The tubular hypothesis of nephron filtration and diabetic kidney disease (original) (raw)
. Author manuscript; available in PMC: 2021 Jun 1.
Published in final edited form as: Nat Rev Nephrol. 2020 Mar 9;16(6):317–336. doi: 10.1038/s41581-020-0256-y
Abstract
Kidney size and glomerular filtration rate (GFR) often increase with the onset of diabetes, and elevated GFR is a risk factor for the development of diabetic kidney disease. Hyperfiltration mainly occurs in response to signals passed from the tubule to the glomerulus: high levels of glucose in the glomerular filtrate drive increased reabsorption of glucose and sodium by the sodium–glucose cotransporters SGLT2 and SGLT1 in the proximal tubule. Passive reabsorption of chloride and water also increases. The overall capacity for proximal reabsorption is augmented by growth of the proximal tubule, which (alongside sodium–glucose cotransport) further limits urinary glucose loss. Hyperreabsorption of sodium and chloride induces tubuloglomerular feedback from the macula densa to increase GFR. In addition, sodium–glucose cotransport by SGLT1 on macula densa cells triggers the production of nitric oxide, which also contributes to glomerular hyperfiltration. Although hyperfiltration restores sodium and chloride excretion it imposes added physical stress on the filtration barrier and increases the oxygen demand to drive reabsorption. Tubular growth is associated with the development of a senescence-like molecular signature that sets the stage for inflammation and fibrosis. SGLT2 inhibitors attenuate the proximal reabsorption of sodium and glucose, normalize tubuloglomerular feedback signals and mitigate hyperfiltration. This tubule-centred model of diabetic kidney physiology predicts the salutary effect of SGLT2 inhibitors on hard renal outcomes, as shown in large-scale clinical trials.
Approximately 40% of patients with type 2 diabetes mellitus (T2DM) and 30% of those with type 1 diabetes mellitus (T1DM) will eventually develop chronic kidney disease (CKD), which is defined by evidence of kidney damage or a glomerular filtration rate (GFR) <60 ml/min/1.73 m2 for ≥3 months and/or elevated urinary albumin excretion1,2. About 50% of patients with end-stage renal disease (ESRD) in developed countries have diabetes1,3, and the presence of mild or moderate CKD doubles the risk of all-cause mortality associated with diabetes4.
No available model can accurately predict who will develop diabetic kidney disease (DKD) or how it will progress, although various studies have identified associations of DKD with genetic factors, socioeconomic status, obesity, smoking, poor glycaemic control, age, sex, hypertension, dyslipidaemia, other microvascular complications of diabetes, markers of oxidative stress, markers of inflammation and hyperfiltration5–8. Cell and molecular theories propose that the underlying pathogenesis of DKD involves complex alterations in metabolic processes, including elevated blood glucose concentrations, changes in fatty acid metabolism, oxidative stress, changes in energy utilization and mitochondrial dysfunction2,9, which cause endothelial dysfunction followed by podocyte loss and microvascular rarefaction2,9–13. An alternative theory proposes that DKD develops as a cellular response to the mechanical stress imposed by glomerular capillary hypertension or hyperfiltration14. This theory, which focuses on the physical consequences of hyperglycaemia, provides a concise explanation for the salutary effects of pharmacological agents that inhibit the renin–angiotensin system and the sodium–glucose transporter SGLT2, which to date are the only proven disease-modifying treatments for DKD. A growing body of literature demonstrating an important role for tubule function in regulating glomerular filtration in the context of diabetes lends further support to this theory15–17.
Although not all patients with diabetes develop DKD and not all patients with DKD follow the same trajectory18–20, in general, the presence of hyperfiltration in the early stages of diabetes is associated with the development of more severe kidney damage in the later stages of this disease9,12,21,22. Hence, the origins of hyperfiltration are worth considering, with the aim of identifying interventions that could prevent the development of DKD. Although hyperfiltration occurs at similar rates in patients with T1DM and T2DM22, most of the mechanistic data are derived from experimental models and patients with T1DM.
In this Review, we discuss changes that occur in the renal tubule early in the course of diabetes, focusing on how those changes affect interactions between tubule and glomerulus and influence the development of DKD. These events underlie the tubular hypothesis of nephron filtration and DKD.
The tubular model of hyperfiltration
A change in GFR can be defined simply as a sum of ‘vascular’ and ‘tubular’ events. A vascular event is anything that causes GFR to change by affecting the pre-glomerular or post-glomerular resistance vessels, the glomerular ultrafiltration coefficient or blood pressure. For example, GFR is reduced by afferent arteriolar vasoconstriction or by a fall in blood pressure. A tubular event is anything that directly affects tubular reabsorption in a manner that alters the concentration of sodium, chloride and potassium at the macula densa ([Na+/Cl−/K+] MD) and induces a change in GFR via tubuloglomerular feedback. For example, a reduction in GFR occurs in response to inhibition of tubular reabsorption upstream of the macula densa. Vascular events cause GFR and [Na+/Cl−/K+]MD to change in the same direction (because a change in reabsorption resulting from a change in filtered load cannot exceed the change in filtered load), in contradistinction to tubular events, which cause GFR and [Na+/Cl−/K+]MD to change in opposite directions as a result of negative feedback via the tubuloglomerular feedback system. When vascular events and tubular events occur simultaneously, the tubular event is declared to be dominant if GFR and [Na+/Cl−/K+]MD change in opposite directions.
Several lines of evidence — including the onset of hyperfiltration itself, vasodilatory failure in response to amino acid infusion, the paradoxical effect of dietary NaCl on renal haemodynamics and the reversible decrease in GFR that is induced by SGLT2 inhibitors — indicate that tubular events are dominant in the context of diabetes mellitus23. The observed predilection for GFR and [Na+/Cl−/K+]MD to change in opposite directions in patients with diabetes has led us to propose a systems model of the diabetic kidney that centres on the proximal tubule (FIGS 1,2). This model proposes that the increased reabsorption of glucose and other solutes occurring as a result of excess glucose filtration enhances glomerular filtration rate through tubuloglomerular feedback and by lowering tubular back pressure, which increases glomerular filtration pressure. Mathematical modelling predicts that tubuloglomerular feedback and decreased tubular back pressure might have a similar magnitude of effect on diabetic glomerular hyperfiltration24. Of note, although this model centres on the tubule, it does not ignore vascular events. Nuances in macula densa signalling, including modulation of the traditional tubuloglomerular feedback response by glucose, are also accommodated.
Fig. 1 |. Tubular hypothesis of glomerular filtration and nephropathy in diabetes mellitus.
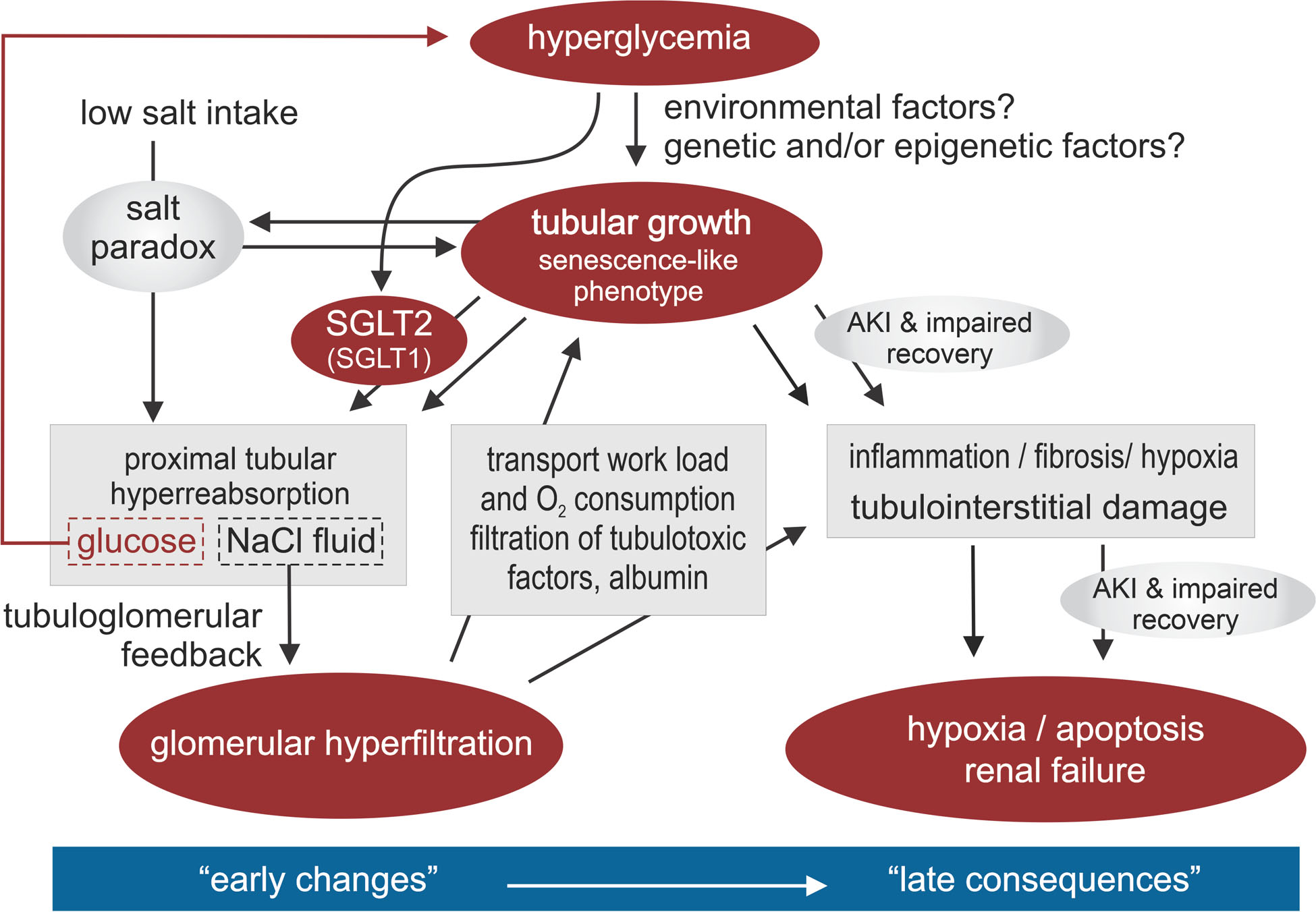
Hyperglycaemia activates renal mechanisms that act to retain the increased amounts of filtered glucose. Tubular growth and the activity of sodium–glucose cotransporters 2 and 1 (SGLT2 and SGLT1) in the proximal tubule increase glucose reabsorption. These adaptations also induce reabsorption of sodium; as a result, the glomerular filtration rate (GFR) also increases, to restore sodium excretion. However, the onset of tubular growth, hyperreabsorption and glomerular hyperfiltration promotes the development of diabetic kidney disease. The increase in GFR increases renal transport and oxygen requirements, which (together with enhanced glomerular filtration of albumin and other tubulotoxic factors) promotes hypoxia, inflammation, fibrosis and tubulointerstitial damage. The molecular signature of tubular growth in the context of diabetes is characterized by a senescence-like cellular phenotype, which is associated with the release of pro-inflammatory and pro-fibrotic factors, and probably also explains the ‘salt paradox’ of the diabetic kidney, whereby renal vasodilation unexpectedly occurs in response to a low dietary NaCl intake. Together, these changes promote hypoxia and apoptosis, facilitate episodes of acute kidney injury (AKI) and eventually lead to kidney failure. We propose that the tubular growth response to hyperglycaemia differs from patient to patient, in part because of unidentified genetic and environmental influences, which may determine not only the extent of tubular sodium and glucose hyperreabsorption and glomerular hyperfiltration in early diabetes mellitus but also the subsequent progression of renal disease. Adapted with permission from REF.205
Fig. 2 |. Primary tubular hyperreabsorption drives hyperfiltration in diabetes.
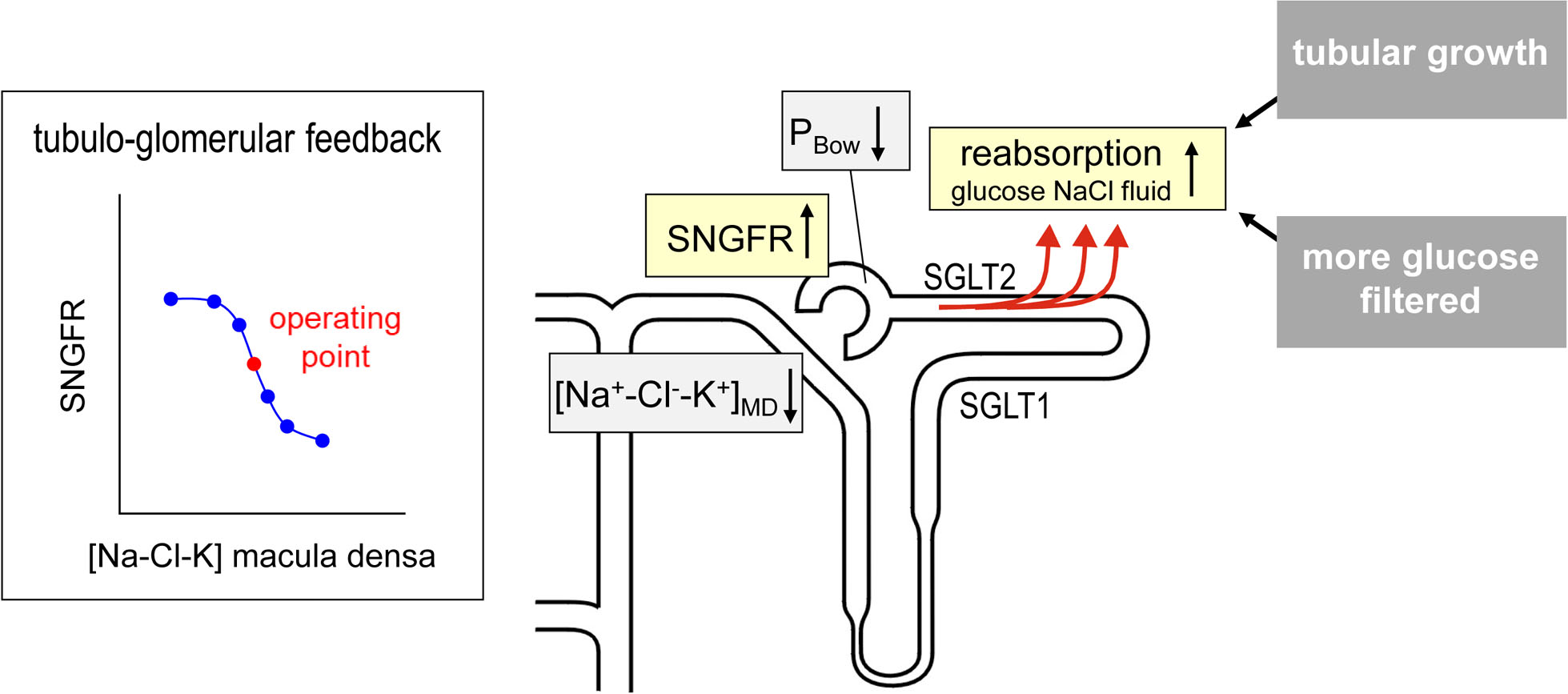
Diabetes induces hyperreabsorption of glucose in the proximal tubule resulting from enhanced glucose reabsorption via sodium–glucose cotransporters 2 and 1 (SGLT2 and SGLT1) and growth of the proximal tubule, which also leads to enhanced reabsorption of sodium, chloride and fluid in the proximal tubule. The reduced levels of sodium, chloride and potassium delivered to the macula densa ([Na+/Cl−K+]MD) induce glomerular hyperfiltration via tubuloglomerular feedback. The reduced fluid delivery to the distal nephron increases hyperfiltration via lowering hydrostatic back pressure in the Bowman’s space (PBOW). SGLT inhibition mitigates diabetic proximal tubular hyperreabsorption and thereby attenuates glomerular hyperfiltration. The graph illustrates how tubuloglomerular feedback results in an inverse relationship between [Na+/Cl−/K+]MD and the single nephron glomerular filtration rate (SNGFR). A red dot indicates the natural operating point of tubuloglomerular feedback, which lies in the steepest part of the curve. NaCl, sodium chloride (table salt).
Supporting evidence
One of the first clues that tubular reabsorption could be key to the development of hyperfiltration in diabetes came from a 1994 study in diabetic rats25, which showed an association between diabetic hyperfiltration and reduced activity of adenosine, which is a constrictor of the glomerular afferent arteriole, a dilator of its efferent arteriole and a mediator of tubuloglomerular feedback26. As explained below, a decreased tonic influence of adenosine over glomerular haemodynamics would be expected in the context of reduced [Na+/Cl−/K+]MD. In support of the tubular hypothesis of glomerular hyperfiltration, studies have consistently shown that either [Na+/Cl−/K+] MDNaClK or fractional lithium excretion (a surrogate of [Na+/Cl−/K+]MD) is lower than normal in animals16,27–29 and humans30–32 with diabetes and hyperfiltration, suggesting the existence of a primary increase in proximal tubule reabsorption. Further evidence for the involvement of a tubuloglomerular feedback mechanism in the regulation of hyperfiltration came from several studies showing that hyperfiltration resolves following normalization of [Na+/Cl−/K+]MD in rats28,33,34 or after ablation of the tubuloglomerular feedback mechanism in dogs35 or mice36. Some details of these early studies are presented below.
Role of adenosine signalling and hyperfiltration.
Increased delivery of sodium, chloride and potassium to the macula densa (that is, an increase in [Na+/Cl−/K+]MD) induces activation of the sodium–potassium–chloride transporter NKCC2 in macula densa cells, which increases both the influx of chloride and depolarization of the macula densa cell basolateral membrane. This depolarization induces the release of ATP, which is converted into adenosine by ecto-5′ nucleotidase37.
Adenosine mediates the tubuloglomerular feedback response by activating adenosine receptor A1 (A1R) on smooth muscle cells of the afferent arteriole, causing vasoconstriction and a reduction in GFR; mice lacking this receptor have no acute tubuloglomerular feedback response37. Under some conditions, the tubuloglomerular feedback-induced formation of adenosine can also reduce filtration by activating vasodilatory adenosine receptor A2 (A2R) on the efferent arteriole, which in turn reduces the filtration fraction38. Whether efferent vasodilation increases or reduces filtration depends on the net effect of these mechanisms, which act in opposing directions on glomerular capillary pressure and renal plasma flow. In the diabetic kidney, however, efferent vasodilation is expected to reduce filtration.
In the early stages of diabetes, hyperreabsorption of glucose and sodium in the proximal tubule reduces [Na+/Cl−/K+]MD, which increases GFR through tubuloglomerular feedback15. Thus, the tubular hypothesis proposes that tubuloglomerular feedback is the main controller of GFR in the early stages of diabetes.
We acknowledge that primary vascular effects might also contribute, which might be unmasked by eliminating tubuloglomerular feedback. In fact, evidence for glomerular hyperfiltration from diabetic_Adora1_-knockout mice (which lack A1R) is inconclusive on this point. One study found that such mice do not exhibit glomerular hyperfiltration, which is consistent with the tubular hypothesis;36 however, two other studies39,40 reported increased GFR in diabetic _Adora1_-knockout mice, although in one of these studies39differences in blood pressure between wild type and knockout mice might have been a confounding factor, explaining the differences in GFR, and in the other40 glucose levels were 600–900 mg/dl (33–50 mmol/l), too high to expect a net increase in proximal reabsorption, according to a mathematical modelling study41. Furthermore, the influence of changes in tubuloglomerular feedback on A2R activation on efferent arterioles is not eliminated in the absence of A1R. A reduction in tubuloglomerular feedback-mediated A2R activation could be of particular relevance in the context of diabetes, in which glomerular hyperfiltration is associated with increases in both efferent arteriolar tone and filtration fraction. With regard to the filtration fraction, a 2017 study reported that it tended to be higher among female patients with T1DM and hyperfiltration than among male patients matched for blood pressure and GFR42, which might imply that sex is a modifier of renal haemodynamics in the diabetic kidney.
Increased fractional proximal tubular reabsorption.
Fractional excretion of lithium in the urine gives a reasonable approximation of the fraction of filtered sodium reaching the macula densa. As an increase in GFR cannot induce a decrease in lithium clearance, a change in proximal reabsorption is implied whenever GFR and lithium clearance change in opposite directions. Indeed, lithium clearance rates reported for patients with T1DM and T2DM over the past three decades are consistent with the tubular hypothesis of hyperfiltration30–32,43,44.
Studies in the 1980s showing that lithium clearance is reduced in patients with diabetes led researchers to propose that increased proximal reabsorption was a cause of hypertension in individuals with diabetes30–32. However, most patients with diabetes are not hypertensive in the early phase of the disease, and reduced lithium clearance is a predictor of higher GFR in these individuals43,44. Thus, and consistent with the physiological role of tubuloglomerular feedback, the increase in GFR in response to tubular hyperreabsorption in diabetes is expected to normalize salt balance and mitigate the need to increase blood pressure24. The suggestion that increased proximal reabsorption might reduce Na+ delivery to the macula densa and that a reduction in tubuloglomerular feedback stimulus might cause glomerular hyperfiltration in patients with T1DM was first made in 1990 (REF.44). A 2010 study in patients of African heritage with newly discovered T2DM found a strong correlation between glomerular hyperfiltration and decreased lithium clearance, which is consistent with increased proximal reabsorption also driving hyperfiltration in patients with T2DM45. Similar observations have been made in rats with streptozotocin (STZ)-induced diabetes, in which glomerular hyperfiltration was associated with increased fractional reabsorption of sodium, chloride and fluid within the proximal tubule16,27,29 (FIG. 2). Use of micropuncture experiments to manipulate single-nephron GFR (SNGFR) also demonstrated that diabetes induces a substantial increase in proximal reabsorption28,46.
Importantly, as the kidney adapts to chronic hyperglycaemia, the size and overall transport capacity of the proximal tubule increases as a result of tubular growth as well as increased expression of SGLT2 (FIGS 1,2). These two mechanisms are discussed in more detail in the following sections.
Hyperglycaemia: effect on glucose filtration and reabsorption.
The daily glomerular filtrate of a normal human adult contains ~180 g glucose, which provides sufficient energy to account for about one-third of the body’s caloric expenditure. One function of the proximal tubule is to prevent this glucose from being lost into the urine. Glucose reabsorption is a two-step process that involves glucose uptake into the proximal tubular cell by SGLT2 and SGLT1 (FIG. 3), followed by its passive exit from the cell via GLUT2 in the basolateral membrane.
Fig. 3 |. Renal glucose reabsorption in the proximal tubule.
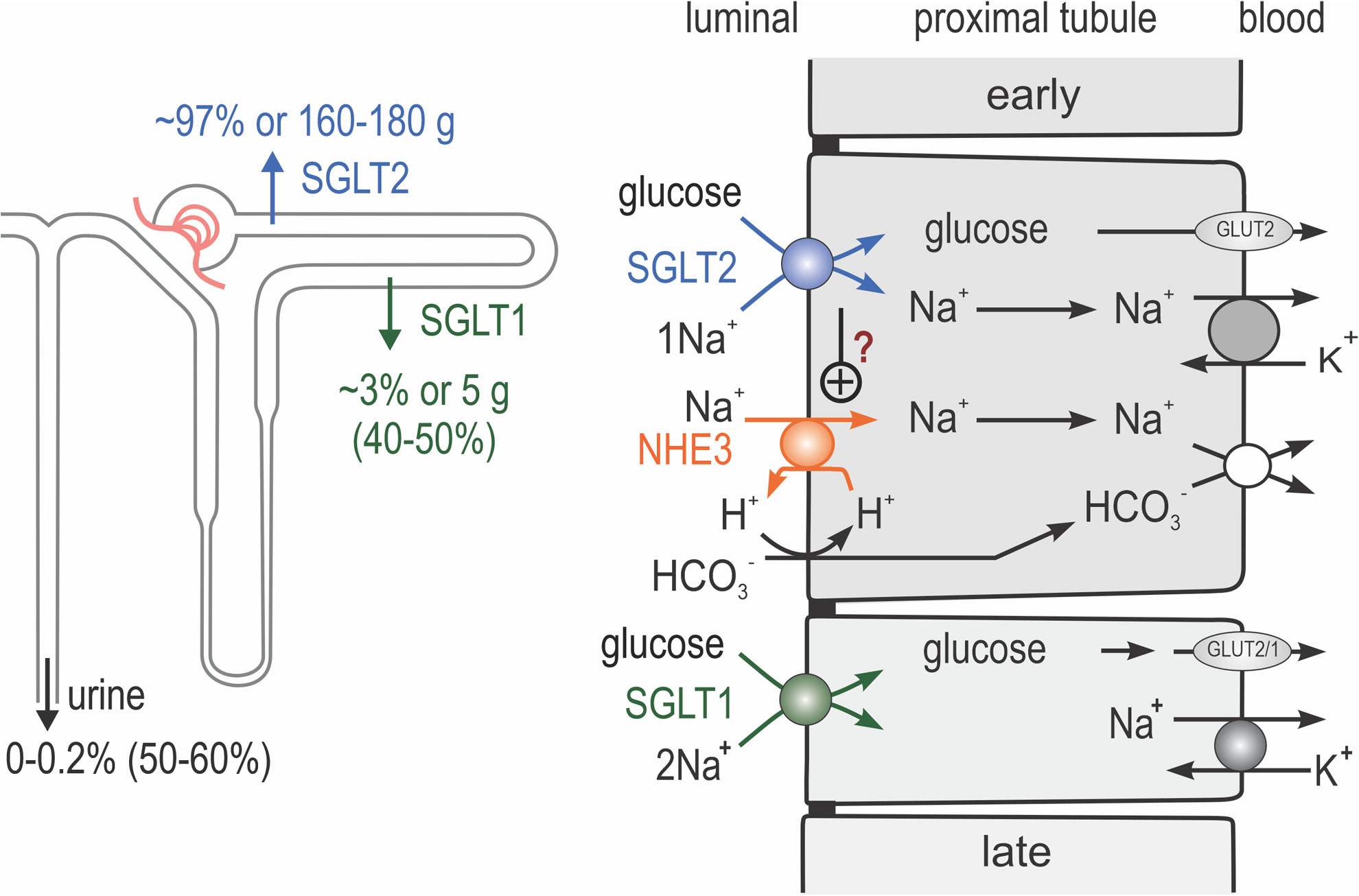
Na+/K+-ATPase in the basolateral membrane lowers intracellular Na+ concentrations and thereby provides the driving force for sodium uptake across the apical membrane. Sodium–glucose cotransporters 2 and 1 (SGLT2 and SGLT1) are expressed in the apical membrane of the early and late proximal tubule, respectively. SGLT2 reabsorbs one glucose molecule along with one Na+ ion, whereas SGLT1 reabsorbs one glucose molecule along with two Na+ ions. In individuals with euglycaemia, SGLT2 and SGLT1 reabsorb ~97% and ~3%, respectively, of filtered glucose. Glucose leaves the cells via basolateral facilitated glucose transporter 2 (GLUT2), and potentially in part by GLUT1 in the late proximal tubule. Inhibition of SGLT2 increases the delivery of glucose to the late proximal tubule and unmasks the high capacity of SGLT1 for glucose reabsorption (which rises from 3% to 40–50% of filtered glucose); thus, only ~50–60% of filtered glucose is excreted. Na+/H+-exchanger 3 (NHE3) in the apical membrane is important for sodium and bicarbonate (HCO3−) reabsorption in the proximal tubule. SGLT2 might be positively linked to NHE3 and therefore to sodium and bicarbonate reabsorption in the proximal tubule. However, the implications of this link (in terms of the effects of SGLT2 inhibition on tubular reabsorption, glomerular filtration rate (GFR) and blood pressure) remain to be determined. NBC1, electrogenic sodium bicarbonate cotransporter 1. Adapted with permission from Annual Review of Medicine volume 66 ©2015 (REF.138).
Studies in the 1980s suggested that two different glucose transporters are expressed on the apical surface of the proximal tubule47,48; the transporters were subsequently cloned and named SGLT1 (encoded by SLC5A1) and SGLT2 (encoded by SLC5A2) (reviewed elsewhere)49. These studies established that the bulk of tubular glucose uptake occurs in the early proximal tubule via the high-capacity transporter SGLT2, whereas the low-capacity transporter SGLT1 was thought to ‘mop up’ most of the remaining luminal glucose in the late proximal tubule (FIG. 3). The logic of positioning SGLT2 upstream of SGLT1 is apparent from an assessment of their stoichiometry and thermodynamic parameters. The Gibbs free energy required for sodium–glucose cotransport comes from the electrochemical potential of sodium ions. SGLT2 uses one sodium ion to transport one glucose molecule and SGLT1 uses two sodium ions to transport one glucose molecule. Thus, SGLT2 uses half the energy capital of SGLT1, but SGLT1 can drive the tubular glucose concentration to half that achieved by SGLT2. Hence, the most efficient route to achieving the lowest possible tubular fluid glucose concentration is to first use SGLT2 to reabsorb glucose up to its thermodynamic limit, and then to use SGLT1 to reabsorb the remainder. These calculations also suggest that the most efficient way to adapt to hyperglycaemia is to increase the protein expression of SGLT2.
Confirmation that SGLT2 and SGLT1 are confined mainly to early and late proximal tubules, respectively, was provided by immunostaining of human and rodent kidneys50–53. Moreover, renal clearance and micropuncture studies in euglycaemic_Sglt1_-knockout, Sglt2_-knockout and_Sglt1–Sglt2 double-knockout mice have shown that SGLT2 accounts for all glucose reabsorption in the early proximal tubule51 and for ~97% of total renal glucose reabsorption, whereas SGLT1 reabsorbs the remaining ~2–3% of glucose51,54,55 (FIG. 3). Further evidence for the dominant role of SGLT2 in glucose reabsorption is provided by the observation that individuals with loss-of-function mutations in SLC5A2 develop familial renal glucosuria, a renal tubular disorder resulting in excretion of 60–120 g per day of glucose in the urine56. By contrast, individuals with loss-of-function mutations in SLC5A1 excrete relatively little glucose in the urine, although they demonstrate malabsorption of intestinal glucose and galactose49,57.
The SGLT2–SGLT1 system has high affinity but limited capacity for reabsorbing glucose. Hence, glucose only appears in the urine when the filtered amount exceeds the capacity of this system for reabsorption, but once that capacity is exceeded, all additional filtered glucose is excreted in the urine. For an individual who is not chronically hyperglycaemic, this capacity is typically reached at a blood glucose concentration >12 mmol/l (>200 mg/dl)58. Hyperglycaemia (irrespective of whether it is acute or chronic) increases the amount of glucose filtered by the kidneys, as long as GFR is preserved. At the same time, the tubular glucose reabsorption capacity increases from ~400–450 g per day to ~500–600 g per day in patients with T2DM59 and T1DM60, to handle the increased load of filtered glucose (FIG. 4), thus setting a new glucose transport maximum. The increase in tubular glucose reabsorption capacity helps to conserve this valuable energy substrate, but becomes maladaptive by sustaining hyperglycaemia (FIGS 1,4). When the filtered load of glucose exceeds the new transport maximum, the kidney serves as a safety valve to forestall extreme hyperglycaemia by allowing excess glucose to escape into the urine. This mechanism postpones the development of severe hyperglycaemia until the osmotic diuresis associated with glucose excretion causes sufficient volume depletion to reduce both GFR and filtered glucose, which has the consequence that the safety valve opens at higher blood glucose levels.
Fig. 4 |. Diabetes induces the hyperreabsorption of glucose and sodium in the proximal tubule.
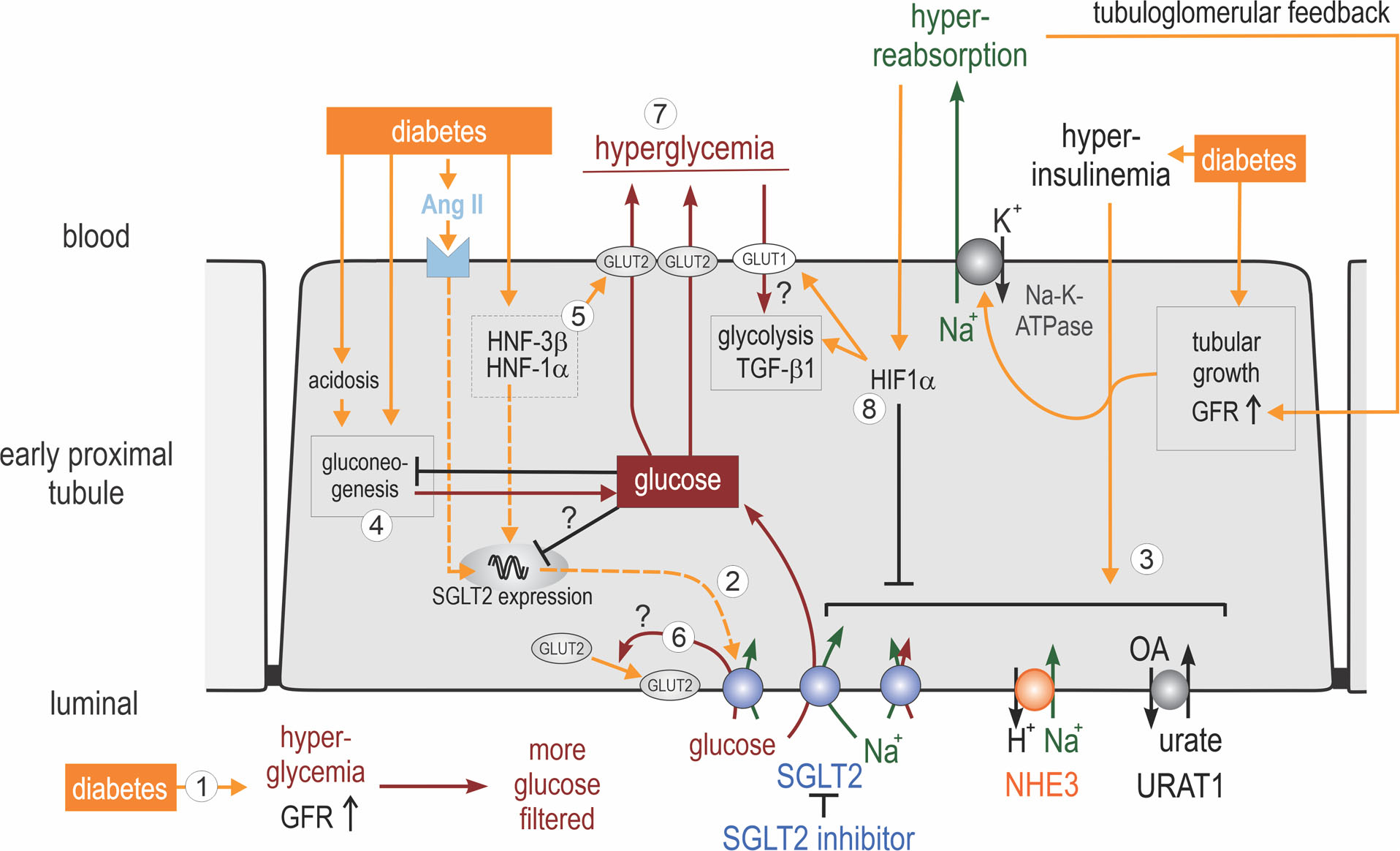
(1) Hyperglycaemia enhances filtered glucose and, via sodium–glucose cotransporter 2 (SGLT2; and also SGLT1, not shown), increases the reabsorption of glucose and sodium in the proximal tubule. (2) Diabetes can increase renal membrane expression of SGLT2; proposed mechanisms include activation of angiotensin II (AngII), hepatocyte nuclear factor 1α (HNF1α) and tubular growth. (3) Hyperinsulinaemia and tubular growth might induce the coordinated upregulation of proximal tubular transporters, including SGLT2, Na+/H+-exchanger 3 (NHE3), solute carrier family 22 member 12 (URAT1) and Na+/K+-ATPase. The resulting increase in proximal tubular Na+ retention enhances the glomerular filtration rate (GFR) via tubuloglomerular feedback, which, by increasing flow rate and thereby torque, may further increase luminal membrane transporter density in the early proximal tubule. (4) Diabetes, in part because of its associated acidosis, can enhance gluconeogenesis in the early proximal tubule. The resulting increase in intracellular glucose may inhibit SGLT2 expression through negative feedback mechanisms. (5) HNF1α and HNF3β have been implicated in stimulating the basolateral exit of glucose through upregulation of basolateral facilitated glucose transporter 2 (GLUT2). (6) The relevance of the apical translocation of GLUT2 in diabetes remains to be determined, but may be secondary to enhanced SGLT2-mediated glucose uptake. (7) Increased glucose reabsorption maintains hyperglycaemia. The induction of transforming growth factor (β1 (TGFβ1) and tubular growth might be particularly sensitive to the basolateral uptake of glucose via GLUT1.(8) Hyperreabsorption of sodium might induce hypoxia-inducible factor 1α (HIF1α), which inhibits apical transporters, including SGLT2 and NHE3, and facilitates basolateral glucose uptake and a metabolic shift to glycolysis.
Cotransport of glucose and sodium means that as the proximal tubule reabsorbs more glucose, it also reabsorbs more sodium. Sodium reabsorption by SGLT1 and SGLT2 is electrogenic and is thought to drive chloride reabsorption by electrodiffusion. Sodium, chloride and glucose reabsorption each contributes osmotic potential for water reabsorption up to a point beyond the glucose transport maximum, after which glucose becomes an osmotic diuretic in the proximal tubule. At normal blood glucose concentrations, the contribution of SGLT1 and SGLT2 to overall proximal reabsorption of NaCl and fluid is relatively small. Under conditions of moderate hyperglycaemia, however, the amount of sodium reabsorption tied to glucose becomes similar to the amount tied to bicarbonate transport, which is indirectly mediated by Na+/H+ exchange and is the benchmark measure of proximal tubular sodium reabsorption. The increase in fluid and electrolyte reabsorption linked to sodium–glucose cotransport in the proximal tubule induces a decrease in [Na+/Cl−/K+]MD, which is mitigated, in turn, by tubuloglomerular feedback, which causes SNGFR to increase (FIG. 2). However, tubuloglomerular feedback is not able to completely normalize [Na+/Cl−/K+]MD; the continued deficit in the amount of sodium and chloride delivered to the macula densa must therefore be compensated for by decreasing sodium and chloride reabsorption downstream of the macula densa or by pressure natriuresis to maintain long-term whole-body fluid and electrolyte balance17,24,61. At very high glucose levels, an osmotic diuresis overrides the SGLT-mediated increase in sodium reabsorption and the [Na+/Cl−/K+]MDstimulus for hyperfiltration disappears41 (as discussed in the section on macula densa SGLT1 and hyperfiltration). The capacity for glucose and sodium retention depends on the level of expression and activity of the glucose transporters, which varies from patient to patient.
Effect of diabetes on SGLT2 expression
In the context of normal blood glucose levels and GFR, the glucose transport capacity of SGLT2 is not saturated. Any acute rise in blood glucose levels prompts an increas in filtered glucose and increased glucose reabsorption by SGLT2 (FIG. 4). Additional rises in blood glucose levels that exceed the transport maximum of glucose result in glucosuria; however, as already mentioned, the tubular glucose reabsorption capacity increases substantially in the setting of chronic hyperglycaemia, possibly due to upregulation of SGLT2 protein expression in the proximal tubule (FIG. 4). Indeed, studies in genetic mouse models of T2DM (db/db mice) and T1DM (Akita mice) have shown that membrane protein expression of renal SGLT2 is increased by 40–80% in the early stages of hyperglycaemia62,63. Moreover, upregulation of the glucose transporter GLUT2 has been reported in proximal tubules of diabetic rats64–67, indicating a concerted upregulation of luminal and basolateral glucose transport.
Studies in rats and mice with STZ-induced diabetes show that, in addition to its expression at the basolateral membrane, GLUT2 is expressed on the brush border membrane of proximal tubules68–70, suggesting that facilitated diffusion via GLUT2 could contribute to glucose reabsorption if the electrochemical potential of luminal glucose rises above that in the cell and interstitium (FIG. 4). On the other hand, SGLT2 and SGLT1 seem to explain net renal glucose reabsorption in genetic mouse models of T1DM and T2DM, in that renal glucose reabsorption could be eliminated by administration of a selective SGLT2 inhibitor to mice lacking SGLT1 (REF.71). Notably, in the small intestine, increases in luminal glucose concentrations are sensed by SGLT1, which is required for the insertion of GLUT2 into the brush border membrane72. If SGLT2 has a similar glucose-sensing role in the early proximal tubule, SGLT2 inhibitors might lower renal glucose reabsorption during severe hyperglycaemia in part by inhibiting the apical translocation of GLUT2 (FIG. 4). Alternatively, glucose may leak back into the lumen via apical GLUT2. Such apical recycling of glucose would promote sodium reabsorption via SGLT1 and SGLT2.
Very little is known about changes in glucose transporter expression in patients with diabetes. One study showed that glucose uptake and protein expression of SGLT2 and GLUT2 were elevated in primary cultures of human exfoliated proximal tubular epithelial cells harvested from fresh urine of patients with T2DM73. Increased SGLT2 protein expression was also reported in fresh kidney biopsy samples from patients with T2DM and advanced DKD74. Thus, the available rodent and human studies indicate that diabetes can be associated with enhanced function of the renal glucose transport machinery.
Mechanisms that enhance SGLT2 expression and activity.
Increased expression of SGLT2 at the apical membrane might occur as a consequence of the overall growth and hypertrophy of the diabetic proximal tubule23,75,76 (FIG. 4). The mechanisms leading to tubular growth are discussed further below. In addition, glomerular hyperfiltration might further increase luminal membrane transporter density in the early proximal tubule by flow-dependent recruitment of transport proteins into the brush border77. Both processes could be exaggerated in the context of advanced DKD, in which nephron loss is compensated for by hyperfiltration and tubular growth of the remaining nephrons78. In the kidneys of diabetic rats, upregulation of SGLT2 protein expression has been linked to activation of type 1 angiotensin II (AngII) receptors in the basolateral membrane of the proximal tubule79 and the transcription factor, hepatocyte nuclear factor 1α (HNF1α)80. Also in diabetic rats, HNF1α and HNF3β have been implicated in the upregulation of GLUT2 (REF.65) (FIG. 4). Finally, insulin has been proposed to phosphorylate SGLT2 at Ser624, to increase its transport capacity81. Thus, postprandial insulin release might increase proximal tubular SGLT2 activity in order to retain increased amounts of filtered glucose. However, in the context of hyperinsulinaemia associated with obesity and T2DM, this increased activity might be maladaptive82 (FIG. 4).
In addition to stimulating SGLT2 activity, hyperinsulinaemia might coordinate the stimulation of other apical transporters in the proximal tubule, including the Na+/H+-exchanger NHE3 (REFS9,82) and the luminal urate transporter URAT1 (REFS83,84). Furthermore, SGLT2 might be functionally coupled to NHE3 and URAT1 in the proximal tubule. Pharmacological blockade of SGLT2 partially inhibits the activity of NHE3 (REFS85–88) and URAT1 (REF.84) (FIG. 4); conversely, tubular knockdown of NHE3 can reduce SGLT2 protein expression89. Such coordinated regulation of apical transporters involved in Na+, bicarbonate, glucose and urate reabsorption in the early proximal tubule might facilitate the appropriate regulation of tubular reabsorption in response to changes in GFR, for example, in the postprandial phase. A similar co-regulation of SGLT1 and NHE3 has been proposed to occur in the small intestine in response to post-prandial stimuli90.
Cell membrane SGLT2 protein expression in the kidney is also increased in response to pharmacological inhibition of SGLT2 (REFS63,91), which might reflect the negative feedback regulation of SGLT2 expression by intracellular glucose concentrations. In line with this proposal, conditions associated with enhanced proximal tubule gluconeogenesis (such as the increased bicarbonate formation occurring in the absence of tubular NHE3) were associated with reduced SGLT2 expression89(FIG. 4). Thus, renal SGLT2 expression might be reduced in some diabetic kidneys as a consequence of enhanced proximal tubular gluconeogenesis (for example, due to metabolic acidosis89). Alternatively, factors associated with severe tubular hypoxia, injury, ketoacidosis, cachexia or inflammation might also reduce SGLT2 levels92–94.
Rationale for SGLT2 inhibition in DKD
Several SGLT2 inhibitors have been approved as glucose-lowering agents in patients with T2DM and preserved kidney function23. The principle underlying SGLT2 inhibition in the diabetic kidney relates to its role in tubular glucose reabsorption and in maintaining hyperglycaemia (FIGS 1,5). SGLT2 inhibitors act on their target proteins in the extracellular surface of the cell membrane95, which they reach by glomerular filtration and, in the case of empagliflozin, also by tubular secretion96. Inhibition of SGLT2 induces a fall in the renal glucose reabsorption capacity down to the residual capacity of SGLT1 — that is, to ~80 g of glucose daily (as further discussed below). In other words, SGLT2 inhibition lowers the threshold at which the renal safety valve opens, thereby inducing a sustained urinary glucose loss of 40–80 g daily. In patients with T2DM, this glucosuria is associated with a sustained decrease in HbA1C levels of 0.5–0.7%23. SGLT2 inhibition also reduces body weight owing to its diuretic effect and as a result of renal glucose and calorie loss, which shifts energy substrate utilization from carbohydrates to lipids and thereby reduces visceral and subcutaneous body fat23. Free fatty acids released from adipose tissue can also be taken up by the liver to form ketone bodies, which can be used as an additional energy substrate by the kidneys, heart, brain and muscle97,98. Moreover, by lowering blood glucose levels and body weight, SGLT2 inhibitors improve pancreatic β-cell function and sensitivity to insulin in patients and rodent models of T2DM99–103. By reducing hyperglycaemia, SGLT2 inhibitors also have the potential to reduce glucotoxicity in the kidney and extrarenal organs104 (FIG. 5). Most importantly, however, the rationale for inhibiting SGLT2 in the diabetic kidney is based on the central role of this transporter in the tubular hypothesis of glomerular hyperfiltration and nephropathy.
Fig. 5 |. Mechanisms of kidney protection in response to SGLT2 inhibition.
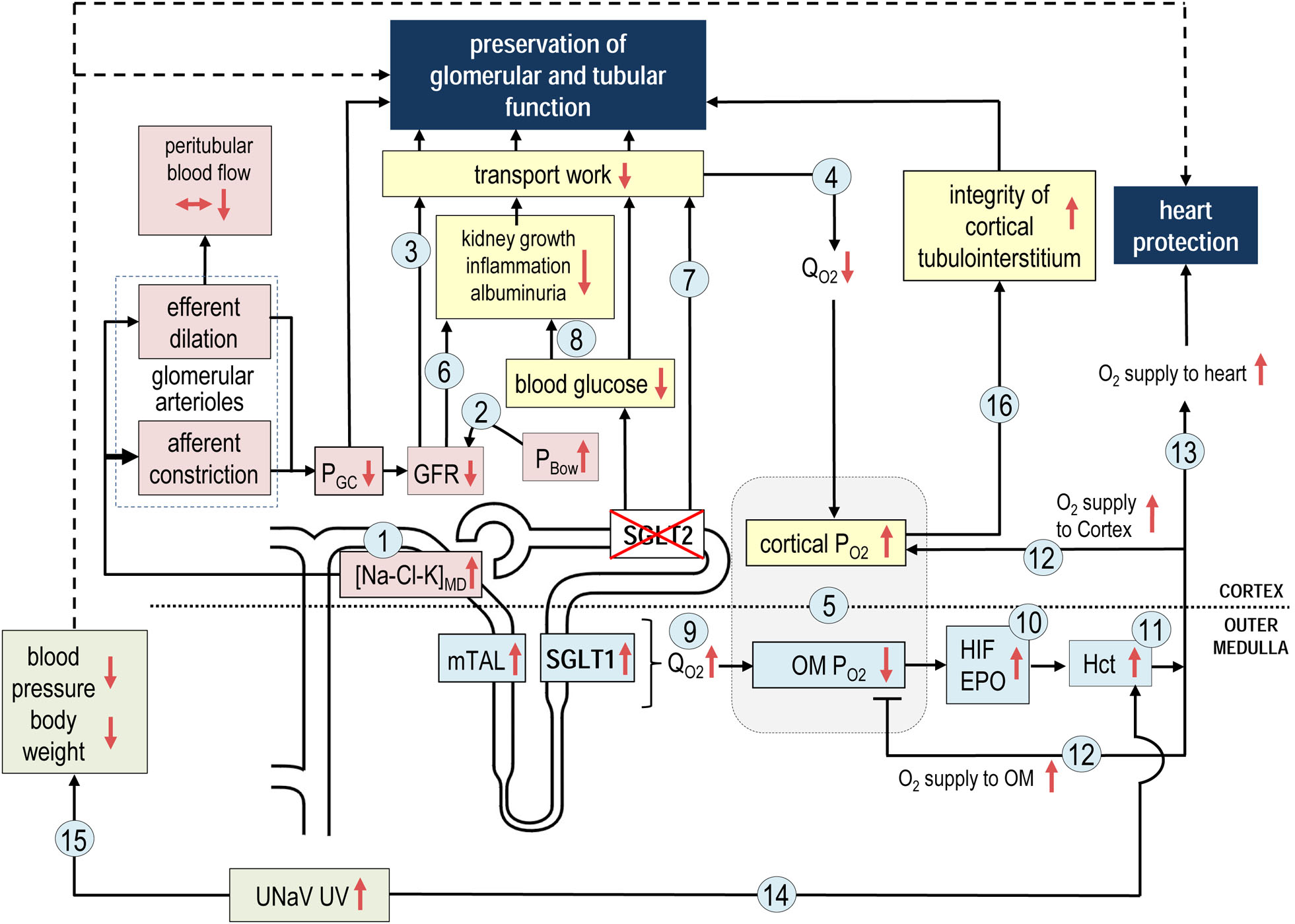
Sodium–glucose cotransporter 2 (SGLT2) inhibition reduces the diabetes-induced hyperreabsorption of glucose and sodium in the early proximal tubule, lowering hyperglycaemia and increasing delivery of sodium chloride (NaCl) and fluid to the macula densa, resulting in higher macula densa concentrations of sodium, chloride and potassium ([Na+/Cl−/K+]MD). The increase in [Na+/Cl−/K+]MDreduces the glomerular filtration rate (GFR) through tubuloglomerular feedback by inducing afferent arteriole constriction and potentially also efferent arteriole dilation, which both reduce glomerular capillary pressure (PGC) (1). Increased delivery of fluid to the distal nephron reduces GFR by increasing hydrostatic back pressure in the Bowman’s space (PBOW) (2). The reduction in GFR is the primary mechanism for reducing tubular transport work (3), particularly in the proximal convoluted tubule, which thereby lowers cortical oxygen demand (QO2) (4) and increases cortical oxygen tension (PO2) (5). Lowering of GFR attenuates tubular growth and albuminuria and consequently kidney inflammation (6). Tubular transport work is further reduced by lowering blood glucose levels and by cellular SGLT2 blockade itself (7). The reduction in hyperglycaemia attenuates tubular growth, albuminuria and inflammation (8). SGLT2 inhibition also shifts glucose reabsorption downstream, particularly to the S3 segment of the proximal tubule, where glucose uptake by SGLT1 compensates for the inhibition of SGLT2 and reduces the risk of hypoglycaemia. Shifting glucose and sodium reabsorption to the S3 segment and medullary thick ascending limb (mTAL) raises oxygen demand (9) and lowers PO2 in the outer medulla (OM) (5). Conversely, lower medullary PO2 might stimulate pathways induced by hypoxia-inducible factors (HIFs), including erythropoietin (EPO) production (10), thereby increasing the haematocrit levels (11), which improves O2 delivery to the kidney medulla and cortex (12) and increases O2 supply to the heart (13). The diuretic and natriuretic effects of SGLT2 inhibition further increase the haematocrit (14) and reduce circulating volume, blood pressure and body weight (15), which might protect the failing heart. The overall reduction in and increased distribution of renal transport activity increases cortical oxygen availability, which improves the cortical energy balance and tubular integrity, thereby enabling the kidney to maintain tubular transport capacity and GFR in the long term (16). UNaV, urinary sodium excretion; UV, urinary flow rate. Adapted with permission from REF.104, Portland Press on behalf of The Biochemical Society.
Effects of SGLT2 inhibition on GFR
Consistent with the prominent role of SGLT2 in the tubular hypothesis of glomerular filtration, inhibition of this transporter attenuates proximal tubule hyperreabsorption in the diabetic kidney and thereby lowers diabetes-associated glomerular hyperfiltration (FIG. 1). In 1999, micropuncture studies of superficial glomeruli of hyperfiltering rats with STZ-induced diabetes showed that concentrations of sodium, chloride and potassium at the macula densa were ~25% lower than those in non-diabetic control rats, implying that reabsorption of these electrolytes is increased upstream of the macula densa16(FIG. 2). Phlorizin (an inhibitor of both SGLT2 and SGLT1) delivery into the Bowman’s space of nephrons from these rats normalized (within minutes) the macula densa concentrations of these electrolytes and also normalized SNGFR in diabetic rats, but had minimal effect on non-diabetic animals. Similar results were obtained with acute or chronic systemic application of a selective SGLT2 inhibitor to diabetic rats33. Moreover, pharmacological inhibition or genetic ablation of SGLT2 reduced whole-kidney hyperfiltration in diabetic mice62,63,71. The suppression of diabetic hyperfiltration in response to SGLT2 inhibition was also associated with (and indeed was attributable to) an increase in the hydrostatic back pressure in the Bowman’s space, due to more fluid being left in the tubular lumen16. Most importantly, the hyperfiltration-suppressing effect of SGLT2 inhibition was independent of changes in blood glucose level16,33,62. A study from 2019 confirmed a role of A1R-mediated afferent arteriolar vasoconstriction in the GFR-lowering effect of SGLT2 inhibition in mice105. These findings are all consistent with the hypothesis that hyperfiltration results from a primary increase in proximal tubular reabsorption that is dependent on sodium–glucose cotransport.
This short-term (that is, observable within a few minutes to hours and weeks of treatment initiation) GFR-lowering effect of SGLT2 inhibition has been confirmed in humans, and also shown in long-term studies (that is, with durations of months to years) that demonstrated a biphasic GFR response characterized by an initial reduction in GFR followed by preservation of GFR at the new, lower level. Briefly, treatment of patients with T1DM and hyperfiltration with the SGLT2 inhibitor empagliflozin for 8 weeks decreased GFR independently of its effects on blood glucose levels106. The trial researchers suggested that empagliflozin exerted a dominant effect on the afferent arteriole, whereas a preliminary study in patients with T2DM proposed that another SGLT2 inhibitor, dapagliflozin, reduced GFR by reducing efferent arteriolar resistance107 (FIG. 5). As described earlier, adenosine produced at the macula densa acts to both constrict the afferent arteriole and dilate the efferent arteriole, and can therefore mediate both of the above-described effects. The short-term and long-term effects of SGLT2 inhibition were first demonstrated in the EMPA-REG OUTCOME108 trial conducted in patients with T2DM and preserved kidney function, in which empagliflozin induced an initial reduction in estimated GFR (eGFR) compared with placebo over the first 4 weeks of treatment. Throughout the remainder of the study (until week 192), eGFR remained stable in the patients treated with empagliflozin, whereas those receiving placebo experienced a progressive decline in eGFR. Accordingly, kidney function was better preserved in patients receiving the SGLT2 inhibitor108. A similar time course of GFR change was observed in clinical trials of canagliflozin109–111 and dapagliflozin112. Importantly, discontinuation of SGLT2 inhibitor treatment restored eGFR to pretreatment levels among patients in the SGLT2 inhibitor group, whereas eGFR remained below pretreatment levels in patients who had received placebo108,109.
The beneficial renal effects of SGLT2 inhibitors seem to be preserved in patients with CKD and diabetes mellitus. Indeed, studies in patients with T2DM and stage 2–3 CKD show that despite the blood glucose-lowering effect of SGLT2 inhibition being attenuated in these individuals owing to their reduced total rate of filtered glucose, the short-term GFR-lowering effect of SGLT2 inhibition is retained111,113,114, as is the long-term preservation of GFR111 and the full reversibility of the GFR-lowering effect after treatment discontinuation114. The expected GFR-preserving effect in patients with CKD was based on the assumption that surviving nephrons hyperfilter as a compensatory mechanism and therefore maintain a high glucose load at the single-nephron level.
The observation that SGLT2 inhibition induces an initial reduction in GFR that is reversible after discontinuation of treatment indicates that the GFR reduction following SGLT2 inhibition has a functional rather than a structural cause, which is consistent with the tubular hypothesis. The demonstration in large clinical trials that long-term SGLT2 inhibition preserves eGFR and renal function in patients with T2DM108,115,116, including in those with CKD111, is also consistent with a tubular origin of DKD (FIGS 1,5). Further details of these trials can be found elsewhere117.
Effect of SGLT2 inhibition on heart function
Given the role of SGLT2 in diabetes-induced proximal tubular hyperreabsorption of glucose, sodium and fluid, inhibition of SGLT2 has a modest diuretic and natriuretic effect that reduces body weight and thereby decreases systolic blood pressure by 3–6 mmHg (REF.23). This blood pressure-lowering effect is expected to have cardiovascular protective consequences, particularly in patients with cardiovascular risk factors118 (FIG. 5). The reduction in blood pressure and modest reduction in plasma volume119 resulting from SGLT2 inhibition can rapidly (that is, within days to weeks) reduce cardiac preload and afterload and thereby contribute to the reduction in heart failure observed in large outcome trials111,116,120,121. The beneficial renal and cardiovascular effects of SGLT2 inhibition might also result from an increase in ketone bodies produced from free fatty acids (as mentioned above), a potential uricosuric and plasma uric acid-lowering effect122 related to increased tubular or urinary glucose delivery123,124 and inhibitory effects on URAT1 (REF.84). Many of the above-described effects of SGLT2 inhibition can occur independently of blood glucose lowering, suggesting that SGTL2 inhibitors might exert beneficial effects in patients with normoglycaemia125. Indeed, the DAPA-HF trial126showed that, compared with placebo, dapagliflozin reduced the risk of worsening heart failure or death from cardiovascular causes among patients with heart failure and a reduced ejection fraction, irrespective of their diabetes status126. A secondary analysis of the CREDENCE trial data127 indicated that canagliflozin reduced the risk of cardiovascular and renal events in patients with T2DM and CKD across the spectrum of baseline HbA1c values, including in patients with baseline HbA1c of 6.5–7.0%127. Ongoing trials are further assessing the effects of SGLT2 inhibitors in non-diabetic patients with heart failure and/or CKD.
Effects on sodium and glucose reabsorption
Hyperfiltration imposes the highest transport burden on the early proximal tubule. Attenuation of glucose reabsorption in the early proximal tubule induced by SGLT2 inhibition increases the load of glucose and sodium delivered to downstream segments of the nephron (FIG. 5). By shifting some of the sodium and glucose reabsorption processes downstream of the proximal tubule, SGLT2 inhibition distributes this transport burden more equally along the tubular and collecting duct system, which (in our opinion) could help to preserve overall renal epithelial integrity and function. Furthermore, the SGLT2 inhibitor ipragliflozin lowered protein kinase C (PKC) activity in distal convoluted tubule cells and reduced the renal phosphorylation of Kelch-like protein 3 and expression of the Na+Cl− cotransporter NCC in diabetic ob/ob mice128 — mechanisms that might facilitate Na+ excretion.
SGLT2 inhibitors do not increase the incidence of hypoglycaemia108,116,120,129 because they become ineffective at lowering blood glucose levels once the filtered glucose load falls to ~80 g daily — the level that can be handled by SGLT1. However, the shift in glucose transport to the S3 segment and mTAL associated with SGLT2 inhibition might further reduce the already physiologically low availability of O2 in the renal outer medulla130–132 (FIG. 5) through mechanisms that include enhanced glucose transport via SGLT1 (which, as mentioned earlier, uses twice as much energy per glucose molecule transported as does SGLT2)49.
The increase in medullary transport and oxygen consumption in response to SGLT2 inhibition is mitigated by the concomitant reduction in blood glucose and GFR, which lowers the tubular glucose, sodium and chloride load and thereby reduces the tubular transport burden130,132 (FIG. 5). Furthermore, the reduction in oxygen pressure in the deep cortex and outer medulla could stimulate hypoxia-inducible factors HIF1 and HIF2, as demonstrated by findings in_Sglt2_-knockout mice, which have increased renal mRNA expression of HMOX1 (encoding haem-oxygenase 1)62, a tissue-protective enzyme that is induced by HIF1α. Moreover, activation of HIF2 might explain the enhanced release of erythropoietin from renal interstitial cells observed in response to SGLT2 inhibition133. Together with extracellular fluid volume loss due to the diuretic effect of SGLT2 inhibition, which increases the concentration of red blood cells, the increase in erythropoietin might contribute to the modest increase in haematocrit and haemoglobin observed in response to SGLT2 inhibition (FIG. 5). These changes are expected to improve oxygenation of the renal outer medulla and cortex and facilitate oxygen delivery to the heart and other organs104. In this regard, almost half of the beneficial effect of the SGLT2 inhibitor empagliflozin on the risk of cardiovascular death is explained by changes in haematocrit (51.8%) and haemoglobin levels (48.9%), respectively134. In other words, in addition to its effect on extracellular volume as a result of osmotic diuresis and natriuresis, SGLT2 inhibition might simulate systemic hypoxia at the oxygen sensors in the deep cortex and outer medulla of the kidney, which has beneficial effects on the kidney and heart135. Modelling studies predict that these effects and the natriuretic effect of SGLT2 inhibition will be preserved in patients with CKD135.
In accordance with their overall nephroprotective effect, an analysis of >3,000 patients with T2DM showed that SGLT2 inhibitor use also reduced the risk of acute kidney injury (AKI) by ~50%136. Nevertheless, caution is warranted in some patient groups. For example, in elderly individuals excessive volume depletion and the shift in transport from the proximal tubule to the outer medulla could increase the risk of AKI.
The role of SGLT1 in diabetes
SGLT1 is expressed in many organs and has a major role in intestinal glucose absorption (reviewed elsewhere137). In the kidney, the role of SGLT1 was thought to be limited to mopping up the residual amounts of glucose in the late proximal tubule that escaped SGLT2, and this activity was thought to have no major physiological or pathophysiological implications. New studies have begun to revise this understanding.
SGLT1 effects on glucose reabsorption
To recapitulate, SGLT1 in the late proximal tubule reabsorbs the glucose that has not been reabsorbed by upstream SGLT2, which under normal conditions equates to about 3% of filtered glucose54,55. However, this value is well below the maximal glucose reabsorption capacity of SGLT1, as is demonstrated when large amounts of glucose are delivered to the late proximal tubule. For example, fractional glucose reabsorption is maintained at 40–50% in healthy humans and rodents following application of a selective SGLT2 inhibitor54,138 (FIG. 3). A similar phenomenon is observed in mice lacking SGLT2 (REF.51). This persistence of substantial glucose reabsorption is due to SGLT1, the sizable glucose transport capacity of which is unmasked by SGLT2 inhibition51,54. As a consequence, and as observed in both non-diabetic and diabetic mice, combined inhibition of renal SGLT2 and SGLT1 is more glucosuric than is inhibition of SGLT2 alone54,71,139. The available evidence suggests that the basal overall glucose reabsorption capacity of SGLT2 is 3–5-fold that of SGLT1 in a non-diabetic mouse kidney140. The contribution of SGLT1 to renal glucose reabsorption is quantitatively increased in the context of diabetes, particularly when the filtered glucose load overwhelms the transport capacity of SGLT2. In this situation, SGLT1 increasingly contributes to proximal tubular hyperreabsorption, a circumstance with implications for the tubular hypothesis similar to those described for SGLT2 (FIGS 1,2,6).
Fig. 6 |. The role of SGLT1 in the diabetic kidney.
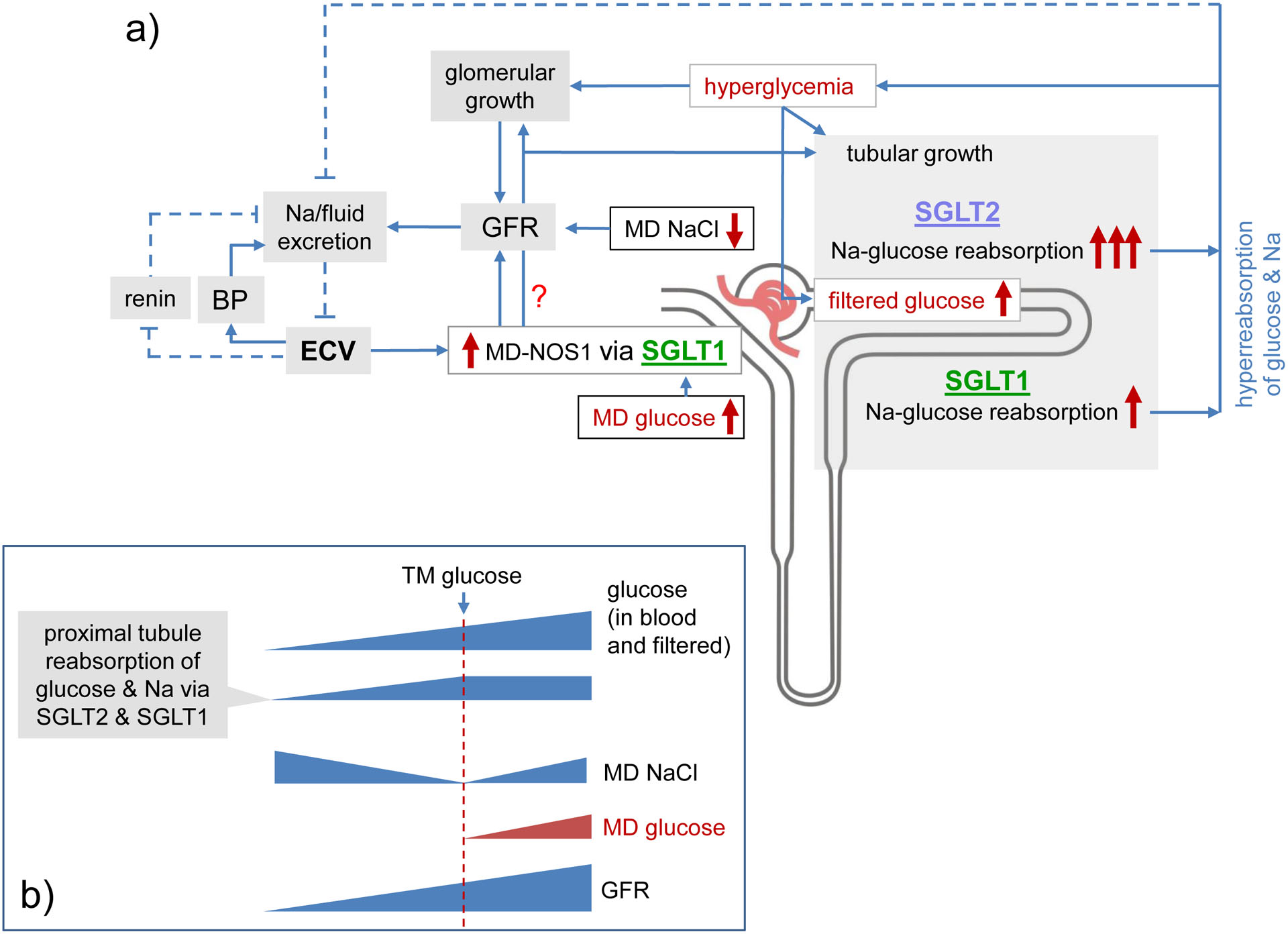
a | Hyperglycaemia enhances filtered glucose, induces tubular growth and increases sodium–glucose cotransport in the proximal tubule, thereby reducing urinary glucose, sodium (Na) and fluid excretion, and maintaining hyperglycaemia. The contribution of sodium–glucose cotransporter 2 (SGLT2) to this process is greater than that of SGLT1. A reduction in urinary sodium and fluid excretion would retain sodium and fluid, increase the effective circulating volume (ECV) and induce a compensating natriuresis by increasing blood pressure (BP). However, tubular hyperreabsorption of glucose also lowers tubular backpressure in the Bowman’s space (PBOW, not shown) and the concentration of sodium chloride (NaCl) at the macula densa, which increases the glomerular filtration rate (GFR) to restore urinary sodium and fluid excretion. An increase in glucose delivery to the macula densa indicates that upstream sodium–glucose cotransport is saturated. This saturation is sensed by SGLT1 in the macula densa (MD) and, by stimulating macula densa nitric oxide synthase 1 (NOS1) to produce nitric oxide (NO), further increases GFR to compensate for maximized sodium–glucose cotransport.As a consequence of hyperfiltration, ECV and BP remain near normal. SGLT1-mediated glucose sensing might also trigger glomerular and tubular growth, and the latter enhances tubular glucose transport capacity. Inhibition of SGLT1 has a smaller effect than SGLT2 inhibition on tubular hyperreabsorption and thus induces little natriuresis and diuresis (asterisks indicate small changes). By inhibiting macula densa glucose-sensing and upregulation of macula densa NOS1 and lowering of hyperfiltration, however, the inhibition of SGLT1 induces a relatively larger net antinatriuretic and antidiuretic effect in individuals with diabetes. As a consequence, and to limit the increase in ECV, SGLT1 inhibition suppresses renin expression and increases BP, in an attempt to restore renal sodium and fluid excretion. b | Sensing of proximal tubular hyperreabsorption, via the sensing of changes in concentrations of NaCl and glucose at the macula densa, enables the kidney to induce adaptive increases in GFR over a wide range of amounts of filtered glucose. An increase in filtered glucose up to the glucose transport maximum (TM glucose) of the proximal tubule is accompanied by an increase in sodium and glucose reabsorption, which lowers the NaCl concentration at the macula densa. When filtered glucose levels exceed TM glucose, glucose concentrations start to rise at the macula densa. The osmotic effect of non-reabsorbed glucose also enhances NaCl delivery to the macula densa.
Effect of diabetes on SGLT1 expression
In contrast to the increase in SGLT2 protein and mRNA expression that occurs in patients with diabetes highlighted above, a biopsy study showed that SGLT1 mRNA expression was not significantly changed in the kidneys of patients with T2DM and CKD compared with their levels in non-diabetic control kidneys74. These findings are in contrast to those from a study showing that SGLT1 protein expression was increased in leptin-deficient, diabetic ob/ob mice141 (a model of T2DM), although a direct effect of leptin on SGLT1 expression cannot be excluded. Moreover, another study showed that renal SGLT1 protein expression was reduced in Akita mice (a model of T1DM)62. In contrast to SGLT2, the activity of which is increased by insulin, SGLT1-mediated sodium–glucose transport in HEK-293T cells was slightly decreased by insulin81. Together, these data indicate differences in the regulation of SGLT2 and SGLT1 in the kidney.
Why diabetes leads to reduced renal SGLT1 expression is not entirely clear, but some insight is offered by the observation that renal SGLT1 protein expression is also reduced in non-diabetic mice by genetic ablation or pharmacological inhibition of SGLT2 (REFS51,63). Both SGLT2 inhibition and diabetes increase glucose delivery to the late proximal tubule and thereby increase SGLT1-mediated glucose reabsorption. Mathematical modelling and experimental studies indicate that diabetes increases the consumption of (and thereby reduces the availability of) oxygen in the outer medulla130–132. Studies in LLC-PK1 pig proximal tubule cells show that hypoxia reduces the protein expression of both SGLT1 and SGLT2, which was associated with activation of HIF1α93. In vitro studies further indicate that glucose-associated oxidative stress also reduces SGLT1 and SGLT2 expression and Na–glucose cotransport in proximal tubule cells142. Furthermore, the absence of SGLT1 can improve renal recovery in a model of renal ischaemia–reperfusion-induced AKI143
Thus, an increased glucose load to the outer medullary S3 segment enhances sodium–glucose reabsorption, which might downregulate SGLT1 in order to limit oxygen-consuming transport and glucotoxicity in this segment, which is known for its high sensitivity to AKI92.
Role of SGLT1 in the macula densa
As described above, SGLT1 in the late proximal tubule contributes to glucose and sodium handling, particularly in the setting of diabetes and/or SGLT2 inhibition. However, SGLT1 located at the macula densa acts as a glucose sensor to regulate the production of nitric oxide (NO) by NO synthase 1 (NOS1) (FIG. 6).
The production of NO following activation of NOS1 in macula densa cells alters the sensitivity of TGF to accommodate an increased [Na+/Cl−/K+]MD, thereby contributing to the overall maintenance of sodium balance144–146. Studies in rat and mouse models of diabetes indicate that NO produced by macula densa NOS1 is involved in mediating the increase in GFR that occurs in response to acute hyperglycaemia147–150. A 2019 study revealed that the binding of sodium and glucose to SGLT1 at the macula densa provides the stimulus for increased macula densa NOS1 activity in hyperglycaemia149, demonstrating that SGLT1 in the macula densa serves as a glucose sensor (FIG. 6). Another study showed that deletion of SGLT1 reduced glomerular hyperfiltration in diabetic Akita mice and mice with STZ-induced diabetes but did not markedly change their blood glucose levels71. Furthermore, the increase in macula densa NOS1 expression observed in diabetic Akita mice was abolished in the absence of SGLT1 (REF.71) (FIG. 6). These findings support the notion that increased tubular glucose delivery is sensed by SGLT1 in the luminal membrane of macula densa cells, which respond by increasing NOS1-dependent NO formation and reducing the vasoconstrictor tone at the afferent arteriole set by tubuloglomerular feedback, thereby contributing to glomerular hyperfiltration (FIG. 6).
The absence of SGLT1 not only lowers glomerular hyperfiltration in Akita mice but also reduces their kidney weight, glomerular size and albuminuria71. These findings suggest that targeting SGLT1 might have implications for renal integrity beyond the transport of glucose in the late proximal tubule, thick ascending limb and macula densa. This suggestion is in line with the finding that the absence of SGLT1 improved renal recovery in both kidney cortex and medulla following ischaemia–reperfusion injury143, as discussed earlier in this Review. The macula densa and the juxtaglomerular apparatus can be thought of as the anatomical centre of a single nephron involved in the regulation of renin and GFR by tubuloglomerular feedback, but this centre might have a much wider role. Sensing of increased glucose delivery at the macula densa might in fact signal the need for increased upstream glucose transport capacity and trigger tubular growth, although the underlying mechanism is not known (FIG. 6).
Mouse studies also indicate that inhibition of SGLT1 and SGLT2 can independently improve early changes in the diabetic kidney, via effects on blood glucose control, GFR, renal glucose reabsorption, kidney weight and glomerular size71.
Rationale for increased GFR in response to macula densa glucose delivery.
In the context of diabetes, GFR rises (at least in part) to stabilize body fluid volume in response to the increased tubular reabsorption of sodium, glucose and fluid resulting from tubular growth and enhanced sodium–glucose cotransport15,24. The delivery of glucose to the macula densa indicates that upstream sodium–glucose cotransporters are saturated and thus that hyperreabsorption of sodium, glucose and fluid has occurred. The macula densa senses the increased luminal glucose via SGLT1, which activates NOS1 to produce NO (and thereby to increase GFR, as described above) in order to maintain urinary sodium and fluid excretion and volume balance (FIG. 6). Blunting this compensatory increase in GFR without inducing a robust effect on hyperreabsorption would be expected to increase blood pressure, which is a first-order mechanism of sodium homeostasis151. In fact, deletion of SGLT1 not only blunted the diabetes-induced hyperfiltration but also suppressed renal mRNA expression of renin — a marker of volume overload — and increased systolic blood pressure (FIG. 6). Macula densa NOS1 has a decisive role in the maintenance of long-term sodium balance152–157. The response of diabetic mice to SGLT1 deletion resembles the effect of high doses of a selective NOS1 inhibitor in diabetic rats147. In both situations, GFR was reduced and accompanied by mild elevations in blood pressure. Thus, we propose that the macula densa SGLT1–NOS1–GFR pathway complements the classic pathways of tubuloglomerular feedback and tubular back pressure in the compensatory adaptation of GFR to tubular hyperreabsorption in the diabetic kidney. In contrast to this new pathway, the two classic mechanisms primarily operate when tubular glucose reabsorption is below the glucose transport maximum15 (FIG. 6). Finally, the activity of the macula densa SGLT1–NOS1–GFR pathway might have a critical role in the transition of diabetic patients from a hyperfiltering but normotensive phenotype to the late-stage presentations of reduced GFR and hypertension71,149.
Role of the macula densa SGLT1–NOS1–GFR pathway in the response to SGLT2 inhibition.
As described above, SGLT2 inhibition lowers GFR by enhancing delivery of sodium, chloride and potassium to the macula densa23. This effect on GFR can be attenuated by increasing glucose delivery to the macula densa, for example, as occurs with SGLT2 inhibition-induced glucosuria, which activates the macula densa SGLT1–NOS1–GFR pathway. Support for these mechanisms is provided by studies in non-diabetic mice, in which the increased expression of macula densa NOS1 induced by SGLT2 inhibition was prevented by the absence of SGLT1 (REF.71). By contrast, inhibition of SGLT2 in severely hyperglycaemic Akita mice did not further increase glucosuria from baseline levels (and probably did not alter glucose delivery to the macula densa either, because the reduction in filtered glucose offsets the inhibition of glucose reabsorption62,63), and actually reduced macula densa NOS1 expression71. This reduction in NOS1 expression might reflect the inhibitory influence of the volume loss induced by SGLT2 inhibition on NOS1 expression (FIG. 6). Thus, multiple inputs affect the expression and activity of macula densa NOS1 in response to SGLT2 inhibition, and the natriuretic and diuretic activity of SGLT2 inhibitors might reduce hyperfiltration in part by suppressing the glucose-independent component of the macula densa NOS1 pathway. Mouse studies have further shown that SGLT2 inhibition prevents the blood pressure increase observed in Akita diabetic mice lacking SGLT1, potentially because of the additive effects of SGLT2 and SGLT1 inhibition on renal glucose, sodium and fluid excretion71. Thus, findings from mouse studies provide supportive evidence for the use of combined SGLT2 and SGLT1 inhibition in the treatment of diabetes. However, further studies are required to more precisely define the role of macula densa glucose sensing and the therapeutic potential of dual SGLT2–SGLT1 inhibitors such as sotagliflozin, as well as the potential beneficial effects of such treatment (resulting mainly from SGLT1 inhibition) on intestinal glucose handling137,158–160.
Diabetes: effect on proximal tubule growth
Some patients with diabetes mellitus will experience overall growth of the kidney, particularly of the proximal tubules9,15,76. Increased kidney size has been linked to the development of DKD161–165. The mechanisms and implications of this kidney growth have been studied extensively in experimental models and patients with T1DM, but although an increase in kidney size has also been documented in experimental models of T2DM, little information is available about this phenomenon in patients with T2DM.
Tubular growth enhances the expression of tubular transporters, including glucose transporters, and, as mentioned earlier, is part of the tubular hypothesis of glomerular hyperfiltration. In addition, the molecular pathways that contribute to tubular growth in the setting of diabetes are linked to inflammation and fibrosis and might in consequence also contribute to renal damage9,15,76. Thus, genetic and/or environmental factors that affect the tubular growth response to the diabetic milieu might not only determine the extent of tubular sodium and glucose hyperreabsorption and glomerular hyperfiltration in early diabetes but might also affect the subsequent progression of renal disease9,15,76 (FIG. 1).
Mechanisms of proximal tubule growth
The growth of the proximal tubule in response to diabetes is a multistep process, in which cells undergo proliferation followed by cell cycle arrest and hypertrophy. The final stages of this process are characterized by a molecular signature characteristic of cellular senescence (reviewed elsewhere9,15,76,166,167).
Proliferation of tubule epithelial cells occurs early in diabetes in response to a number of stimuli, including high levels of glucose-induced oxidative stress, the tubular renin–angiotensin system, enhanced filtration and tubular expression of growth factors (including insulin-like growth factor I (IGF1), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF)), inhibition of AMPK and activation of PKCβ, the JAK–STAT pathway, mTORC1 and ornithine decarboxylase (ODC), the latter being the rate-limiting enzyme in polyamine synthesis; ODC is required for hyperplasia and also most likely for hypertrophy of the proximal tubule in diabetes9,15,76,166,167 (FIG. 7).
Fig. 7 |. Mechanisms and consequences of tubular growth in diabetes.
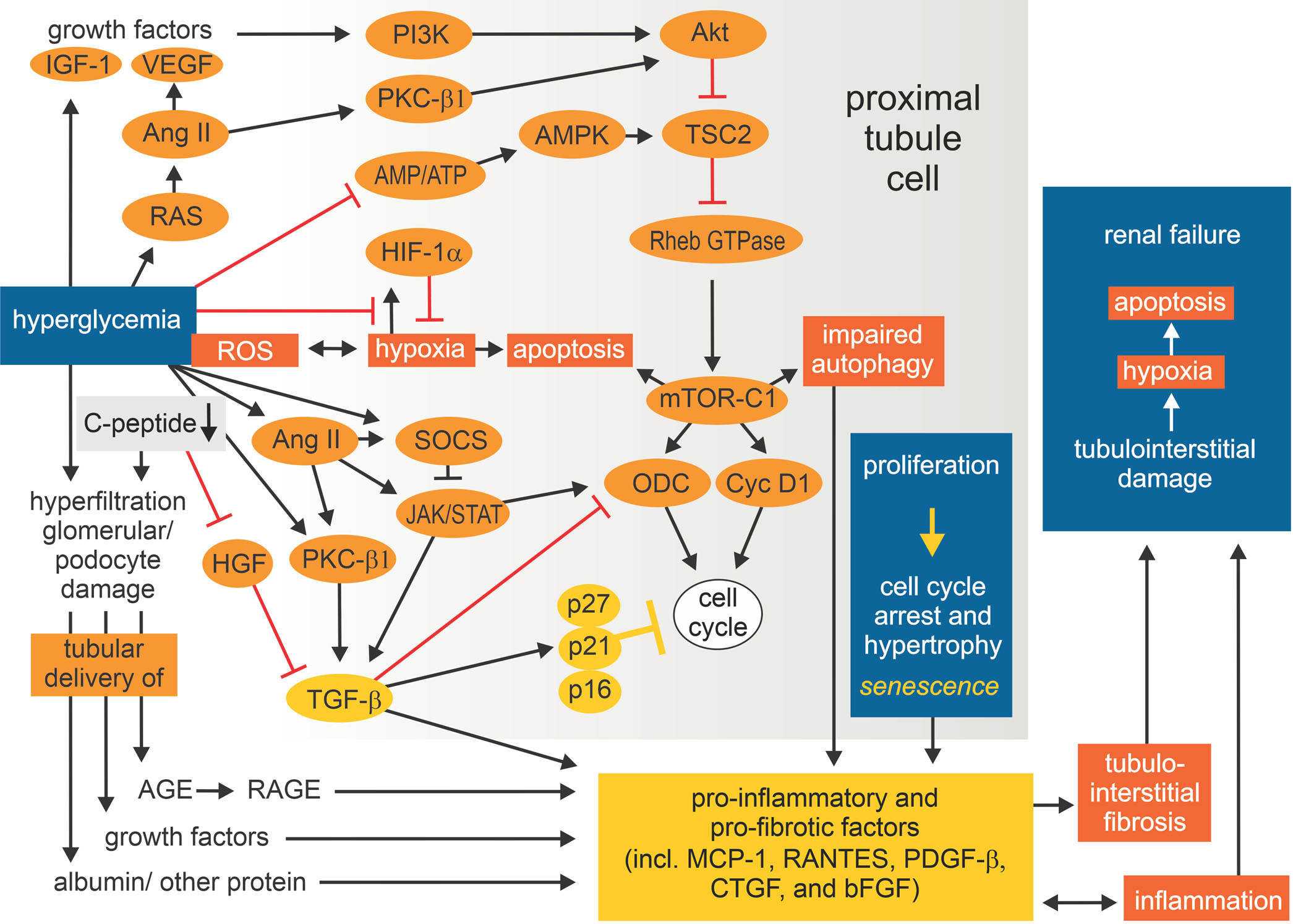
Growth of the proximal tubule in response to diabetes involves an initial phase of tubular cell proliferation, followed by G1 cell cycle arrest, hypertrophy and finally the development of a senescence-like cellular phenotype. The stimuli for the initial proliferation phase include filtered and locally produced growth factors, angiotensin II (AngII)-induced activation of protein kinase C β1 (PKCβ1), and inhibition of 5′-AMP-activated protein kinase (AMPK) owing to excess energy substrate. Hyperglycaemia also enhances reactive oxygen species (ROS), and ROS-induced hypoxia and activation of mechanistic target of rapamycin complex 1 (mTORCi) can promote apoptosis. Activation of mTORC1 also impairs autophagy and promotes cell proliferation. Induction of transforming growth factor β (TGFβ) drives the expression of cyclin-dependent kinase inhibitors that induce G1 cell cycle arrest and the transition from proliferation to hypertrophy and a senescence-like phenotype associated with the secretion of pro-inflammatory and pro-fibrotic factors. Thus, the molecular pathways involved in and activated by tubular growth in the diabetic kidney are linked to impaired autophagy, inflammation and tubulointerstitial fibrosis and might set the stage for the later development of diabetic kidney disease. AGE, advanced glycation end product; AKT, protein kinase B; bFGF, basic fibroblast growth factor; CTGF, connective tissue growth factor; CycD1, cyclin D1; EGF, epidermal growth factor; HGF, hepatocyte growth factor; HIF1α, hypoxia-inducible factor 1α; IGF1, insulin-like growth factor 1; JAK–STAT, Janus kinase–signal transducer and activator of transcription; MCP1, monocyte chemoattractant protein 1; p16, p16INK4a; ODC, ornithine decarboxylase; p21, p21Cip1; p27, p27Kip1; PDGF, platelet-derived growth factor ; PI3K , phosphoinositide 3-kinase; RAS, renin–angiotensin system; RHEB, Ras homologue enriched in brain; SOCS, suppressor of cytokine signalling protein; TSC1, tuberous sclerosis complex 1; VEGF, vascular endothelial growth factor. Adapted with permission from REF.9, Wiley-VCH.
The transition of the diabetic kidney from hyperplastic to hypertrophic growth also occurs early — within a few days in rodents with STZ-induced diabetes9,15,76,166,167. This transition is mediated by TGFβ1, which can be induced by the JAK–STAT signalling pathway, PKCβ, ERK and p38. PKCs (most likely PKCβ, but this pathway has only been confirmed using non-selective PKC inhibitors) and TGFβ can induce cell cycle arrest in the G1 phase by inducing p27Kip1 (p27, also known as cyclin-dependent kinase (CDK) inhibitor 1B)168,169. A role for oxidative stress and p21Cip1 (p21, also known as CDK inhibitor 1) has also been implicated in the hypertrophic response to diabetes170. Thus, signalling pathways that initially induce proliferation subsequently induce a switch to cell hypertrophy through induction of TGFβ and CDK inhibitors in the diabetic tubule (FIG. 7). Tubule cell hypertrophy is sustained through mechanisms involving the mTORC1–S6K1 pathway171, impaired autophagy172 and decreased proteolysis.
Diabetes-induced cell senescence
The cellular senescence programme is a tumour-suppression mechanism involving cell cycle arrest, which stops cells from replicating and passing on a damaged genome. Prototypical cellular senescence involves transient induction of p21, p16INK4a (p16, also known as CDK inhibitor 2A) and/or p27. An analysis of kidneys from rats with STZ-induced diabetes showed an early transient induction of growth phase components followed by their suppression by day 10, which occurred concurrently with the induction of p16, p21 and p27 and the expression of senescence-associated β-galactosidase activity in cortical tubules173 (FIG. 7). Moreover, cultured proximal tubule cells transitioned to a senescence-like phenotype in response to oxidative stress173. More importantly, an accelerated senescence-like phenotype was also observed in tubule cells of patients with T2DM and DKD174.
Glucose sensing triggers tubular growth
The glucose-lowering effect of SGLT2 inhibition has been demonstrated in experimental models and clinical studies. Our group showed that mice with genetic deletion of SGLT2 and STZ-induced diabetes had lower blood glucose levels than did wild type mice with STZ-induced diabetes (~300 mg/dl (17 mmol/l) versus ~470 mg/dl (26 mmol/l)) but had similarly increased kidney weight and expression of kidney growth markers62, indicating that SGLT2-mediated glucose reabsorption itself was not critical for inducing changes in kidney growth. In another study, administration of the SGLT2 inhibitor empagliflozin to diabetic Akita mice induced a strong blood glucose-lowering effect: their blood glucose decreased from >500 mg/dl (28 mmol/l) to ~200 mg/dl (11 mmol/l). Although kidney growth was not entirely prevented in empagliflozin-treated mice, the drug strongly attenuated kidney growth and reduced molecular markers of renal growth in proportion to the reduction in hyperglycaemia63. These results indicate that SGLT2 inhibition can reduce diabetic kidney growth secondary to a strong blood glucose-lowering effect (FIGS 1,5).
The observation that empagliflozin attenuated but did not completely prevent kidney growth or upregulation of p27 in diabetic Akita mice63 might reflect the fact that the drug did not fully normalize blood glucose levels and/or that the treatment-related reductions in GFR and blood glucose were insufficient to reduce glucose delivery downstream of the early proximal tubule, which itself could be a tubular growth stimulus. Although SGLT2 inhibition might be expected to increase glucose delivery to the downstream proximal tubule, the net effect of this treatment depends on the degree of drug-related reductions in GFR and blood glucose. Thus, in these mice, SGLT2 inhibition did not increase glucosuria because their pretreatment blood glucose levels were already very high; instead, SGLT2 inhibitor-related reductions in GFR and blood glucose resulted in a decrease in filtered glucose levels matching the inhibition of proximal tubule glucose reabsorption. The finding that empagliflozin reduced kidney growth in Akita mice without lowering urinary glucose excretion indicates that the increase in luminal glucose delivery downstream of the early proximal tubule is not by itself sufficient to explain the majority of the kidney growth that occurs in diabetes. These studies leave open the possibility that glucose delivery downstream of the proximal tubule might have a minor role in diabetic tubular growth — particularly given the role of SGLT1 in diabetic kidney growth71 described earlier (FIG. 6) — but indicate the need for additional stimuli that are independent of SGLT2 and luminal glucose delivery.
Role of basolateral GLUT1 in tubular growth.
Studies in LLC-PK1 proximal tubule cells showed that exposure to 25 mmol/l d-glucose induced glucose uptake via basolateral (but not apical) GLUT1 and that the subsequent intracellular metabolism of glucose enhanced the synthesis and secretion of TGFβ175,176. Thus, hyperglycaemia-induced basolateral uptake of glucose via GLUT1, and potentially also GLUT2, might be the driving force that enhances proximal tubular synthesis of TGFβ and thereby promotes tubular hypertrophy (FIG. 4). In line with this hypothesis, administration of metformin to rats with STZ-induced diabetes prevented the upregulation of renal GLUT1 protein expression177; moreover, administration of the AMPK activators metformin and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) inhibited renal hypertrophy without affecting hyperglycaemia178.
Alterations in energy metabolism might also contribute to the development of renal injury in diabetes. A metabolic shift from oxidative phosphorylation to glycolysis has been proposed to occur in regenerating proximal tubule cells after AKI, as well as in proximal tubule cells undergoing atrophy179. This metabolic switch is reportedly reversed following successful tubular recovery, but persists and becomes progressively more severe in tubule cells that fail to redifferentiate179. Upregulation of HIF1α in the tubules of non-diabetic mice enhanced renal Glut1 mRNA expression, was associated with a reduction in oxygen consumption and increased glycolysis180(FIG. 4). Further studies are needed to test the hypothesis that proximal tubular hypoxia in the diabetic kidney increases basolateral GLUT1-mediated facilitative uptake of glucose, which is then used for glycolysis and linked to tubular growth or pro-fibrotic pathways. Notably, a prominent role of GLUT1 in diabetic glomerulosclerosis has also been proposed181.
Tubular growth: effect on hyperreabsorption
An increase in proximal tubular length and diameter enhances tubule reabsorptive capacity. Difluoromethylornithine (DFMO, an ODC inhibitor), had no effect on kidney weight or GFR in non-diabetic rats, but attenuated kidney growth46,182 and reduced kidney weight and glomerular hyperfiltration proportionally in rats with STZ-induced diabetes46. Moreover, renal micropuncture studies showed that DFMO eliminated the primary increase in proximal tubular reabsorption in rats with STZ-induced diabetes46. That is, for a given level of SNGFR, proximal fluid reabsorption was lower in DFMO-treated rats than in control rats. Thus, attenuation of tubular growth is expected to have secondary effects on diabetic hyperreabsorption and hyperfiltration. This mechanism probably also involves Na+/K+-ATPase in the basolateral membrane of tubular epithelial cells, which lowers intracellular sodium concentrations and thereby provides the driving force for sodium uptake across the apical membrane. In fact, inhibition of PKCβ reduces diabetic hyperfiltration183, which might reflect the above-described role of this factor in both diabetic tubular growth184 and activation of Na+/K+-ATPase — and thereby in sodium transport in proximal tubules185. A 2019 study also confirmed the role of the Na+/H+-exchanger NHE3 as a determinant of tubular reabsorption, kidney weight and GFR in both non-diabetic and diabetic Akita mice: genetic knockdown of NHE3 in the tubular system was associated with reduced kidney weight and GFR89.
Tubular growth and the salt paradox
In the 1990s our group made the surprising finding that in rats with STZ-induced diabetes, a low-salt (NaCl) diet reduced renal vascular resistance and increased renal blood flow, GFR and kidney weight186, whereas a high NaCl diet induced renal vasoconstriction187. An inverse relationship between dietary NaCl and GFR is counterintuitive, given that GFR determines the tubular NaCl reabsorption needed to maintain sodium homeostasis; in other words, the kidneys increase GFR (and thus increase the tubular delivery of NaCl) under conditions when urinary NaCl excretion has to be reduced to match dietary intake and vice versa. This discovery prompted us to coin the term ‘salt paradox’ of the diabetic kidney. The phenomenon was subsequently confirmed in other rodent models of diabetes36,188 and in patients with uncomplicated T1DM189.
The salt paradox is unique to the diabetic kidney, and is a manifestation of the tubular hypothesis of glomerular filtration. This paradox results from the growth phenotype of the proximal tubule, which enhances the sensitivity of proximal tubule reabsorption to changes in dietary NaCl. The result is that increased ingestion of NaCl suppresses proximal tubular reabsorption and increases NaCl delivery at the macula densa (and vice versa for a decreased NaCl intake, FIG. 1). Tubuloglomerular feedback then adjusts GFR to stabilize the delivery of sodium and chloride to the early distal tubule28,36,190. The salt paradox occurs independently of renal innervation191 or type 1 AngII receptor activation186. Inhibition of tubular growth by ODC, however, abolishes the salt paradox190, highlighting the importance of tubular growth to this phenomenon.
As described above, hypertrophic proximal tubular cells in the diabetic kidney are continuously exposed to mitogens, but are prevented from entering the cell cycle owing to the presence of CDK inhibitors. These cells also demonstrate a senescence-like phenotype, which could alter their cellular responses (FIG. 7). More specifically, these tubular cells might have lost the cellular programmes that normally tell them not to respond to moderate changes in dietary NaCl. Thus, diabetes not only increases proximal tubular reabsorption but also increases the responsiveness of the proximal tubule to dietary salt28.
The salt paradox raises the question of whether a low-salt diet might increase the vulnerability of the diabetic kidney to chronic damage as a result of the associated increase in GFR and augmented kidney growth (FIG. 1). Two prospective studies have assessed the association of salt intake, based on 24-h urine collection, with outcomes in patients with diabetes over a median follow-up of 10 years. One of these studies, conducted in patients with T2DM, found that survival was monotonically associated with urinary sodium, such that a daily increase in sodium intake of 100 mmol predicted a 30% reduction in cardiovascular and all-cause mortality192. In the second study, conducted in patients with T1DM, the relationship of all-cause mortality to sodium intake was biphasic: mortality was lowest for patients with sodium intakes of 100–200 mmol daily, but increased gradually for patients with higher sodium intakes, and increased even more notably in patients with a sodium intake below this range193. Moreover, the cumulative incidence of ESRD was monotonically inversely associated with sodium intake, such that reducing sodium intake from the 90th to the 10th percentile equated to a 10-fold increase in the likelihood of developing ESRD193. These two studies question the view that a low salt intake is similarly beneficial for patients with T1DM and T2DM, and indicate the need for intervention studies.
Effects of hyperfiltration on the kidney
As 99% of filtered sodium is normally reabsorbed by the kidney, urinary sodium excretion is matched to dietary intake. Therefore, GFR is the primary determinant of renal sodium reabsorption and transport work, as well as renal oxygen consumption. Mathematical modelling predicts that sodium transport increases by 50%, and sodium transport-dependent oxygen consumption (active QO2) increases by 100%, in the diabetic rat kidney from non-diabetic levels, primarily as a consequence of glomerular hyperfiltration132. The proximal tubule segments in the renal cortex actively reabsorb most of the glomerular filtrate and therefore show the largest increases in QO2 in response to increases in GFR130,132. In accordance with these findings, oxygen tension (PO2) in the renal cortex was reduced in a rat model of T1DM131.
Reducing the hyperfiltration and therefore the oxygen-consuming transport work of single nephrons has the potential to preserve the integrity of the remaining nephrons and overall kidney function in the long term (FIG. 1). Such a strategy has been proposed to underlie the nephroprotective effects of renin–angiotensin system blockers194,195 and SGLT2 inhibitors (FIG. 5). The observation that ~80% of the patients with initial GFRs ≥30 ml/min/1.73 m2 who were included in clinical trials and showed beneficial effects of SGLT2 inhibitors on heart and kidney outcomes were also receiving a form of renin–angiotensin system blockade provides evidence that the two strategies are additive in such patients108,111,116. Mathematical modelling predicted that inhibition of SGLT2 in the diabetic kidney reduces oxygen consumption in the proximal convoluted tubule and renal cortex, in large part by lowering GFR130,132. The predicted increase in cortical O2 pressure has been observed in diabetic rats treated with the non-specific SGLT inhibitor phlorizin131. Preservation of renal cortical oxygenation might be critical for the preservation of kidney function in patients with CKD196, a hypothesis consistent with the notion that glomerular hyperfiltration primarily induces a burden on the proximal tubule.
Glomerular hyperfiltration can also damage the diabetic kidney by exposing glomerular capillaries to an increased hydrostatic filtration pressure and by enhancing the glomerular filtration of factors that are potentially deleterious for the tubulointerstitium (FIGS 1,7). Albumin, growth hormones, advanced glycation end products and other factors in the glomerular filtrate interact with the tubular system and have various consequences that contribute to the development and progression of DKD: increased energy consumption, renal oxidative stress and cortical interstitial inflammation; impairment of autophagy; and stimulation of hypoxia and tubulointerstitial fibrosis9,76,172,176,197–199.
Consequences of tubular growth
As described above, tubular growth and hyperreabsorption increase renal oxygen consumption. In addition, the molecular mechanisms involved in diabetic tubular growth are thought to set the stage for long-term progression of DKD76, in accordance with the observation that kidney enlargement is linked to the development of DKD161–165. Tubule cells that develop a senescence-like phenotype remain differentiated to some extent, but exhibit several abnormalities (including increased secretion of pro-inflammatory cytokines, increased production of growth factors and extracellular matrix, and resistance to apoptotic remodelling), changes that are independent of diabetes200. The senescence-like cell cycle arrest of tubular cells might act to prevent excessive proliferation of damaged cells, but also alters proximal tubular function and sets the stage for inflammation and fibrosis (FIGS 1,7).
TGFβ probably has a prominent role in linking the tubular growth phenotype of the diabetic kidney to inflammation, fibrosis, scarring and impairment of renal function176,201. Other prominent pathways include growth factors (such as PDGF, IGF1 and EGF), AngII, oxidative stress, the JAK–STAK pathway, the TSC2–mTOR pathway, PKCβ and epigenetic changes that have been implicated in renal growth and fibrosis of the diabetic kidney, reviewed elsewhere15,76,202(FIG. 7). For example, a 2011 study indicated that the TSC2–mTOR pathway, which is involved in tubular proliferation and hypertrophy, also promotes apoptosis of tubular epithelial cells in diabetes203. Findings from an extensive set of studies also suggest that the diabetes-induced inhibition of AMPK and hyperactivation of the mTOR signalling pathway have (via negative regulation of autophagy) a critical role in the pathogenesis of DKD172. Experimental studies indicate that SGLT2-mediated glucose uptake might contribute to the inhibition of autophagy in proximal tubules in diabetes, thereby contributing to the renoprotective effects of SGLT2 inhibitors62,204.
Conclusions
Diabetes mellitus is a leading cause of CKD that affects the kidney differently at different disease stages. In a subset of patients, the onset of diabetes mellitus induces kidney growth and glomerular hyperfiltration, which are risk factors for the development of DKD later in life. We propose a unifying tubular hypothesis of DKD based on the concept that the kidney aims to retain glucose in both euglycaemic and hyperglycaemic states. The glucose-retaining process involves sodium and glucose cotransport in the proximal tubule, which is augmented by diabetes-induced tubular growth. Both mechanisms, however, also increase the proximal tubular reabsorption of NaCl and fluid, which in turn induces glomerular hyperfiltration through tubuloglomerular feedback and the lowering of tubular back pressure to normalize renal NaCl and fluid excretion. The increase in GFR, however, is a burden for the kidney on multiple levels, including enhanced physical intraglomerular stress, increased renal transport and oxygen requirement, and enhanced exposure of the tubular system to tubulotoxic compounds.
The relevance of SGLT2 for glomerular hyperfiltration and the development of DKD has been demonstrated in patients with T2DM in large clinical outcome studies, although the therapeutic potential of SGLT2 inhibitors in patients with T1DM remains to be established. Experimental studies show that SGLT1 in the apical membrane of the macula densa senses enhanced luminal glucose delivery and increases GFR by enhancing the formation of NO, thereby extending the tubular hypothesis of nephron filtration to tubular segments downstream of the proximal tubule. The therapeutic potential of SGLT1 inhibition and of the additive effects of SGLT1 and SGLT2 inhibition remains to be determined. The molecular signature of diabetic tubular growth is also unique, and involves a senescence-like phenotype that is linked to pro-inflammatory and pro-fibrotic signalling that can set the stage for tubulointerstitial injury and development of DKD. Unidentified genetic and environmental factors might modulate the tubular response to hyperglycaemia, thereby contributing to the variation of kidney growth, tubular hyperreabsorption, glomerular hyperfiltration and the development of DKD in patients with diabetes (FIG. 1).
Hyperfiltration and/or an increase in blood pressure compensate for proximal hyperreabsorption of sodium via the SGLTs and tubular growth. Why there is no relevant neurohumoral downregulation of renal sodium reabsorption in the diabetic setting that could have a similar effect on sodium balance without burdening the kidney is unclear24. Further experimental and clinical studies are needed to fully understand the mechanisms that trigger diabetic tubular growth and the link between the molecular signature of tubular growth and the development and progression of DKD, including studies in patients with T2DM. Several questions remain to be answered, including the cellular consequences of tubular glucose uptake via apical and basolateral transporters and the extent to which the macula densa is involved in the phenotypic switch from hyperfiltration to hypertension in diabetes. Mathematical modelling suggests that transport mechanisms downstream of the macula densa affect diabetic hyperfiltration24, but the therapeutic potential of targeting these mechanisms remains to be tested. Finally, we need improved understanding of the tubular protective mechanisms that explain why the development of DKD occurs typically 10–20 years after the onset of diabetes, when many of the proposed deleterious molecular pathways are activated rapidly by elevated blood glucose levels, and why they eventually fail.
Key points.
- Rising blood glucose levels drive increased delivery of glucose into the Bowman’s space by glomerular filtration and cause increased reabsorption of glucose and sodium by the proximal tubule sodium–glucose cotransporters, SGLT2 and SGLT1, up to a transport maximum.
- Diabetes mellitus causes the kidney tubule to grow and increase its capacity for transport; most of this growth occurs in the proximal tubule, which increases glucose reabsorption and contributes to hyperglycaemia, and induces abnormally high reabsorption of filtered sodium chloride (NaCl).
- The increase in proximal reabsorption reduces NaCl and fluid delivery to the macula densa and induces glomerular hyperfiltration through tubuloglomerular feedback and a decline in tubular back pressure, which helps to normalize renal NaCl and fluid excretion.
- Glomerular hyperfiltration places physical stress on the filtration barrier and increases the demand for oxygen to reabsorb the filtered load; hypertrophic tubular cells exhibit a senescence-like phenotype, with pro-inflammatory and pro-fibrotic consequences that might promote the development of diabetic kidney disease (DKD).
- Not all patients with diabetes develop DKD, suggesting that the tubular growth and hyperreabsorption response must be subject to genetic or environmental influences; one such environmental factor is dietary NaCl, which mitigates hyperfiltration in diabetes by suppressing proximal reabsorption and thereby increasing the tubuloglomerular feedback signal.
- The clinical relevance of increased proximal reabsorption and hyperfiltration in diabetes is demonstrated by the ability of SGLT2 to improve renal outcomes in patients with diabetes, which has been verified in large-scale clinical trials.
- SGLT1 is a positive regulator of NOS1 in the macula densa, and acts as a glucose sensor to stimulate hyperfiltration and potentially regulate tubule growth in the diabetic kidney.
Glomerular ultrafiltration coefficient
A measure of the glomerular capillary membrane’s permeability to water; specifically, the volume of fluid filtered in unit time through a unit area of glomerular capillary membrane per unit pressure difference.
Tubuloglomerular feedback
A mechanism that inversely relates single nephron glomerular filtration rate to the NaCl concentration at the macula densa of the same nephron.
Tubular back pressure
The hydrostatic pressure in the Bowman’s space.
Filtration fraction
The fraction of renal plasma flow that is filtered by the glomeruli.
Gibbs free energy
The thermodynamic potential (or available energy) associated with a chemical reaction that can be used to do reversible work at a constant temperature and pressure.
Electrogenic
A type of transport process that leads to the translocation of net charge across the membrane and produces a change in the electrical potential of a cell.
Facilitated diffusion
The process of spontaneous passive transport of molecules or ions across a biological membrane along their electrochemical gradient, usually via a transmembrane integral protein.
Acknowledgements
The authors are supported by NIH grants R01DK112042 (V.V., S.C.T.), R01DK106102, R01HL142814, RF1AG061296 (V.V.), the UAB/UCSD O’Brien Center of Acute Kidney Injury NIH-P30DK079337 (V.V., S.C.T.) and the Department of Veterans Affairs (V.V., S.C.T.).
Footnotes
Competing interests
V.V. declares that he has served as a consultant and received honoraria from Astra-Zeneca, Bayer, Boehringer Ingelheim, Janssen Pharmaceutical, Eli Lilly, Merck and Retrophin, and has received grant support for investigator-initiated research from Astra-Zeneca, Bayer, Boehringer Ingelheim, Fresenius and Janssen. S.C.T. declares that he has received grant support for investigator-initiated research from Merck.
References
- 1.United States Renal Data System. 2015 Annual Data Report: Epidemiology of kidney disease in the United States, Bethesda, MD. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2015, https://www.usrds.org/2015/view/(2015). [Google Scholar]
- 2.Thomas MC et al. Diabetic kidney disease. Nat. Rev. Dis. Prim 1, 15018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jha V et al. Chronic kidney disease: global dimension and perspectives. Lancet 382, 260–272 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Papademetriou V et al. Chronic kidney disease and intensive glycemic control increase cardiovascular risk in patients with type 2 diabetes. Kidney Int 87, 649–659 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Lanting LC, Joung IM, Mackenbach JP, Lamberts SW & Bootsma AH Ethnic differences in mortality, end-stage complications, and quality of care among diabetic patients: a review. Diabetes Care 28, 2280–2288 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Salem RM et al. Genome-wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J. Am. Soc. Nephrol 30, 2000–2016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Zuydam NR et al. A genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes 67, 1414–1427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gheith O, Farouk N, Nampoory N, Halim MA & Al-Otaibi T Diabetic kidney disease: world wide difference of prevalence and risk factors. J. Nephropharmacol 5, 49–56 (2016). [PMC free article] [PubMed] [Google Scholar]
- 9.Vallon V & Komers R Pathophysiology of the diabetic kidney. Compr. Physiol 1, 1175–1232 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reidy K, Kang HM, Hostetter T & Susztak K Molecular mechanisms of diabetic kidney disease. J. Clin. Invest 124, 2333–2340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu J, Lee K, Chuang PY, Liu Z & He JC Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Renal Physiol 308, F287–F297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanwar YS, Sun L, Xie P, Liu FY & Chen S A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol 6, 395–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziyadeh FN & Wolf G Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr. Diabetes Rev 4, 39–45 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Hostetter TH Diabetic nephropathy. Metabolic versus hemodynamic considerations. Diabetes Care 15, 1205–1215 (1992). [DOI] [PubMed] [Google Scholar]
- 15.Vallon V & Thomson SC Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu. Rev. Physiol 74, 351–375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vallon V, Richter K, Blantz RC, Thomson S & Osswald H Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J. Am. Soc. Nephrol 10, 2569–2576 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Thomson SC, Vallon V & Blantz RC Kidney function in early diabetes: the tubular hypothesis of glomerular filtration. Am. J. Physiol. Renal Physiol 286, F8–F15 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Macisaac RJ et al. Nonalbuminuric renal insufficiency in type 2 diabetes. Diabetes Care 27, 195–200 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Mogensen CE, Christensen CK & Vittinghus E The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes 32, 64–78 (1983). [DOI] [PubMed] [Google Scholar]
- 20.Kramer HJ, Nguyen QD, Curhan G & Hsu CY Renal insufficiency in the absence of albuminuria and retinopathy among adults with type 2 diabetes mellitus. JAMA 289, 3273–3277 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Magee GM et al. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 52, 691–697 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Tonneijck L et al. Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J. Am. Soc. Nephrol 28, 1023–1039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vallon V & Thomson SC Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 60, 225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hallow KM, Gebremichael Y, Helmlinger G & Vallon V Primary proximal tubule hyperreabsorption and impaired tubular transport counterregulation determine glomerular hyperfiltration in diabetes: a modeling analysis. Am. J. Physiol. Renal Physiol 312, F819–F835 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallon V & Osswald H Dipyridamole prevents diabetes-induced alterations of kidney function in rats. Naunyn Schmiedebergs Arch. Pharmacol 349, 217–222 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Thomson S, Bao D, Deng A & Vallon V Adenosine formed by 5’-nucleotidase mediates tubuloglomerular feedback. J. Clin. Invest 106, 289–298 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollock CA, Lawrence JR & Field MJ Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am. J. Physiol 260, F946–F952 (1991). [DOI] [PubMed] [Google Scholar]
- 28.Vallon V et al. Salt-sensitivity of proximal reabsorption alters macula densa salt and explains the paradoxical effect of dietary salt on glomerular filtration rate in diabetes mellitus. J. Am. Soc. Nephrol 13, 1865–1871 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Vallon V, Blantz RC & Thomson S Homeostatic efficiency of tubuloglomerular feedback is reduced in established diabetes mellitus in rats. Am. J. Physiol 269, F876–F883 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Ditzel J, Lervang HH & Brochner-Mortensen J Renal sodium metabolism in relation to hypertension in diabetes. Diabete Metab 15, 292–295 (1989). [PubMed] [Google Scholar]
- 31.Mbanya JC, Thomas TH, Taylor R, Alberti KG & Wilkinson R Increased proximal tubular sodium reabsorption in hypertensive patients with type 2 diabetes. Diabet. Med 6, 614–620 (1989). [DOI] [PubMed] [Google Scholar]
- 32.Brochner-Mortensen J, Stockel M, Sorensen PJ, Nielsen AH & Ditzel J Proximal glomerulo-tubular balance in patients with type 1 (insulin-dependent) diabetes mellitus. Diabetologia 27, 189–192 (1984). [DOI] [PubMed] [Google Scholar]
- 33.Thomson SC et al. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol. Regul. Integr. Comp. Physiol 302, R75–R83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blantz RC, Singh P, Deng A, Thomson SC & Vallon V Acute saline expansion increases nephron filtration and distal flow rate but maintains tubuloglomerular feedback responsiveness: role of adenosine A(1) receptors. Am. J. Physiol. Renal Physiol 303, F405–F411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woods LL, Mizelle HL & Hall JE Control of renal hemodynamics in hyperglycemia: possible role of tubuloglomerular feedback. Am. J. Physiol 252, F65–F73 (1987). [DOI] [PubMed] [Google Scholar]
- 36.Vallon V et al. Adenosine A(1) receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus. Nephron Physiol 111, 30–38 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vallon V, Muhlbauer B & Osswald H Adenosine and kidney function. Physiol. Rev 86, 901–940 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Ren Y, Garvin JL, Liu R & Carretero OA Possible mechanism of efferent arteriole (Ef-Art) tubuloglomerular feedback. Kidney Int 71, 861–866 (2007) [DOI] [PubMed] [Google Scholar]
- 39.Sallstrom J et al. Diabetes-induced hyperfiltration in adenosine A(1)-receptor deficient mice lacking the tubuloglomerular feedback mechanism. Acta Physiol 190, 253–259 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Faulhaber-Walter R et al. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J. Am. Soc. Nephrol 19, 722–730 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinstein AM Osmotic diuresis in a mathematical model of the rat proximal tubule. Am. J. Physiol 250, F874–F884 (1986). [DOI] [PubMed] [Google Scholar]
- 42.Skrtic M et al. Influence of sex on hyperfiltration in patients with uncomplicated type 1 diabetes. Am. J. Physiol. Renal Physiol 312, F599–F606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vervoort G, Veldman B, Berden JH, Smits P & Wetzels JF Glomerular hyperfiltration in type 1 diabetes mellitus results from primary changes in proximal tubular sodium handling without changes in volume expansion. Eur. J. Clin. Invest 35, 330–336 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Hannedouche TP et al. Renal hemodynamics and segmental tubular reabsorption in early type 1 diabetes. Kidney Int 37, 1126–1133 (1990). [DOI] [PubMed] [Google Scholar]
- 45.Pruijm M et al. Glomerular hyperfiltration and increased proximal sodium reabsorption in subjects with type 2 diabetes or impaired fasting glucose in a population of the African region. Nephrol. Dial. Transplant 25, 2225–2231 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Thomson SC et al. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J. Clin. Invest 107, 217–224 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barfuss DW & Schafer JA Differences in active and passive glucose transport along the proximal nephron. Am. J. Physiol 241, F322–F332 (1981). [DOI] [PubMed] [Google Scholar]
- 48.Turner RJ & Moran A Heterogeneity of sodium-dependent D-glucose transport sites along the proximal tubule: evidence from vesicle studies. Am. J. Physiol 242, F406–F414 (1982). [DOI] [PubMed] [Google Scholar]
- 49.Wright EM, Loo DD & Hirayama BA Biology of human sodium glucose transporters. Physiol. Rev 91, 733–794 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Vrhovac I et al. Localizations of Na-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 467, 1881–1898 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Vallon V et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol 22, 104–112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sabolic I et al. Expression of Na+-D-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am. J. Physiol. Cell Physiol 302, C1174–C1188 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balen D et al. Revised immunolocalization of the Na+-D-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am. J. Physiol. Cell Physiol 295, C475–C489 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Rieg T et al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Renal Physiol 306, F188–F193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gorboulev V et al. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61 , 187–196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santer R & Calado J Familial renal glucosuria and SGLT2: from a Mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol 5, 133–141 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Martin MG, Turk E, Lostao MP, Kerner C & Wright EM Defects in Na+/glucose cotransporter (SGLT1) trafficking and function cause glucose-galactose malabsorption. Nat. Genet 12, 216–220 (1996). [DOI] [PubMed] [Google Scholar]
- 58.DeFronzo RA et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 36, 3169–3176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farber SJ, Berger EY & Earle DP Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J. Clin. Invest 30, 125–129 (1951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mogensen CE Maximum tubular reabsorption capacity for glucose and renal hemodynamcis during rapid hypertonic glucose infusion in normal and diabetic subjects. Scand. J. Clin. Lab. Invest 28, 101–109 (1971). [DOI] [PubMed] [Google Scholar]
- 61.Vallon V, Blantz RC & Thomson S Glomerular hyperfiltration and the salt paradox in early type 1 diabetes mellitus: a tubulo-centric view. J. Am. Soc. Nephrol 14, 530–537 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Vallon V et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Renal Physiol 304, F156–F167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vallon V et al. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Renal Physiol 306, F194–F204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kamran M, Peterson RG & Dominguez JH Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. J. Am. Soc. Nephrol 8, 943–948 (1997). [DOI] [PubMed] [Google Scholar]
- 65.Freitas HS et al. SLC2A2 gene expression in kidney of diabetic rats is regulated by HNF-1alpha and HNF-3beta. Mol. Cell Endocrinol 305, 63–70 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Chin E et al. Changes in facilitative glucose transporter messenger ribonucleic acid levels in the diabetic rat kidney. Endocrinol 138, 1267–1275 (1997). [DOI] [PubMed] [Google Scholar]
- 67.Dominguez JH, Camp K, Maianu L, Feister H & Garvey WT Molecular adaptations of GLUT1 and GLUT2 in renal proximal tubules of diabetic rats. Am. J. Physiol 266, F283–F290 (1994). [DOI] [PubMed] [Google Scholar]
- 68.Marks J, Carvou NJ, Debnam ES, Srai SK. & Unwin RJ Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J. Physiol 553,137–145 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hinden L et al. Modulation of renal GLUT2 by the. cannabinoid-1 receptor: implications for the treatment of diabetic nephropathy. J. Am. Soc. Nephrol 29, 434–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goestemeyer AK, Marks J, Srai SK, Debnam ES & Unwin RJ GLUT2 protein at the rat proximal tubule brush border membrane correlates with protein kinase C (PKC)-betal and plasma glucose. concentration. Diabetologia 50, 2209–2217 (2007) [DOI] [PubMed] [Google Scholar]
- 71.Song P et al. Knockout of Na-glucose-cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase-1 in macula densa and glomerular hyperfiltration. Am. J. Physiol. Renal Physiol 317, F207–F217 (2019).. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kellett GL, Brot-Laroche E, Mace OJ & Leturque A Sugar absorption in the intestine: the role of GLUT2. Annu. Rev. Nutr 28, 35–54 (2008). [DOI] [PubMed] [Google Scholar]
- 73.Rahmoune H et al. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54, 3427–3434 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Wang XX et al. SGLT2 expression is increased in human diabetic nephropathy: SGLT2 inhibition decreases renal lipid accumulation, inflammationand the development of nephropathy in diabetic mice. J. Biol. Chem 292, 5335–5348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seyer-Hansen K Renal hypertrophy in experimental diabetes: some functional aspects. J. Diabet. Complications 1, 7–10 (1987). [DOI] [PubMed] [Google Scholar]
- 76.Vallon V The proximal tubule in the pathophysiology of the diabetic kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol 300, R1009–R1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weinstein AM et al. Flow-dependent transport in a mathematical model of rat proximal tubule. Am. J. Physiol. Renal Physiol 292, F1164–F1181 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Fattah H, Layton A & Vallon V How do kidneys adapt to a deficit or loss in nephron number? Physiol 34, 189–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Osorio H et al. Effect of treatment with losartan on salt sensitivity and SGLT2 expression in hypertensive diabetic rats. Diabetes Res. Clin. Pract 86, e46–e49 (2008). [DOI] [PubMed] [Google Scholar]
- 80.Freitas HS et al. Na(+)-glucose transporter-2 messenger ribonucleic acid expression in kidney of diabetic rats correlates with glycemic levels: involvement of hepatocyte nuclear factor-1alpha expression and activity. Endocrinol 149, 717–724(2008). [DOI] [PubMed] [Google Scholar]
- 81.Ghezzi C & Wright EM Regulation of the human Na+dependent glucose cotransporter hSGLT2. Am. J. Physiol. Cell Physiol 303, C348–C354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tiwari S, Riazi S & Ecelbarger CA Insulin’s impact on renal sodium transport and blood pressure in health, obesity, and diabetes. Am. J. Physiol. Renal Physiol 293, F974–F984 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Toyoki D et al. Insulin stimulates uric acid reabsorption via regulating urate transporter 1 and ATP-binding cassette subfamily G member 2. Am. J. Physiol. Renal Physiol 313, F826–F834 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Novikov A et al. SGLT2 inhibition and renal urate excretion: role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol. Renal Physiol 316, F173–F185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC & Malnic G Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+exchanger isoform 3 activity in the renal proximal tubule. J. Am. Soc. Nephrol 25, 2028–2039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang W et al. Tubular NHE3 is a determinant of the acute natriuretic and chronic blood pressure lowering effect of the SGLT2 inhibitor empagliflozin. FASEB J 32, (Supplement No 1), 620.17 (2018). [Google Scholar]
- 87.Coady MJ et al. MAP17 is a necessary activator of renal Na+/glucose cotransporter SGLT2. J. Am. Soc. Nephrol 28, 85–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fu Y, Gerasimova M, Mayoux E, Masuda T & Vallon V SGLT2 inhibitor empagliflozin increases renal NHE3 phosphorylation in diabetic Akita mice: possible implications for the prevention of glomerular hyperfiltration. Diabetes 63, (supplement 1), A132 (2014). [Google Scholar]
- 89.Onishi A et al. Effect of renal tubule-specific knockdown of the Na(+)/H(+) exchanger NHE3 in Akita diabetic mice. Am. J. Physiol. Renal Physiol 317, F419–F434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin R et al. D-glucose acts via sodium/glucose cotransporter 1 to increase NHE3 in mouse jejunal brush border by a Na+/H+exchange regulatory factor 2-dependent process. Gastroenterology 140, 560–571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Masuda T et al. Unmasking a sustained negative effect of SGLT2 inhibition on body fluid volume in the rat. Am. J. Physiol. Renal Physiol 315, F653–F664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vallon V Tubular transport in acute kidney injury: relevance for diagnosis, prognosis and intervention. Nephron 134, 160–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zapata-Morales JR, Galicia-Cruz OG, Franco M & Martinez YM Hypoxia-inducible factor-1alpha (HIF-1alpha) protein diminishes sodium glucose transport 1 (SGLT1) and SGLT2 protein expression in renal epithelial tubular cells (LLC-PK1) under hypoxia. J. Biol. Chem 289, 346–357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schmidt C, Hocherl K, Schweda F, Kurtz A & Bucher M Regulation of renal sodium transporters during severe inflammation. J. Am. Soc. Nephrol 18, 1072–1083 (2007). [DOI] [PubMed] [Google Scholar]
- 95.Ghezzi C et al. SGLT2 inhibitors act from the extracellular surface of the cell membrane. Physiol. Rep 2, e12058 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fu Y et al. The organic anion transporter OAT3 enhances the glucosuric effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal Physiol 31 5, F386–F394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qiu H, Novikov A & Vallon V Ketosis and diabetic ketoacidosis in response to SGLT2 inhibitors: basic mechanisms and therapeutic perspectives. Diabetes Metab. Res. Rev 33, 5 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Ferrannini E, Mark M & Mayoux E CV Protection in the EMPA-REG OUTCOME trial: a “Thrifty Substrate” hypothesis. Diabetes Care 39, 1108–1114 (2016). [DOI] [PubMed] [Google Scholar]
- 99.Jurczak MJ et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes 60, 890–898 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Merovci A et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Invest 124, 509–514 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferrannini E et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Invest 124, 499–508 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hansen HH et al. The sodium glucose cotransporter type 2 inhibitor empagliflozin preserves beta-cell mass and restores glucose homeostasis in the male zucker diabetic fatty rat. J. Pharmacol. Exp. Ther 350, 657–664 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Macdonald FR et al. The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes. Metab 12, 1004–1012 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Nespoux J & Vallon V SGLT2 inhibition and kidney protection. Clin. Sci 132, 1329–1339 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kidokoro K et al. Evaluation of glomerular hemodynamic function by empagliflozin in diabetic mice using in vivo imaging. Circulation 140, 303–315 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Cherney DZ et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129, 587–597 (2014). [DOI] [PubMed] [Google Scholar]
- 107.van Bommel EJ, Muskiet MH, & van Baar MJB Dapagliflozin reduces measured GFR by reducing renal efferent arteriolar resistance in type 2 diabetes. Diabetes American Diabetes Association 79th Scientific Sessions, S-157 (Abstract) (2019).
- 108.Wanner C et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med 375, 323–334 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Perkovic V, Jardine M, Vijapurkar U & Meininger G Renal effects of canagliflozin in type 2 diabetes mellitus. Curr. Med. Res. Opin 31, 2219–2231 (2015). [DOI] [PubMed] [Google Scholar]
- 110.Heerspink HJ et al. Canagliflozin slows progression of renal function decline independently of glycemic effects. J. Am. Soc. Nephrol 28, 368–375 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perkovic V et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med 380, 2295–2306 (2019). [DOI] [PubMed] [Google Scholar]
- 112.Kohan DE et al. The effect of dapagliflozin on renal function in patients with type 2 diabetes. J. Nephrol 29, 391–400 (2016). [DOI] [PubMed] [Google Scholar]
- 113.Yale JF et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab 15, 463–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barnett AH et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2, 369–384 (2014). [DOI] [PubMed] [Google Scholar]
- 115.Zelniker TA et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet 393, 31–39 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Neal B et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med 377, 644–657 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Thomson SC & Vallon V Renal effects of sodium-glucose Co-transporter inhibitors. Am. J. Cardiol 124, S28–S35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Foote C, Perkovic V & Neal B Effects of SGLT2 inhibitors on cardiovascular outcomes. Diab. Vasc. Dis. Res 9, 117–123 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Lambers Heerspink HJ, de ZD, Wie L, Leslie B & List J Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes. Metab 15, 853–862 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zinman B et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med 373, 2117–2128 (2015). [DOI] [PubMed] [Google Scholar]
- 121.Kato ET et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation 139, 2528–2536 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Kanbay M et al. Uric acid in metabolic syndrome: from an innocent bystander to a central player. Eur. J. Intern. Med 29, 3–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lytvyn Y et al. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am. J. Physiol. Renal Physiol 308, F77–F83 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Chino Y et al. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug. Dispos 35, 391–404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vaduganathan M & Januzzi JL Jr. Preventing and treating heart failure with sodium-glucose co-transporter 2 inhibitors. Am. J. Cardiol 124, S20–S27 (2019). [DOI] [PubMed] [Google Scholar]
- 126.McMurray JJV et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med 381, 1995–2008 (2019). [DOI] [PubMed] [Google Scholar]
- 127.Cannon CP et al. Evaluating the effects of canagliflozin on cardiovascular and renal events in patients with type 2 diabetes and chronic kidney disease according to baseline HbA1c, Including those with HbA1c<7%: results from the CREDENCE trial. Circulation. 10.1161/CIRCULATIONAHA.119.044359 (2019). [DOI] [PubMed] [Google Scholar]
- 128.Ishizawa K et al. Inhibition of sodium glucose cotransporter 2 attenuates the dysregulation of Kelch-Like 3 and NaCl cotransporter in obese diabetic mice. J. Am. Soc. Nephrol 30, 782–794 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Monami M, Nardini C & Mannucci E Efficacy and safety of sodium glucose co-transport-2 inhibitorsin type 2 diabetes: a meta-analysis of randomized clinical trials. Diabetes Obes. Metab 16, 457–466 (2014). [DOI] [PubMed] [Google Scholar]
- 130.Layton AT, Vallon V & Edwards A Modeling oxygen consumption in the proximal tubule: effects of NHE and SGLT2 inhibition. Am. J. Physiol. Renal Physiol 308, F1343–F1357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Neill O et al. Acute SGLT inhibition normalizes oxygen tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Renal Physiol 309, F227–F234 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Layton AT, Vallon V & Edwards A Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am. J. Physiol. Renal Physiol 310, F1269–F1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sano M, Takei M, Shiraishi Y & Suzuki Y Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J. Clin. Med. Res 8, 844–847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Inzucchi SE et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care 41, 356–363 (2018). [DOI] [PubMed] [Google Scholar]
- 135.Layton AT & Vallon V SGLT2 inhibition in a kidney with reduced nephron number: modeling and analysis of solute transport and metabolism. Am. J. Physiol. Renal Physiol 314, F969–F984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nadkarni GN et al. Acute kidney injury in patients. on SGLT2 inhibitors: a propensity-matched analysis. Diabetes Care 40, 1479–1485 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Song P, Onishi A, Koepsell H & Vallon V Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert. Opin. Ther. Targets. 20, 1109–1125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vallon V The mechanisms and therapeutic potential. of SGLT2 inhibitors in diabetes mellitus. Annu. Rev. Med 66, 255–270 (2015). [DOI] [PubMed] [Google Scholar]
- 139.Powell DR et al. Improved glycemic control in mice. lacking Sglt1 and Sglt2. Am. J. Physiol. Endocrinol. Metab 304, E117–E130 (2013). [DOI] [PubMed] [Google Scholar]
- 140.Gallo LA, Wright EM & Vallon V Probing SGLT2 as a therapeutic target for diabetes: Basic physiology. and consequences. Diab. Vasc. Dis. Res 12, 78–89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gembardt F et al. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in. BTBR ob/ob type 2 diabetic mice with and without hypertension. Am. J. Physiol. Renal Physiol 307, F317–F325 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Han HJ, Lee YJ, Park SH, Lee JH & Taub M High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am. J. Physiol. Renal Physiol 288, F988–F996. (2005). [DOI] [PubMed] [Google Scholar]
- 143.Nespoux J et al. Gene deletion of the Na-glucose cotransporter SGLT1 ameliorates kidney recovery in a murine model of acute kidney injury induced by ischemia-reperfusion. Am. J. Physiol. Renal Physiol 316, F1201–F1210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wilcox CS et al. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc. Natl Acad. Sci. USA 89, 11993–11997 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu R, Carretero OA, Ren Y & Garvin JL Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int 67, 1837–1843 (2005). [DOI] [PubMed] [Google Scholar]
- 146.Vallon V et al. Feedback control of glomerular vascular tone in neuronal nitric oxide synthase knockout mice. J. Am. Soc. Nephrol 12, 1599–1606 (2001). [DOI] [PubMed] [Google Scholar]
- 147.Komers R, Lindsley JN, Oyama TT, Allison KM & Anderson S Role of neuronal nitric oxide synthase (NOS1) in the pathogenesis of renal hemodynamic changes in diabetes. Am. J. Physiol. Renal Physiol 279, F573–F583 (2000). [DOI] [PubMed] [Google Scholar]
- 148.Tolins JP, Shultz PJ, Raij L, Brown DM & Mauer SM Abnormal renal hemodynamic response to reduced renal perfusion pressure in diabetic rats: role of NO. Am. J. Physiol 265, F886–F895 (1993). [DOI] [PubMed] [Google Scholar]
- 149.Zhang J et al. Macula densa SGLT1-NOS1-TGF pathway — a new mechanism for glomerular hyperfiltration during hyperglycemia. J. Am. Soc Nephrol 30, 578–593 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thomson SC et al. Early diabetes as a model for testing the regulation of juxtaglomerular NOS I. Am. J. Physiol. Renal Physiol 287, F732–F738 (2004). [DOI] [PubMed] [Google Scholar]
- 151.Guyton AC Blood pressure control — special role of the kidneys and body fluids. Science 252, 1813–1816 (1991). [DOI] [PubMed] [Google Scholar]
- 152.Lu D et al. Salt-sensitive splice variant of nNOS expressed in the macula densa cells. Am. J. Physiol. Renal Physiol 298, F1465–F1471 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lu Y et al. Macula Densa nitric oxide synthase 1beta protects against salt-sensitive hypertension. J. Am. Soc. Nephrol 27, 2346–2356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang X et al. Inhibition of nitric oxide synthase 1 induces salt-sensitive hypertension in nitric oxide synthase 1alpha knockout and wild-type mice. Hypertension 67, 792–799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Welch WJ & Wilcox CS Role of nitric oxide in tubuloglomerular feedback: effects of dietary salt. Clin. Exp. Pharmacol. Physiol 24, 582–586 (1997). [DOI] [PubMed] [Google Scholar]
- 156.Thomson SC Nitric oxide mediates anomalous tubuloglomerular feedback in rats fed high-NaCl diet after subtotal nephrectomy. Am. J. Physiol. Renal Physiol 316, F223–F230 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Thomson SC et al. Temporal adjustment of the juxtaglomerular apparatus during sustained inhibition of proximal reabsorption. J. Clin. Invest 104, 1149–1158 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rieg T & Vallon V Development of SGLT1 and SGLT2 inhibitors. Diabetologia 61, 2079–2086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sims H, Smith KH, Bramlage P & Minguet J Sotagliflozin: a dual sodium-glucose co-transporter-1 and -2 inhibitor for the management of Type 1 and Type 2 diabetes mellitus. Diabet. Med 35, 1037–1048 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Cefalo CMA et al. Sotagliflozin, the first dual SGLT inhibitor: current outlook and perspectives. Cardiovasc. Diabetol 18, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zerbini G et al. Persistent renal hypertrophy and faster decline of glomerular filtration rate precede the development of microalbuminuria in type 1 diabetes. Diabetes 55, 2620–2625 (2006). [DOI] [PubMed] [Google Scholar]
- 162.Rigalleau V et al. Large kidneys predict poor renal outcome in subjects with diabetes and chronic kidney disease. BMC. Nephrol 11, 3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lawson ML, Sochett EB, Chait PG, Balfe JW & Daneman D Effect of puberty on markers of glomerular hypertrophy and hypertension in IDDM. Diabetes 45, 51–55 (1996). [DOI] [PubMed] [Google Scholar]
- 164.Bognetti E, Zoja A, Meschi F, Paesano PL & Chiumello G Relationship between kidney volume, microalbuminuria and duration of diabetes mellitus. Diabetologia 39, 1409 (1996). [PubMed] [Google Scholar]
- 165.Baumgartl HJ, Sigl G, Banholzer P, Haslbeck M & Standl E On the prognosis of IDDM patients with large kidneys. Nephrol. Dial. Transplant 13, 630–634 (1998). [DOI] [PubMed] [Google Scholar]
- 166.Wolf G & Ziyadeh FN Molecular mechanisms of diabetic renal hypertrophy. Kidney Int 56, 393–405 (1998). [DOI] [PubMed] [Google Scholar]
- 167.Chiarelli F, Gaspari S & Marcovecchio ML Role of growth factors in diabetic kidney disease. Horm. Metab. Res 41, 585–593 (2009). [DOI] [PubMed] [Google Scholar]
- 168.Kamesaki H, Nishizawa K, Michaud GY, Cossman J & Kiyono T TGF-beta 1 induces the cyclin-dependent kinase inhibitor p27Kip1 mRNA and protein in murine B cells. J. Immunol 160, 770–777 (1998). [PubMed] [Google Scholar]
- 169.Wolf G et al. High glucose stimulates expression of p27Kip1 in cultured mouse mesangial cells: relationship to hypertrophy. Am. J. Physiol 273, F348–F356 (1997). [DOI] [PubMed] [Google Scholar]
- 170.Huang JS, Chuang LY, Guh JY & Huang YJ Effects of nitric oxide and antioxidants on advanced glycation end products-induced hypertrophic growth in human renal tubular cells. Toxicol. Sci 111, 109–119 (2009). [DOI] [PubMed] [Google Scholar]
- 171.Chen JK, Chen J, Thomas G, Kozma SC & Harris RC S6 kinase 1 knockout inhibits uninephrectomy- or diabetes-induced renal hypertrophy. Am. J. Physiol. Renal Physiol 297, F585–F593 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yang D et al. Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol. Life Sci 75, 669–688 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Satriano J et al. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am. J. Physiol. Cell Physiol 299, C374–C380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Verzola D et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am. J. Physiol. Renal Physiol 295, F1563–F1573 (2008). [DOI] [PubMed] [Google Scholar]
- 175.Phillips AO, Steadman R, Morrisey K & Williams JD Polarity of stimulation and secretion of transforming growth factor-beta 1 by cultured proximal tubular cells. Am. J. Pathol 150, 1101–1111 (1997). [PMC free article] [PubMed] [Google Scholar]
- 176.Phillips AO & Steadman R Diabetic nephropathy: the central role of renal proximal tubular cells in tubulointerstitial injury. Histol. Histopathol 17, 247–252 (2002). [DOI] [PubMed] [Google Scholar]
- 177.Sokolovska J et al. Influence of metformin on GLUT1 gene and protein expression in rat streptozotocin diabetes mellitus model. Arch. Physiol. Biochem 116, 137–145 (2010). [DOI] [PubMed] [Google Scholar]
- 178.Lee MJ et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Renal Physiol 292, F617–F627 (2007). [DOI] [PubMed] [Google Scholar]
- 179.Lan R et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol 27, 3356–3367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Farsijani NM et al. Renal epithelium regulates erythropoiesis via HIF-dependent suppression of erythropoietin. J. Clin. Invest 126, 1425–1437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Heilig CW et al. GLUT1 regulation of the pro-sclerotic mediators of diabetic nephropathy. Am. J. Nephrol 38, 39–49 (2013). [DOI] [PubMed] [Google Scholar]
- 182.Pedersen SB, Flyvbjerg A & Richelsen B Inhibition of renal ornithine decarboxylase activity prevents kidney hypertrophy in experimental diabetes. Am. J. Physiol 264, C453–C456 (1993). [DOI] [PubMed] [Google Scholar]
- 183.Noh H & King GL The role of protein kinase C activation in diabetic nephropathy. Kidney Int. Suppl S49–S53 10.1038/sj.ki.5002386(2007). [DOI] [PubMed] [Google Scholar]
- 184.Meier M et al. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes 56, 346–354 (2007). [DOI] [PubMed] [Google Scholar]
- 185.Efendiev R, Bertorello AM & Pedemonte CH PKC-beta and PKC-zeta mediate opposing effects on proximal tubule Na+, K+-ATPase activity. FEBS Lett 456, 45–48 (1999). [DOI] [PubMed] [Google Scholar]
- 186.Vallon V, Wead LM & Blantz RC Renal hemodynamics and plasma and kidney angiotensin II in established diabetes mellitus in rats: effect of sodium and salt restriction. J. Am. Soc. Nephrol 5, 1761–1767 (1995). [DOI] [PubMed] [Google Scholar]
- 187.Vallon V et al. Effect of chronic salt loading on kidney function in early and established diabetes mellitus in rats. J. Lab. Clin. Med 130, 76–82 (1997). [DOI] [PubMed] [Google Scholar]
- 188.Lau C et al. Salt-resistant blood pressure and salt-sensitive renal autoregulation in chronic streptozotocin diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol 296, R1761–R1770 (2009). [DOI] [PubMed] [Google Scholar]
- 189.Miller JA Renal responses to sodium restriction in patients with early diabetes mellitus. J. Am. Soc. Nephrol 8, 749–755 (1997). [DOI] [PubMed] [Google Scholar]
- 190.Miracle CM, Rieg T, Mansoury H, Vallon V & Thomson SC Ornithine decarboxylase inhibitor eliminates hyperresponsiveness of the early diabetic proximal tubule to dietary salt. Am. J. Physiol. Renal Physiol 295, F995–F1002 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Birk C et al. The salt paradox of the early diabetic kidney is independent of renal innervation. Kidney Blood Press. Res 26, 344–350 (2003). [DOI] [PubMed] [Google Scholar]
- 192.Ekinci EI et al. Dietary salt intake and mortality in patients with type 2 diabetes. Diabetes Care 34, 703–709 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Thomas MC et al. The association between dietary sodium intake, ESRD, and all-cause mortality in patients with type 1 diabetes. Diabetes Care 34, 861–866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Holtkamp FA et al. An acute fall in estimated glomerular filtration rate during treatment with losartan predicts a slower decrease in long-term renal function. Kidney Int 80, 282–287 (2011). [DOI] [PubMed] [Google Scholar]
- 195.O’Bryan GT & Hostetter TH The renal hemodynamic basis of diabetic nephropathy. Semin. Nephrol 17, 93–100 (1997). [PubMed] [Google Scholar]
- 196.Pruijm M et al. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int 93, 932–940 (2018). [DOI] [PubMed] [Google Scholar]
- 197.Singh DK, Winocour P & Farrington K Mechanisms of disease: the hypoxic tubular hypothesis of diabetic nephropathy. Nat. Clin. Pract. Nephrol 4, 216–226 (2008). [DOI] [PubMed] [Google Scholar]
- 198.Magri CJ & Fava S The role of tubular injury in diabetic nephropathy. Eur. J. Intern. Med 20, 551–555 (2009). [DOI] [PubMed] [Google Scholar]
- 199.Thomas MC, Burns WC & Cooper ME Tubular changes in early diabetic nephropathy. Adv. Chronic. Kidney Dis 12, 177–186 (2005). [DOI] [PubMed] [Google Scholar]
- 200.Ren JL, Pan JS, Lu YP, Sun P & Han J Inflammatory signaling and cellular senescence. Cell Signal 21, 378–383 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ziyadeh FN et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl Acad. Sci. USA 97, 8015–8020 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Brosius FC, Tuttle KR & Kretzler M JAK inhibition in the treatment of diabetic kidney disease. Diabetologia 59, 1624–1627 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Velagapudi C et al. The tuberin/mTOR pathway promotes apoptosis of tubular epithelial cells in diabetes. J. Am. Soc. Nephrol 22, 262–273 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lee YH et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Renal Physiol 317, F767–F780 (2019). [DOI] [PubMed] [Google Scholar]
- 205.Vallon V Do tubular changes in the diabetic kidney affect the susceptibility to acute kidney injury? Nephron Clin. Pract 127, 133–138 (2014). [DOI] [PubMed] [Google Scholar]