribution is attributed to the CB FUS (15). In this study, we used two biochemical approaches, native gel electrophoresis and chemical crosslinking, to examine the nuclear granules and the cytoplasmic inclusions observed in Figure 1. The CB and non-chromatin-bound (NCB) fractions were prepared from lysates of cells expressing wild-type, R521G or R495X FUS as previously described (15) (Fig. 3A). The CB FUS represents the portion of FUS producing nuclear granules, and the NCB FUS includes the cytoplasmic FUS that can form inclusions. The CB wild-type FUS migrated slower than the NCB wild-type FUS (Fig. 3B), which we previously interpreted that the CB FUS was oligomeric, whereas the NCB was largely monomeric (15). Interestingly, R521G and R495X mutants in both CB and NCB fractions migrated similarly to wild-type FUS in the corresponding fraction (Fig. 3B). The results supported that the mutant FUS proteins behaved similarly to wild-type FUS, that is mutant FUS also existed as oligomeric form when bound to chromatin and as monomeric when in cytoplasm. Although mutant FUS showed reduced ability of binding to chromatin (15), the CB mutant FUS protein formed oligomers as wild-type FUS. The lack of oligomeric mutant FUS in the cytoplasmic fraction suggests that the cytoplasmic FUS protein does not need to oligomerize in the process of forming inclusions, which is consistent with the earlier results that the QGSY-rich region is not required for cytoplasmic inclusion formation (Fig. 1B and C, right).
Figure 3.

FUS mutants oligomerized similarly to wild-type FUS. (A) Flow chart of preparing CB and NCB samples, see details in Methods. (B) R521G and R495X mutant FUS proteins migrated similarly to wild-type FUS in native gel in both CB and NCB fractions. In the CB fraction, both mutant and wild-type FUS migrated as oligomers. In the NCB fraction, both mutant and wild-type FUS migrated as monomers. (C) The CB and NCB samples were subjected to crosslinking by 1% formaldehyde at room temperature for 10 min. SDS/PAGE analysis showed crosslinked oligomers for both wild-type and mutant FUS in the CB fraction.
We performed chemical crosslinking to further examine the oligomeric status of FUS proteins. The CB wild-type and mutant FUS proteins all formed oligomeric species as evidenced by the crosslinked signals (Fig. 3C). In contrast, no crosslinked species were detected for either wild-type or mutant FUS proteins in the NCB fraction. The above results consistently support that, regardless wild-type or ALS mutants, the CB FUS protein oligomerizes, whereas the cytoplasmic FUS protein does not.
Chromatin content initiates oligomerization of wild-type and mutant FUS
We next examined what triggers FUS oligomerization. Since the NCB FUS did not oligomerize no matter how long it was incubated in the cell lysate deprived of chromatin content (data not shown), we hypothesized that certain constituents in the CB fraction initiated FUS oligomerization. We incubated the CB fraction with the NCB fraction and analyzed them with native gel electrophoresis. With increasing amounts of chromatin content in the mixture, the wild-type FUS protein shifted from the monomeric position towards the oligomeric position (Fig. 4A). The result demonstrated that the chromatin content was able to initiate oligomerization of NCB FUS. Interestingly, a single band was observed in each sample with a position between the monomeric and oligomeric bands (Fig. 4A), suggesting that FUS protein reached a new equilibrium during incubation. Similarly, the chromatin content initiated oligomerization of mutant FUS in the NCB fraction as well (Fig. 4B).
Figure 4.

The CB fraction induced oligomerization of FUS in the NCB fraction. (A) Incubation of the CB and NCB fractions induced oligomerization of endogenous wild-type FUS in the NCB fraction. Different amounts of CB samples were incubated with the NCB samples on ice for 20 min. The mixtures were subjected to native gel electrophoresis followed by western blot with a FUS antibody. The individual CB and NCB fractions were shown as control. The slower mobility of FUS suggested a high order assembly of FUS. (B) The R495X mutant FUS also oligomerized when incubated with the CB fraction. The experiment was carried out as in (A) except that R495X mutant was tested in (B).
Chromatin-associated RNAs are responsible for initiating FUS oligomerization
The chromatin fraction contains DNAs, RNAs and proteins; thus, we asked what triggers FUS oligomerization. Since FUS can bind nucleic acids, we specifically tested whether DNA or RNA triggered FUS oligomerization. We incubated RNase-free DNase or RNase A along with the CB and NCB fractions. The RNase treatment abolished mutant FUS oligomerization, whereas the DNase treatment had no effect on mutant FUS oligomerization (Fig. 5A). The RNase treatment did not change DNA content in the CB fraction, whereas the DNase treatment digested DNAs (Fig. 5B). The result suggests that RNA molecules associated with chromatin are responsible for initiating FUS oligomerization. To confirm this notion, we extracted nuclear RNAs with TRIzol reagent and incubated them with chromatin-deprived cell extract. The TRIzol-extracted nuclear RNAs indeed triggered mutant FUS oligomerization in a dose-dependent manner (Fig. 5C). As a control, RNAs extracted from the cytoplasmic fraction induced little oligomerization of FUS. The results support the notion that the nuclear RNAs associated with chromatin are responsible for initiating oligomerization of FUS protein.
Figure 5.

Nuclear RNAs triggered FUS oligomerization. (A) Addition of RNase A in the mixture of CB and NCB fractions inhibited the oligomerization of FUS. In contrast, DNase did not have any effect on FUS oligomerization. Fifty units per milliliter RNase-free DNase or 1 ng/ml RNase A was added into mixture of the NCB sample isolated from cells expressing R495X FUS and the CB fraction (1:1 ratio). After 1 h incubation at room temperature, the samples were subjected to native gel electrophoresis and western blot with GFP antibody. (B) Agarose gel electrophoresis followed by ethidium bromide staining was performed to examine the nucleic acid contents in the mixtures from (A). DNA (500–10 000 bp smear) and RNA (visible in the top part of NT lane, >10 000 bp, and in the full DNase lane) were evidently degraded in the presence of DNase and RNase, respectively. (C) The NCB fraction (50 μl) isolated from cells expressing R495X FUS was incubated with 2 or 20 μg of nuclear RNAs extracted with the TRIzol reagent. After incubation at 4°C overnight, the samples were subjected to native gel electrophoresis and western blot.
RNA binding is also required for cytoplasmic FUS inclusions
We next examined whether RNAs are also involved in inclusion formation of cytoplasmic FUS. The RNA recognition motif (RRM) binds to RNA with low affinity (29) and the flanking RGG1, RGG2, Zinc finger (ZnF) and RGG3 motif can potentially enhance the RNA binding. We thus generated a series of FUS truncation mutants with different RNA-binding motifs (Fig. 6A). R495X mutant FUS formed large inclusions in most cells (Fig. 6B, lower right panel). However, after deleting RGG2/ZnF/RGG3 domains, the truncated protein was evenly distributed in the cytoplasm with no inclusions (Fig. 6B, upper left panel). The ΔZnF/RGG3 truncation containing both RRM and RGG2 domains formed cytoplasmic inclusions, but the proportion of inclusion-containing cells is significantly lower than R495X (Fig. 6B, upper right panel; Fig. 6C). The ΔRGG3 truncation containing RRM, RGG2 and ZnF domains formed inclusions in more cells than ΔZnF/RGG3 (Fig. 6B, lower left panel; Fig. 6C). The quantitative results of the percentage of cells containing cytoplasmic inclusions are shown in Figure 6C.
Figure 6.

RNA binding was also required for the formation of FUS cytoplasmic inclusions. (A) Diagram of GST tagged R495X FUS and a series of truncations lacking RNA-binding motifs. QGSY, the QGSY-rich region; G, the glycine-rich region; RRM, the RNA recognition motif; RGG, arginine–glycine–glycine repeat regions; and ZnF, the zinc finger motif. (B) Inclusion formation of the GST tagged R495X FUS and the RNA-binding motif truncation mutants in HEK cells. GST-FUS mutants were immunostained with a GST antibody and the images were acquired with a Nikon A1 confocal microscope. The insets are magnified areas indicated by yellow squares. Green, GST-FUS; Blue, DAPI staining of DNA. (C) The percentages of inclusion-containing cells with different FUS truncation mutants. More than a hundred cells in three random view fields were counted for each different truncation mutant. Data shown represent mean ± SD and _P_-values were calculated using Student's _t_-test. (D) In vitro RNA binding of FUS truncation mutants with different RNA-binding motifs. Indicated concentrations of purified FUS truncation proteins were incubated with 5′-Cy3-labeled RNA probe and the samples were subjected to polyacrylamide gel electrophoresis (see details in Materials and Methods). The images were acquired with a ProteinSimple gel imaging system. The gel shift of the RNA probe illustrates the binding of FUS protein to the RNA probe.
We also examined the RNA-binding abilities of the relevant domains using a gel-shift titration assay. Under the conditions tested, the RRM domain alone did not bind to the RNA probe, which is consistent with our previous publication that the binding affinity between the RRM domain and RNA was weak (_K_d in hundreds micromolar range) (29). With addition of RGG2, ZnF and RGG3 domains, the binding affinity between the truncated FUS and the RNA probe significantly increased as evidenced by the shift of the FUS protein band by the RNA probe (Fig. 6D). The correlation between the stronger RNA binding affinity and the increased formation of cytoplasmic FUS inclusions suggests that RNA binding is also required for cytoplasmic FUS inclusion formation.
Discussion
The architecture of the FUS protein is different in nuclear granules and cytoplasmic inclusions
Since the cytoplasmic inclusions of mutant FUS are a hallmark of familial ALS caused by the FUS mutations, it is critical to understand the structural basis of such inclusions. No cross-β-sheet structure, which is typical in amyloidal aggregates, was identified in FUS inclusions (13,14). The purified FUS protein formed fibrils in vitro, and the N-terminal prion-like domain (amino acids 1–266) was required for the fibril formation (19). Another study also demonstrated that the FUS N-terminal domain itself (amino acids 2–214) formed hydrogel at high concentrations in vitro (30). We also found that the N-terminal QGSY-rich region (amino acids 1–164) was required to form functional oligomers in the nucleus, which can be demonstrated by a granular pattern under confocal microscope and a slower motility in native gel electrophoresis (15). In this study, we started testing whether FUS cytoplasmic inclusions are mediated by the N-terminal domain of FUS.
We first deleted the QGSY-rich region from R495X mutant FUS and examined its distribution under confocal microscope. To our surprise, deletion of the QGSY-rich region did not change cytoplasmic inclusions of R495X (Fig. 1C), suggesting that the N-terminal domain is not required for FUS cytoplasmic inclusion formation. This finding is in contrast to the observation that FUS nuclear granules disappeared after the deletion of the QGSY-rich region (Fig. 1B). Similar observations were found in cells expressing R521G point mutant FUS that was present in both the nucleus and cytoplasm (Fig. 1D). Moreover, native gel electrophoresis (Fig. 3B) and crosslinking (Fig. 3C) results provided additional evidence that the FUS protein in cytoplasmic inclusions mostly existed as the monomeric form. This is in contrast to mutant SOD1 inclusions where oligomeric SOD1 was readily found (31,32). Our results suggest that the cytoplasmic FUS inclusions are not disordered region-mediated fibrillar aggregates (Fig. 7). It is conceivable that FUS inclusions can be amorphous aggregates that can either become amyloidal aggregates through a common intermediate (33) or be disaggregated by heat shock proteins (34).
Figure 7.

The proposed model of FUS distribution and architecture in different cellular compartments. The left side shows that chromatin-associated RNAs initiate FUS oligomerization through the QGSY-rich region in the nucleus. Nuclear soluble and cytoplasmic FUS normally exists as monomers under physiological conditions. The right side shows that ALS mutations cause cytoplasmic retention of FUS. However, mutant FUS in the nucleus was still able to oligomerize similarly to wild-type FUS. The cytoplasm-retained FUS forms relatively disordered inclusions that require binding cytoplasmic RNAs.
Subcellular compartment determines FUS local distribution pattern and behavior
We asked the question whether subcellular compartment or ALS mutation is more important to determine FUS protein architecture and local distribution pattern. By forcing mutant FUS into the nucleus (Fig. 1F) and wild-type FUS out of the nucleus (Fig. 1G), we found that swapping subcellular compartments completely changed the local distribution patterns of FUS protein, regardless of wild-type or mutant. The results suggest that the subcellular localization plays a critical role in determining the architecture and distribution of FUS in different cell compartments. The ALS-causing mutations clearly change the subcellular localization of FUS, but mutations appear to have less influence on the local distribution pattern of FUS protein inside the specific compartment. An additional implication is that the ALS mutations within the C-terminal NLS have no significant impact on the overall conformation of FUS. However, the approaches used in this study are not quantitative; thus, it is possible that the nucleus-redistributed mutant FUS may not restore its chromatin binding ability to the full extent as the wild-type FUS.
Moreover, we demonstrated that mutant FUS protein localized in the nucleus behaves similarly to the wild-type FUS protein. Specifically, the nucleus-localized mutant FUS protein formed granules (Fig. 1D–F), oligomerized (Fig. 3) and responded to transcription inhibition (Fig. 2) in a similar fashion to the wild-type FUS. The results support the critical significance to restore the nuclear localization of mutant FUS that accumulates in cytoplasm aberrantly. Several studies have aimed to redirect mutant FUS into the nucleus as a potential therapeutic intervention (20–24), and this intervention showed beneficial effects in model organisms (21,22,24). Additional work is needed to test whether this approach can be an effective therapy in mammalian models.
Local RNAs are critical for FUS protein architecture
We demonstrated that chromatin-associated RNAs initiated FUS oligomerization (Figs 4 and 5), which is essential to the granular pattern of FUS in the nucleus. We have previously shown that FUS nuclear granules are strongly correlated with the CB FUS that was particularly required for its function in regulating gene transcription (15). This notion is supported by another study that FUS nuclear granules are colocalized with RNA polymerase II (35). In addition, FUS nuclear granules were also reported to colocalize with paraspeckles that play an important role in transcription and splicing regulation (28). It is evident that RNAs are involved in FUS-mediated transcription regulation and mRNA splicing, but the identities of the RNAs involved in FUS oligomerization and function are unknown. A previous study using a chimeric protein of the N-terminal prion-like domain of FUS and a DNA-binding domain of a transcription factor FLI showed that microsatellite DNA fragments with repetitive sequence significantly enhanced the oligomerization of the prion-like domain of FUS (36). It is conceivable that the proximal binding of multiple FUS chimeric proteins increased the local concentration of N-terminal FUS and triggered its oligomerization. This observation suggests that nascently transcribed non-coding RNAs, which are still attached on chromatin, from microsatellite sites may trigger the oligomerization of full-length FUS in the nucleus. Indeed, chromatin-associated non-coding RNA was reported to recruit FUS to chromatin to regulate gene transcription (37). A separate genome-wide study identified a substantial portion of FUS-binding RNAs as long non-coding RNAs (38). However, the specific nuclear RNAs initiating oligomerization of FUS in the nucleus remain to be experimentally determined.
Interestingly, we also found that the formation of FUS inclusions in the cytoplasm also required RNA-binding capability (Fig. 6). FUS has been reported to play a role in the formation of stress granules (4,5,7,8), a dynamic cytoplasmic structure containing proteins and messenger RNAs when translation halts in response to a variety of stresses. Different from the dynamic stress granules that are normally temporary under stresses, the cytoplasmic inclusions of FUS were observed persistently in this study. Thus, the RNAs sequestered in the persistent inclusions could result in a loss of function of the RNAs and the corresponding proteins. Knowing their identities can help us determine what cellular processes might be impaired by mutant FUS. The cytoplasmic inclusions are highly dynamic and difficult to isolate biochemically, excluding us to test the RNA dependence of such inclusion directly using the method as shown in Figure 5C. A biochemical approach needs to be developed to isolate FUS-containing cytoplasmic inclusions to determine the oligomeric status of FUS and the identities of RNAs in these inclusions.
Furthermore, we speculate that the RNAs required for nuclear FUS oligomerization and cytoplasmic FUS inclusions are likely two separate cohorts, although a partial overlap of these two pools is conceivable. In summary, the subcellular localization and the local pool of RNAs play a critical role in determining the specific architecture of FUS protein (Fig. 7). Such protein architecture may be crucial in distinguishing RNA-binding protein pathogenic mechanism from non-RNA-binding protein misfolding/aggregation mechanism in ALS. Moreover, the findings from this study support the feasibility of restoring the nuclear localization of mutant FUS as a potential therapeutic strategy of ALS.
Materials and Methods
Plasmids and reagents
The pEBG-FUS (GST-FUS) and pEGFP-C3-FUS plasmids were constructed in a previously published study (5). Truncated FUS coding sequences were amplified by PCR from pEBG-FUS or pEGFP-C3-FUS R521G plasmid template and subcloned into pEBG (restriction sites: Bam HI and Kpn I) or pEGFP-C3 (restriction sites: Bgl II and Kpn I) vectors. pEGFP-C3-FUS-NES and pEGFP-C3-FUS R495X-NLS were constructed in another previously published study (39). FUS antibody (sc-47711) and GFP antibody (sc-8334) were purchased from Santa Cruz Biotechnology. Nop56 antibody was provided by Dr Stefan Stamm at University of Kentucky. RNase-free DNase (catalog no. M6101) was purchased from Promega. Proteinase K (catalog no. P8102) was purchased from New England BioLabs. RNase A (catalog no. 19101) and QIAquick PCR Purification Kit (catalog no. 28104) were purchased from QIAGEN.
Cell culture and transfection
HEK cells were grown in Dulbecco's Modified Eagle's Medium (Sigma-Aldrich, catalog no. D5796) plus 10% fetal bovine serum (Sigma-Aldrich, catalog no. F2442) and 1% penicillin–streptomycin (Sigma-Aldrich, catalog no. P4333). Cell transfection was performed using 0.5 μg plasmid for cells cultured in 6-well plates following the standard protocol as previously described unless otherwise noted (5). Cells were collected 48 h after transfection.
Punch skin biopsies were obtained after informed consent from symptomatic familial ALS patients carrying the R521G FUS mutation and healthy control subjects as previously published (16). The study was approved by the Institutional Review Board of the University of Kentucky, and details of the patients were described in (16). Fibroblast cells were generated from the skin biopsies and cultured in Minimum Essential Medium Eagle (Sigma-Aldrich, catalog no. M5650) plus 20% FBS, 2 mm l-glutamine (Sigma-Aldrich, catalog no. G7513) and 1% penicillin–streptomycin.
Immunostaining and confocal microscopy
Cells were seeded on gelatin-covered 18 mm glass coverslips in 12-well plates and transfected with 0.25 μg plasmids. Forty-eight hours after transfection, cells were fixed with 4% paraformaldhyde at 37°C for 15 min and permeabilized with 0.25% Triton-X100 at room temperature for 5 min. Permeabilized cells and nuclei were blocked with 10% bovine serum albumin (BSA) at 37°C for 1 h, and immunostaining was performed with primary antibodies at 4°C overnight and secondary antibodies at 37°C for 1 h. Both primary and secondary antibodies were diluted in 3% BSA in 1× PBS. During secondary antibody incubation, 2 μg/ml DAPI was added to stain nuclear DNA. Cells were subsequently mounted with Vectashield mounting medium (Vector Laboratories, catalog no. H-1000) and observed under a Nikon A1 confocal microscope.
CB protein isolation
The CB protein isolation was performed as previously described (15). Briefly, cells were lysed in a detergent-free lysis buffer (50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA) and homogenized by passing through a 23G needle several times. After centrifugation at 1000_g_ at 4°C, chromatin and cell debris were in the pellet and NCB proteins were in the supernatant. The pellet was resuspended in the detergent-free lysis buffer, and CB proteins were released by breaking chromatin DNA with sonication. After centrifugation again at 1000_g_ at 4°C, CB proteins were retained in the supernatant. Both CB proteins and NCB proteins were subjected to 8% native polyacrylamide gel electrophoresis and western blot as described below.
Initiation of FUS oligomerization by chromatin content
Different amount of chromatin content, which was released by sonicating the re-suspended chromatins in the detergent-free lysis buffer, was incubated with the NCB sample for 20 min on ice. The mixture was then subjected to native gel electrophoresis and western blot.
During the incubation of the chromatin content and the NCB sample, 50 U/ml RNase-free DNase or 1 ng/ml RNase A was added in the mixture. After incubation, the effect of DNase or RNase on the oligomerization of NCB FUS was assessed by native gel electrophoresis. The nucleic acid contents in each sample were purified with QIAquick PCR Purification Kit after proteinase K digestion and subjected to agarose gel electrophoresis.
Native gel electrophoresis
The native gel electrophoresis was performed as previously described (15). Briefly, the samples prepared as above were loaded on a 8% (wt/vol) polyacrylamide gel soaked in detergent-free running buffer (25 mm Tris, 192 mm glycine). After electrophoresis, the gel was incubated with transfer buffer [25 mm Tris, 192 mm glycine, 10% (vol/vol) methanol] supplemented with 0.25% SDS at 70°C for 10 min. After the denaturation step, the gel was ready for transferring and routine western blotting.
Formaldehyde crosslinking
The CB and NCB proteins were prepared as above and separately incubated with formaldehyde at a final concentration of 1% at room temperature with rocking for 10 min. Crosslinking was quenched by adding glycine to a final concentration of 137.5 mm. The crosslinked samples were subjected to 10% SDS/PAGE followed by western blot.
Nuclear RNA extraction and the effect of nuclear RNA on FUS oligomerization
Cells were harvested, washed and incubated in nucleus isolation buffer (10 mm HEPES, pH 7.9; 10 mm KCl; 1.5 mm MgCl2; 0.34 m Sucrose; 10% Glycerol; 0.1% Triton X-100) on ice for 8 min. Nuclei were isolated by 1300_g_ centrifugation and resuspended in 0.25 ml nucleus isolation buffer. Total nuclear RNA was extracted by 0.75 ml TRIzol LS Reagent (Life Technologies, catalog no. 10296–010) following manufacturer's instructions. The RNA concentration was determined by NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). Two or 20 μg nuclear RNA was incubated with 50 μl NCB extract at 4°C overnight. These samples were subsequently subjected to native gel electrophoresis and western blot.
In vitro RNA-binding assay
Various FUS truncation proteins were expressed in Escherichia coli and purified as we previously published (29). Briefly, the coding sequences of different FUS truncations were subcloned into pET22b plasmid (Novagen). FUS peptides were expressed in E. coli Rosetta cells (EMD Millipore) and purified with Ni2+ affinity chromatography and cation exchange column (GE Healthcare, catalog no. 17-5157-01). The RNA probe (5′-UUAGGGUUAGGGUUAGGGUUAGGG-3′) was labeled with 5′-Cy3 (Invitrogen). Different concentrations of FUS peptides were incubated with 5 pmol RNA probes in 10 μl reaction buffer (20 mm Tris–HCl, pH 7.5; and 50 mm NaCl) on ice for 30 min. The mixtures were loaded on a 6% polyacrylamide gel and run in ice-cold TBE buffer at 100 V for 45 min. The gel was analyzed by a gel imaging system (ProteinSimple).
Funding
This study was supported in part by the National Institutes of Neurological Disorder and Stroke (grant R01NS077284 to H.Z.), ALS Association (grant 6SE340 to H.Z.), National Science Foundation of China (grant 91219202 to W.G.) and the 973 program from Ministry of Science and Technology of China (2011CB910503 to W.G.).
Acknowledgements
We thank Dr Emilia Galperin for critically reading the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwiatkowski T.J., Jr, Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- 3.Rowland L.P., Shneider N.A. (2001) Amyotrophic lateral sclerosis. N. Engl. J. Med., 344, 1688–1700. [DOI] [PubMed] [Google Scholar]
- 4.Bosco D.A., Lemay N., Ko H.K., Zhou H., Burke C., Kwiatkowski T.J., Jr, Sapp P., McKenna-Yasek D., Brown R.H., Jr, Hayward L.J. (2010) Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet., 19, 4160–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gal J., Zhang J., Kwinter D.M., Zhai J., Jia H., Jia J., Zhu H. (2011) Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging, 32, 2323 e2327–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson P., Kedersha N. (2008) Stress granules: the Tao of RNA triage. Trends Biochem. Sci., 33, 141–150. [DOI] [PubMed] [Google Scholar]
- 7.Sama R.R., Ward C.L., Kaushansky L.J., Lemay N., Ishigaki S., Urano F., Bosco D.A. (2013) FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol., 228, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersson M.K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., Stenman G., Nilsson O., Aman P. (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol., 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baron D.M., Kaushansky L.J., Ward C.L., Sama R.R., Chian R.J., Boggio K.J., Quaresma A.J., Nickerson J.A., Bosco D.A. (2013) Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener., 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takanashi K., Yamaguchi A. (2014) Aggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formation. Biochem. Biophys. Res. Commun., 452, 600–607. [DOI] [PubMed] [Google Scholar]
- 11.Ryu H.H., Jun M.H., Min K.J., Jang D.J., Lee Y.S., Kim H.K., Lee J.A. (2014) Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol. Aging, 35, 2822–2831. [DOI] [PubMed] [Google Scholar]
- 12.Li Y.R., King O.D., Shorter J., Gitler A.D. (2013) Stress granules as crucibles of ALS pathogenesis. J. Cell Biol., 201, 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bigio E.H., Wu J.Y., Deng H.X., Bit-Ivan E.N., Mao Q., Ganti R., Peterson M., Siddique N., Geula C., Siddique T., et al. (2013) Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol., 125, 463–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shelkovnikova T.A., Robinson H.K., Southcombe J.A., Ninkina N., Buchman V.L. (2014) Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum. Mol. Genet., 23, 5211–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L., Gal J., Chen J., Zhu H. (2014) Self-assembled FUS binds active chromatin and regulates gene transcription. Proc. Natl Acad. Sci. USA, 111, 17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhar S.K., Zhang J., Gal J., Xu Y., Miao L., Lynn B.C., Zhu H., Kasarskis E.J., St Clair D.K. (2014) FUsed in sarcoma is a novel regulator of manganese superoxide dismutase gene transcription. Antioxid. Redox Signal., 20, 1550–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Convertini P., Zhang J., de la Grange P., Hayward L.J., Zhu H., Stamm S. (2013) Genome wide array analysis indicates that an amyotrophic lateral sclerosis mutation of FUS causes an early increase of CAMK2N2 in vitro. Biochim. Biophys. Acta, 1832, 1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King O.D., Gitler A.D., Shorter J. (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res., 1462, 61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Z., Diaz Z., Fang X., Hart M.P., Chesi A., Shorter J., Gitler A.D. (2011) Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol., 9, e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dormann D., Madl T., Valori C.F., Bentmann E., Tahirovic S., Abou-Ajram C., Kremmer E., Ansorge O., Mackenzie I.R., Neumann M., et al. (2012) Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J., 31, 4258–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackel S., Summerer A.K., Thommes C.M., Pan X., Voigt A., Schulz J.B., Rasse T.M., Dormann D., Haass C., Kahle P.J. (2014) Nuclear import factor transportin and arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol. Dis., 74C, 76–88. [DOI] [PubMed] [Google Scholar]
- 22.Tradewell M.L., Yu Z., Tibshirani M., Boulanger M.C., Durham H.D., Richard S. (2012) Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet., 21, 136–149. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi A., Kitajo K. (2012) The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS One, 7, e49267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scaramuzzino C., Monaghan J., Milioto C., Lanson N.A., Jr, Maltare A., Aggarwal T., Casci I., Fackelmayer F.O., Pennuto M., Pandey U.B. (2013) Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS One, 8, e61576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waibel S., Neumann M., Rabe M., Meyer T., Ludolph A.C. (2010) Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology, 75, 815–817. [DOI] [PubMed] [Google Scholar]
- 26.Waibel S., Neumann M., Rosenbohm A., Birve A., Volk A.E., Weishaupt J.H., Meyer T., Muller U., Andersen P.M., Ludolph A.C. (2013) Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: a clinico-genetic study in Germany. Eur. J. Neurol., 20, 540–546. [DOI] [PubMed] [Google Scholar]
- 27.Shav-Tal Y., Blechman J., Darzacq X., Montagna C., Dye B.T., Patton J.G., Singer R.H., Zipori D. (2005) Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol. Biol. Cell, 16, 2395–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shelkovnikova T.A., Robinson H.K., Troakes C., Ninkina N., Buchman V.L. (2014) Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet., 23, 2298–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X., Niu C., Ren J., Zhang J., Xie X., Zhu H., Feng W., Gong W. (2013) The RRM domain of human fused in sarcoma protein reveals a non-canonical nucleic acid binding site. Biochim. Biophys. Acta, 1832, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato M., Han T.W., Xie S., Shi K., Du X., Wu L.C., Mirzaei H., Goldsmith E.J., Longgood J., Pei J., et al. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell, 149, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang F., Zhu H. (2006) Intracellular conformational alterations of mutant SOD1 and the implications for fALS-associated SOD1 mutant induced motor neuron cell death. Biochim. Biophys. Acta, 1760, 404–414. [DOI] [PubMed] [Google Scholar]
- 32.Strom A.L., Shi P., Zhang F., Gal J., Kilty R., Hayward L.J., Zhu H. (2008) Interaction of amyotrophic lateral sclerosis (ALS)-related mutant copper-zinc superoxide dismutase with the dynein-dynactin complex contributes to inclusion formation. J. Biol. Chem., 283, 22795–22805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fink A.L. (1998) Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold. Des., 3, R9–R23. [DOI] [PubMed] [Google Scholar]
- 34.DeSantis M.E., Leung E.H., Sweeny E.A., Jackrel M.E., Cushman-Nick M., Neuhaus-Follini A., Vashist S., Sochor M.A., Knight M.N., Shorter J. (2012) Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell, 151, 778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz J.C., Podell E.R., Han S.S., Berry J.D., Eggan K.C., Cech T.R. (2014) FUS is sequestered in nuclear aggregates in ALS patient fibroblasts. Mol. Biol. Cell, 25, 2571–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon I., Kato M., Xiang S., Wu L., Theodoropoulos P., Mirzaei H., Han T., Xie S., Corden J.L., McKnight S.L. (2013) Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell, 155, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X., Arai S., Song X., Reichart D., Du K., Pascual G., Tempst P., Rosenfeld M.G., Glass C.K., Kurokawa R. (2008) Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature, 454, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lagier-Tourenne C., Polymenidou M., Hutt K.R., Vu A.Q., Baughn M., Huelga S.C., Clutario K.M., Ling S.C., Liang T.Y., Mazur C., et al. (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci., 15, 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia R., Liu Y., Yang L., Gal J., Zhu H., Jia J. (2012) Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol. Neurodegener., 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]