Phage P22 tailspike protein: crystal structure of the head-binding domain at 2.3 Å, fully refined structure of the endorhamnosidase at 1.56 Å resolution, and the molecular basis of O-antigen recognition and cleavage (original) (raw)
Abstract
The tailspike protein of Salmonella phage P22 is a viral adhesion protein with both receptor binding and destroying activities. It recognises the O-antigenic repeating units of cell surface lipopolysaccharide of serogroup A, B and D1 as receptor, but also inactivates its receptor by endoglycosidase (endorhamnosidase) activity. In the final step of bacteriophage P22 assembly six homotrimeric tailspike molecules are non-covalently attached to the DNA injection apparatus, mediated by their N-terminal, head-binding domains. We report the crystal structure of the head-binding domain of P22 tailspike protein at 2.3 Å resolution, solved with a recombinant telluromethionine derivative and non-crystallographic symmetry averaging. The trimeric dome-like structure is formed by two perpendicular β-sheets of five and three strands, respectively in each subunit and caps a three-helix bundle observed in the structure of the C-terminal receptor binding and cleaving fragment, reported here after full refinement at 1.56 Å resolution. In the central part of the receptor binding fragment, three parallel β-helices of 13 complete turns are associated side-by-side, while the three polypeptide strands merge into a single domain towards their C termini, with close interdigitation at the junction to the β-helix part. Complex structures with receptor fragments from S. typhimurium, S. enteritidis and S. typhi253Ty determined at 1.8 Å resolution are described in detail. Insertions into the β-helix form the O-antigen binding groove, which also harbours the active site residues Asp392, Asp395 and Glu359. In the intact structure of the tailspike protein, head-binding and receptor-binding parts are probably linked by a flexible hinge whose function may be either to deal with shearing forces on the exposed, 150 Å long tailspikes or to allow them to bend during the infection process.
Keywords: virus protein, protein-carbohydrate recognition, endoglycosidase, telluromethionine, β-helix
Abbreviations: LPS, lipopolysaccharide; MIR, multiple isomorphous replacement; ncs, non-crystallographic symmetry
Introduction
Salmonella phage P22 is a dsDNA phage of the Podoviridae family, which is characterised by a short base plate or tail structure incorporated into one of the 12 5-fold vertices of the icosahedral phage head (Poteete, 1994; Figure 1). Its scaffolding protein assisted assembly pathway proceeds though a DNA-free procapsid structure Earnshaw and Casjens 1980, Casjens and Hendrix 1988. The structure of the scaffolding-containing procapsid, an empty procapsid, and the mature capsid with T = 7 laevo, analysed by cryo-electronmicroscopy at 22, 28 and 28 Å resolution, respectively, revealed a marked structural transition during DNA packing Prasad et al 1993, Thuman-Commike et al 1996. The formation of the short tail is initiated by incorporation of the portal protein 12-mer into one vertex of the procapsid during procapsid assembly together with 10 to 20 copies of each of three pilot proteins, gp7, gp16 and gp20 (Bazinet et al., 1988). The pilot proteins and the portal complex are required for infectivity but not for procapsid assembly (Botstein et al., 1973). Upon DNA packaging, proteins gp4, gp10 and gp26 are attached, forming the slender neck structure of the base plate. The portal vertex is both the site of DNA entry during phage maturation and DNA exit during infection. In the very last step of phage assembly tailspikes are attached to the neck structure. The tailspike protein, a homotrimer of 72 kDa subunits is anchored by its N-terminal, head-binding domain in a non-covalent but irreversible manner Berget and Poteete 1980, Maurides et al 1990. Three tailspikes are required on average to form an infectious phage (Israel, 1978).
Figure 1.
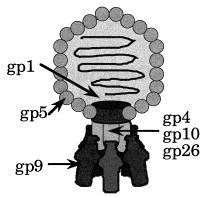
Diagram of Salmonella phage P22. The icosahedral head is formed by approximately 420 molecules gp5. The portal protein (gp1) 12-mer is inserted into one icosahedral facet. Up to six tailspike molecules (gp9) are attached to a slender neck structure formed by gp4, gp10 and gp26. This contact is mediated by the N-terminal, head-binding domain. Minor components (gp7, gp16 and gp20) are associated with the portal protein or the DNA.
The tailspike protein functions as a viral adhesion protein and binds to the O-antigenic repeating units of Salmonella host lipopolysaccharide (LPS), its cellular receptor (Israel et al., 1967). The number of O-antigenic repeating units of LPS varies greatly within the LPS pool of a cell. The distribution of the O-antigen chain length was reported to be unequal in S. typhimurium with 77% having between 19 and 34 repeating units, but two thirds of all LPS molecules being devoid of O side-chains (Palva & Mäkelä, 1980). Additional modifications, such as acetylation or glucosylation, known as O-antigen form variation, also contribute to microheterogeneity Kauffmann 1941, Goldman and Leive 1980, Palva and Makela 1980. The chemical structure of the O-antigen correlates with the serological classification of Salmonella as these are the main antigens upon gastrointestinal infections (Lüderitz et al., 1966).
The interaction of the tailspike protein with its receptor defines the host range of phage P22 comprising serotypes A, B and D1. These share a common trisaccharide repeating unit α-d-mannose-(1–-4)-α-l-rhamnose-(1–3)-α-d-galactose for the O-antigen and differ in their branching carbohydrate moieties, a 3,6-dideoxyhexose α-(1–3)-linked to d-mannose. Dideoxyhexoses are paratose (serogroup A), abequose (serotype B), or tyvelose (serotype D1). In addition, P22 tolerates the O-antigen 122, which shows an additional α-(1–4)-linked d-glucose at d-galactose as in S. typhi 253Ty.
The tailspike protein shows a receptor destroying endorhamnosidase activity cleaving the α(1,3)-_O_-glycosidic bond between rhamnose and galactose of the O-antigenic repeats. It produces mainly octasaccharide fragments of two repeating units with rhamnose at the reducing end Iwashita and Kanegasaki 1976, Eriksson and Lindberg 1977, Eriksson et al 1979.
Receptor destroying enzymatic activities are well known for viruses that use carbohydrates as receptors. Influenza A and B virus or paramyxovirus have neuraminidases (Air & Laver, 1989), whereas influenza virus C and some coronaviruses that recognise an _O_-acetylated sialic acid epitope have a sialate 9-_O_-acetylesterase, removing an acetyl group (Herrler et al., 1985). Endoglycosidase or acetylesterase activities have also been demonstrated for a large number of phages acting on encapsulated Gram-negative bacteria like Escherichia coli, Salmonella or Klebsiella Lindberg 1977, Svenson et al 1979.
Proteolytic fragmentation of intermediates in thermal unfolding and refolding of the tailspike protein suggested a domain border between residues 100 and 120. The C-terminal fragment (residues 109 to 666) maintains LPS binding properties and enzymatic activity of the intact tailspike protein and resembles the complete protein in its folding pathway Chen and King 1991, Danner et al 1993.
The crystal structure of the C-terminal part, lacking the N-terminal head-binding domain has been solved at 2.0 Å resolution (Steinbacher et al., 1994). We have recently analysed the crystal structure of the C-terminal fragment in complex with O-antigen fragments at 1.8 Å resolution comprising two repeating units, i.e. octa- or decasaccharides, derived from S. typhimurium (serogroup B), S. enteritidis (serogroup D1), and S. typhi253Ty (serotype D1, O-antigen 122), which shows additional α-(1–4)-linked d-glucose at d-galactose (Steinbacher et al., 1996).
Here we report on the crystal structure of the head-binding domain (residues 1 to 124 with an additional C-terminal hexa-His tag) at 2.3 Å resolution. The structure solution was mainly based on a telluromethionine containing derivative produced by overexpressing the protein in the Met auxotrophic E. coli strain B834(DE3)(hsd metB) in the presence of Ac-dl-Met(Te)-OH lithium salt (Karnbrock et al., 1996; N.B. et al., unpublished results). We also present the crystal structure of the C-terminal fragment after full refinement at 1.56 Å and O-antigen complexes determined at 1.8 Å in detail.
Results and discussion
Structure determination ofhead-binding domain
Fragmentation of the tailspike protein
The structures of a variety of plant, insect and animal viruses as well as those of a number of bacteriophages have been analysed by high resolution X-ray crystallography Harrison et al 1996, Liljas 1996. These studies mainly focus on icosahedral capsids, taking advantage of their internal symmetry both for crystallisation and structure solution. In contrast, only very limited structural information at atomic resolution is available on tail or fibre components of phages and viruses protruding from symmetric capsids or anchored in lipid membranes, although these are crucial for receptor recognition or viral entry into the host cell. Exceptions are haemagglutinin (Wilson et al., 1981) and neuraminidase (Varghese et al., 1983) from influenza A virus, tick-borne encephalitis envelope protein (Rey et al., 1995) and the receptor binding knob domain of adenovirus type 5 fibre protein (Xia et al., 1994).
The tailspike protein could be crystallised and subsequently analysed by X-ray crystallography by fragmentation into two structurally intact parts based on biochemical studies (Chen & King, 1991) and recombinant overexpression in E. coli. Stable, functional proteins were formed upon expression of residues 1 to 124 comprising the N-terminal, head-binding domain (Miller, 1995) and of the C-terminal part consisting of residues 109 to 666 (Danner et al., 1993).
Telluromethionine as heavy atom derivative
The crystal structure of the head-binding domain was solved by isomorphous replacement methods. Phasing was based on a strong telluromethionine derivative and a weak uranium derivative (Table 1). The incorporation of the amino acid analogues l-selenomethionine and l-telluromethionine into proteins overexpressed by recombinant DNA technology using Met auxotrophic E. coli strains opened the possibility to produce heavy atom derivatives of protein crystals on a rational basis. While use of synchrotron radiation is essential to take advantage of the anomalous contribution of selenium by MAD phasing (Hendrickson, 1991) in Met(Se)-derivatives, Met(Te)-derivatives show good phasing with CuKα-radiation due to the larger number of additional electrons. The incorporation of the tellurium analogue Met(Te) was hampered by the lack of an efficient chemical synthesis and mainly by its very limited stability under aqueous, aerobic conditions as in E. coli cultures. _N_-acetyltelluromethionine and its Te-hydroxylated form, Ac-dl-Met(Te)-OH rapidly formed under aerobic conditions, were demonstrated to have a significantly increased stability. Ac-dl-Met(Te) has a half-life of about 20 hours compared to 30 minutes for free l-Met(Te) in non-degassed aqueous solution at pH 7.0 and room temperature, which is sufficient for bioincorporation (Karnbrock et al., 1996). Both compounds can be used directly for efficient high level incorporation of Met(Te) residues as demonstrated for a variety of proteins (Budisa et al., 1995; and unpublished results). Met(Te) residues are stable when they are buried in the interior of the protein.
Table 1.
Data collection and phasing statistics for the head-binding domain
| Parameters | Native | TEMEa | UOACb | UOTEc |
|---|---|---|---|---|
| Total observations | 48,766 | 64,266 | 33,849 | 35,960 |
| Unique reflections | 20,822 | 28,937 | 18,290 | 18,811 |
| Resolution (Å) | 2.6 | 2.3 | 2.7 | 2.7 |
| Completeness (%)d | 96.5/72.2 | 95.4/82.9 | 94.7/84.0 | 97.4/86.5 |
| Rmerge (%)e | 7.6/29.8 | 5.6/17.4 | 8.2/32.1 | 9.8/28.4 |
| Riso (25–3.0 Å)f | — | 0.21 | 0.21 | 0.25 |
| Heavy atom sites | — | 6 | 9 | 7 |
| RCullisg | — | 0.62/0.69 | 0.93/0.92 | 0.65/0.71 |
| Phasing powerh | — | 2.16/1.41 | 2.0/1.29 | 0.61/0.45 |
For structure determination of the head-binding domain the refined coordinates of Te were used to derive _ncs_-operators for two trimers in the asymmetric unit. The MIR phased electron density was improved by sixfold _ncs_-averaging. The model was subjected to crystallographic refinement with _ncs_-restraints.
The final crystallographic _R_-factor for the head-binding domain was 19.9% for data from 8.0 to 2.3 Å resolution using data set TEME and 18.6% using native data from 8.0 to 2.6 Å resolution for refinement. The _rms_-deviations between _ncs_-related protein molecules were 0.28 and 0.29 Å, respectively for all protein atoms. The quality of the final model is summarised in Table 2.
Table 2.
Parameters of the refined model of the head-binding domain with datset TEME at 2.3 Å and Native at 2.6 Å resolution.
| Parameter | TEME | Native |
|---|---|---|
| Unique reflections I > 2σ(I) | 28,206 | 18,825 |
| Active protein atoms (excl. H) | 4782 | 4782 |
| Active water atoms | 432 | 432 |
| R.m.s.-deviations of _ncs_-related atoms (Å) | 0.28 | 0.29 |
| R.m.s. bonded _B_-factors (Å2) | 3.1 | 3.3 |
| R.m.s.-deviation from target values: | ||
| Bond length (Å) | 0.011 | 0.009 |
| Bond angles (deg.) | 1.55 | 1.49 |
| R-factor (%)a for I > 2σ(I) | 19.9 | 18.6 |
Structure of the head-binding domain
The model of the N-terminal domain comprises residues 5 to 108 according to the numbering of Sauer et al. (1982), which does not include the N-terminal Met. Residues 1 to 4, the preceding Met and residues 109 to 124 as well as the C-terminal hexa-His tag are not visible in the electron density and are probably disordered or mobile. All residues lie within or near energetically allowed regions of (Φ, ψ) values (Ramachandran & Sasisekharan, 1968).
The molecular structure of each monomer of the homotrimer is characterised by two regular β-sheets, A and B, oriented nearly perpendicular to each other and composed of five and three strands, respectively. The direction of the strands with respect to the molecular triad is almost parallel for β-sheet A, whereas β-sheet B is perpendicular to the triad. The topology of the strands is exclusively antiparallel (Figure 2). The N-terminal residues 7 to 9 from a neighbouring subunit extend β-sheet B by one strand forming three regular hydrogen-bonds with residues 81 to 83. Each monomer is stabilised by a hydrophobic core built of eight Val, nine Ile, one Pro, one Phe, one Met and two Leu residues.
Figure 2.
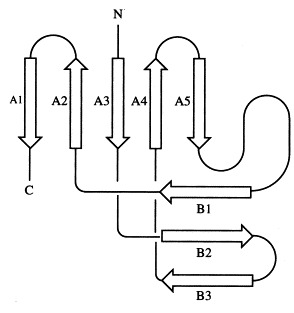
Schematic representation of the topology of one subunit of the head-binding domain. β-Sheet A comprises five strands (A1 from 100 to 109, A2 from 89 to 94; A3 from 29 to 32, A4 from 65 to 67, A5 from 72 to 74), β-sheet B comprises three strands (B1 from 81 to 83, B2 from 49 to 54, B3 from 56 to 62). In the trimer, β-sheet B is extended by the N-terminal strand of a neighbouring subunit (residues 7 to 9). The secondary structure was analysed with DSSP (Kabsch & Sander, 1983).
In the trimer the monomers form a dome-like structure 30 Å high and about 55 Å in diameter (Figure 3(a)). The two β-sheets do not form a barrel structure but the walls of the dome. Prominent features that stabilise the trimer structure are a cluster of residues Phe22 that close the dome at its top (Figure 3(b)) and the N-terminal strand that extends on the inner surface of the molecule to the neighbouring subunit (Figure 3(c)). Polypeptide arms extending over or under domains of neighbouring subunits intertwining with others are a remarkable feature of virus structures (Harrison et al., 1996). The contacts between adjacent subunits are, however, not very extensive and involve a salt bridge formed between Arg13 and Asp100#, a hydrophobic contact between Lys80 and Tyr108 and hydrogen bonds between Lys71 and Thr17# and between Arg20 and the carbonyl group of Gly76# (# denotes residues from a neighbouring subunit).
Figure 3.
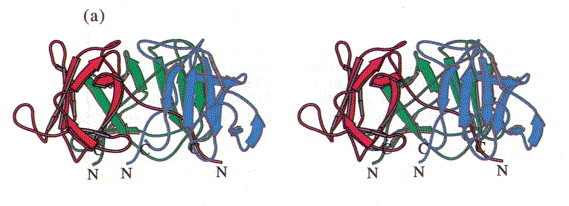


Stereoview of the head-binding domain. The β-sheets A and B form the walls of a dome-like structure. (a) Side view perpendicular to the trimer axis. The side contacts between the subunits are not very extensive. (b) Top view. A cluster of the three Phe22 residues at the molecular triad stabilises the head-binding domain. (c) Bottom view. The N-terminal polypetide strands extend to the neighbouring subunits and shield the single Met89 from the solvent, which was replaced by Met(Te) for heavy atom derivative preparation. The Figure was produced with MOLSCRIPT (Kraulis, 1991).
Implications for trimer stability
During thermal denaturation, the N-terminal domain is the first part of the tailspike protein to unfold (Chen & King, 1991). The treatment of a thermal unfolding intermediate with proteases results in complete destruction of the N-terminal part up to residues 107 to 120, whereas the C-terminal part remains intact. The N-terminal domain is dispensable for thermostability and SDS resistance of the intact protein, and its deletion has only minor effects on tailspike folding kinetics (Danner et al., 1993).
A single point mutation produced by chemical mutagenesis, Asp100Asn has been reported to influence the head-binding properties of tailspike protein. The mutant’s affinity for phage heads is only 1% compared to the wild-type protein and its N-terminal domain is denatured by SDS at room temperature. A set of intragenic second site mutations (Arg13Ser, Arg13Leu and Arg13His) suppresses the Asp100Asn phenotype (Schwarz & Berget, 1989). Both residues, Asp100 and Arg13, are located at the subunit interface and form an inter-subunit salt bridge. Arg13 protrudes from the N-terminal strand running across the inner surface of the trimer. Both residues are partly solvent accessible and located at the bottom of a shallow cleft formed at the subunit interface. The effects of the mutation can be rationalised by the electrostatic interaction within the salt bridge.
Surface properties and implications forhead-binding
A marked clustering of charged residues is present at the interface of the subunits, extending to the top of the molecule. Hydrophilic residues predominate at exposed loop structures, whereas small patches of hydrophobic residues are exposed in the centre of the outer surface of β-sheet A. The electrostatic surface potential of the N-terminal domain is therefore dominated by areas of neutral potential as seen in Figure 4. Regions of negative and only small ones of positive potential are concentrated at the subunit interface.
Figure 4.
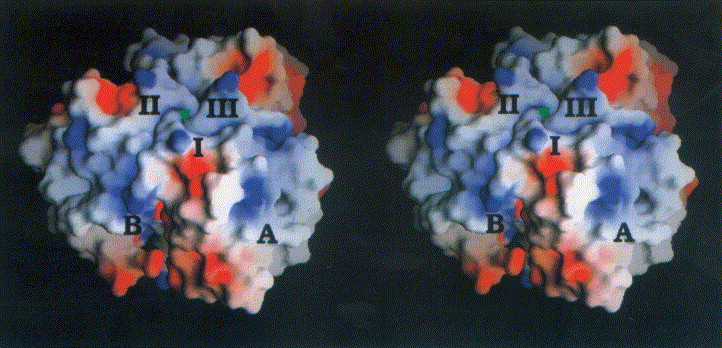
Stereoview of the electrostatic potential of the head-binding domain from −7kT/e− (red) to +7kT/e− (blue). The three monomers are labelled I to III. The view is perpendicular to the molecular triad (green triangle). Charged residues mainly cluster at the domain interface, whereas the centre of β-sheet A is dominated by polar and hydrophobic residues corresponding to a neutral potential. The saltbridge Arg19/Asp100# is located at the subunit interface (B). The Figure was produced with GRASP (Nicholls et al., 1993).
It is not known which protein of the neck structure binds to the N-terminal domain of the tailspike protein. Prominent loop structures and the hydrophobic centre of β-sheet A are likely candidates for head binding.
Structure of the C-terminal fragment: the right-handed parallel β-helix motif
The crystal structure of the C-terminal fragment, resembling the intact protein in enzymatic activity, receptor binding properties and temperature stability was initially refined to a 2.0 Å resolution (Steinbacher et al., 1994). Data to 1.56 Å resolution were collected at the beamline BW6 at the HASYLAB of the DESY in Hamburg. The model was subsequently refined to a final _R_-factor of 17.1% with data between 8 and 1.56 Å resolution (Table 3).
Table 3.
Data collection and refinement of the receptor-binding and catalytic fragment (residues 109 to 666) with synchrotron data
| Data collection (13.2 to 1.56 Å/1.60 to 1.56 Å) | |
|---|---|
| Total measurements | 277,488/8131 |
| Unique reflections | 77,437/4497 |
| _R_merge (%)a | 7.7/16.2 |
| Completeness (%) | 92.1/46.0 |
| Refinement (8.0 to 1.56 Å) | |
| Unique reflections for I > 0σ(I) | 76,961 |
| Active protein atoms (excl. H) | 4107 |
| Active water atoms | 365 |
| R.m.s. bonded _B_-factors (Å2) | 2.7 |
| R.m.s-deviation from target values: | |
| Bond length (Å) | 0.010 |
| Bond angles (deg.) | 1.65 |
| _R_-factor (%)b for I 0σ(I) | 17.1 |
The homotrimeric molecule is 133 Å in length and between 35 and 80 Å in diameter (Figure 5). Each subunit is composed of six segments, together resembling the shape of a fish. They correspond to the mouth, the main body, the dorsal fin, and the first, second and third segment of the caudal fin, respectively. In addition, three insertions in the main body group together to form a ventral fin. In terms of secondary structure, the main body comprises a parallel β-helix (residues 143 to 540) of 13 complete turns, a domain motif also present in pectate lyases, from Erwinia and Bacillus Yoder et al 1993, Pickersgill et al 1994, Yoder and Jurnak 1995, alkaline protease from Pseudomonas aeruginosa (Baumann et al., 1993), and the virulence factor P.69 pertactin from Bordetella pertussis (Emsley et al., 1996). A left-handed β-helix has been identified in UDP-_N_-acetylglucosamine acyltransferase from E. coli (Raetz & Roderick, 1995) and in carbonic anhydrase from Methanosarcina thermophila (Kisker et al., 1996). The β-helix motifs of the tailspike protein, pectate lyase and pertactin share a common triangular cross-section, but differ in their oligomer state as pectate lyases and pertactin are monomeric proteins. Both structures with a left-handed domain motif, which has a much more regular triangular cross-section, are trimeric and share considerable overall similarities despite diverse functions.
Figure 5.
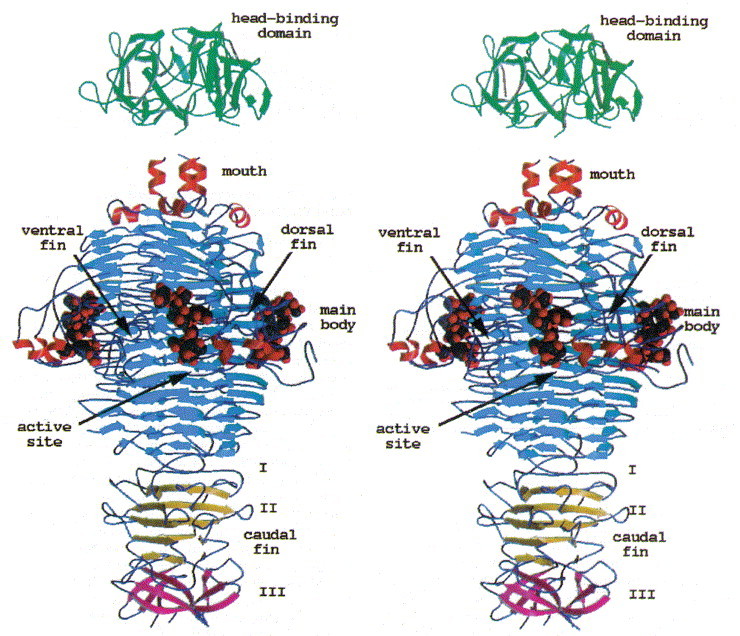
Stereoview of the tailspike protein with bound O-antigen receptor fragment from S. typhi253Ty. The head-binding domain caps a three helix-bundle present at the C terminus of the receptor binding and catalytic fragment. The active site is located 80 Å above the C terminus.
In the trimer of the tailspike protein, adjacent β-helices form a β-sandwich with about 66 residues involved in the intersubunit contact. As the majority of these residues is either polar (18) or charged (18) the interface is hydrophilic. A wide channel along the molecular triad harbours about 132 ordered water molecules, whereas the interior of the β-helix is mainly filled with hydrophobic residues and only 12 ordered water molecules. A complete β-helix turn comprises approximately 22 residues in the N-terminal and central part, decreasing to 16 residues in the C-terminal part. Stacks of hydrophobic side-chains are formed by aliphatic or aromatic residues, but repetitive elements extend only up to three residues (Figure 6). The mouth segment preceding the main body is an extension of the β-helix by one turn, formed by a β-strand and a short seven-residue α-helix. At the N terminus of the endorhamnosidase fragment, a seven-residue α-helix forms a three helix bundle in the trimer. The dorsal fin domain (residues 197 to 259) is inserted into the β-helix and forms a compact domain of mainly irregular structure and a strongly twisted triple-stranded β-sheet of topology 2-1-3 and only four regular hydrogen bonds.
Figure 6.

Structurally based amino acid alignment within the β-helix main body of the tailspike protein. Each row corresponds to a β-helix turn (1 to 13) and each column to a ladder of aligned residues, pointing either into the interior of the β-helix (i) or to the outside (a). Dots indicate insertion or deletions into the β-helix. The sheets termed A (blue), B (yellow) and C (green) (Steinbacher et al., 1994) correspond to the sheets PB2, PB3 and PB1 in pectate lyase C from Ewinia chrysanthemi(Yoder et al., 1993). Only short repetitive elements of identical amino acids are present.
In contrast to the main body with independently folded subunits, the three polypeptides merge into a single domain at the caudal fin. The latter consists of three segments. In the first segment, each polypeptide folds around the molecular triad, interdigitating with the others. The second segment is folded into a five-stranded antiparallel β-sheet, which is extended by two parallel strands from the first caudal fin segments of the other two subunits. The seven-stranded sheets form a prism-like structure in the trimer. The third segment of the caudal fin constitutes the C-terminal end of the tailspike protein, where a propeller is formed by three-stranded antiparallel β-sheets. Each β-sheet participates in two six-stranded β-barrels, one with each of the two remaining polypeptides. Only hydrophobic residues are buried in the interior of the caudal fin domain.
Connection between the head andreceptor-binding parts
The N-terminal fragment (residues 1 to 124) shows electron density for residues 5 to 108, whereas in the structure of the C-terminal fragment (residues 109 to 666) residues 109 to 112 are disordered. The N-terminal three-helix bundle from residues 113 to 120 present at the mouth segment of the C-terminal fragment was chosen as an overlapping segment in the construction of the expression vector for the head-binding domain. It is stabilised by hydrophobic interactions between aromatic and aliphatic residues and was therefore expected to be ordered in the N-terminal fragment as well. Although the absence of electron density for residues 109 to 124 in the N-terminal fragment could be due to complete disorder of this part, it could also be explained by mobility of the stable three-helix bundle with respect to the compact head-binding domain. Simple docking experiments between both fragments show that the three-helix bundle cannot penetrate the N-terminal dome very deeply and therefore only few direct contacts between both fragments are possible. This is suggestive of a hinge function of the connecting part between both fragments, which might be necessary either to deal with shearing forces on the exposed 150 Å long molecule or might have a functional role during the infection process, allowing the tailspikes to bend away from the DNA injection apparatus.
Binding of O-antigen receptor fragments
The infection of Salmonella by phage P22 starts with the specific recognition of O-antigenic repeating units of LPS by the tailspikes. This interaction defines the host range comprising serotypes A, B and D1. The binding cleft and the active site of the tailspike protein have recently been identified by X-ray analyses of complexes with receptor fragments (Steinbacher et al., 1996), which was confirmed by mutational data (Baxa et al., 1996). The bound carbohydrates Figure 7, Figure 8were purified from S. typhimurium (serotype B), S. enteritidis (serotype D1) and S. typhi253Ty (serotype D1, O-antigen 122) after partial digestion with the phage P22 endorhamnosidase. They comprise two O-antigenic repeats and represent the multiple host specificity of phage P22 and its tolerance to O-antigen 122 introduced by random α(1–4)-glucosylation at d-glucose due to form variation. The shape and location of the receptor binding site in the tailspike protein are ideal to recognise an elongated receptor molecule like LPS O-antigen repeats (Figure 9).
Figure 7.
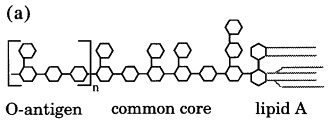
(a) Schematic structure of Salmonella LPS, consisting of a lipid A membrane anchor, a common core of carbohydrates and a varying number of 4 to 30 O-antigenic repeats. (b) Chemical structure of Salmonella O-antigenic repeats of serotypes A (Par, paratose), B (Abe, abequose) and D1 (Tyv, tyvelose) α-(1,3)-linked to Man of the trisaccharide repeat α-Man-(1–4)-α-Rha-(1–3)-α-Gal. Randomly α-(1–4)-linked Glc represents O-antigen 122. The arrow marks the cleavage site.
Figure 8.
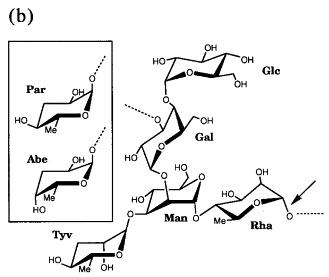
Stereoview of the 2_F_o − _F_c electron density of (a) S. typhimurium and (b) S. enteritidis O-antigen octasaccharides at 1.8 Å resolution contoured at 1σ. Repeating unit I is proximal, repeating unit II distal from the active site.
Figure 9.
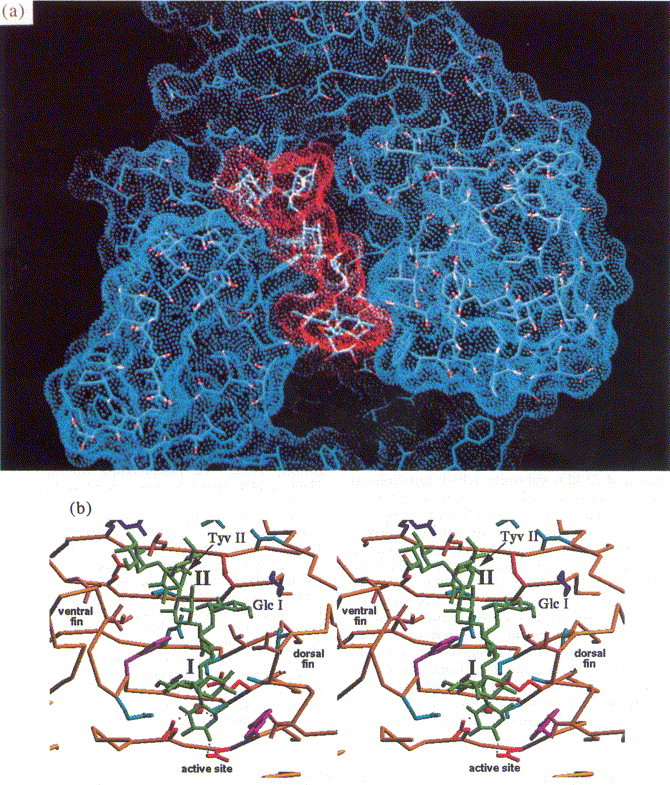
(a) Solvent accessible surface of O-antigen binding site (blue) with bound O-antigen octasaccharide from S. enteritidis (red). (b) Stereoview of the binding site. The O-antigen receptor is recognised in a 20 Å cleft formed between the dorsal and the ventral fin domain.
Active-site topology and enzyme mechanism
The terminal rhamnose at the reducing end of the bound product of the endorhamnosidase is located at the lower part of the binding site towards the C terminus of the protein. The electron density of the terminal rhamnose shows a discontinuity between C2 and C3, which is present in all three complexes determined and probably reflects some conformational flexibility or heterogeneity (Figure 10). However, the terminal rhamnose fits the electron density best in a twisted boat conformation, which is often observed for carbohydrates bound to the active site of a glycosidase. The C1 hydroxyl group is clearly present in α-configuration, as in the substrate. It is hydrogen-bonded to Asp392 with a distance of 2.7 Å. A water molecule is located 2.9 Å below the anomeric C-atom of the terminal rhamnose. This water molecule is H-bonded to Glu359, Ser360 and Asp395 with distances of 2.7, 2.6 and 2.8 Å, respectively. In addition, Glu359 forms a salt bridge with Lys363. Residue Trp391 contributes to the binding of the terminal rhamnose by a hydrophobic contact to the 6-methyl group and it additionally may influence the hydrophobicity in the environment of Asp392, which probably serves as general acid (Figure 11). The mutants Glu359Gln, Asp392Asn and Asp395Asn were shown to be enzymatically inactive without reducing the substrate binding affinity with binding constants of about 1 × 106 M−1 for the octasaccharide product or dodecasaccharide substrate (Baxa et al., 1996).
Figure 10.
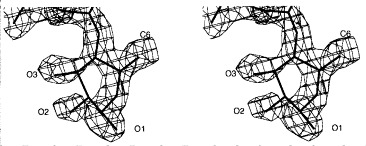
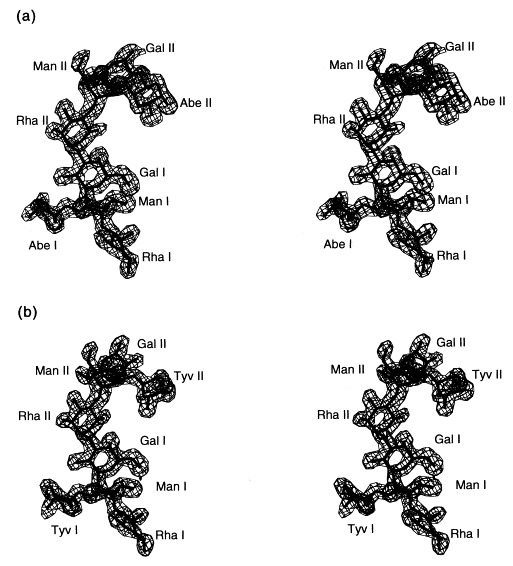
Stereoview of the 2_F_o − _F_c electron density of the terminal rhamnose I (Rha I) of the O-antigen fragment from S. typhi253Ty bound to the active site at 1.8 Å resolution contoured at 1σ. The discontinuity of the electron density between C2 and C3 is also observed for S. typhimurium and S. enteritidis receptor fragments. The main conformation adopted by Rha I corresponds to a twisted boat conformation with C1 in α-configuration as in the substrate.
Figure 11.
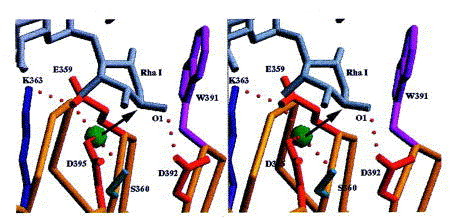
Stereoview of the active site with Rha I of the bound product. The steric situation suggests an inverting mechanism. A water molecule tightly bound also in the absence of the carbohydrate is activated by Glu359 and Asp395 as general bases. Asp392 serves as general acid and is H-bonded to O1 of Rha I.
It is not known to which class of endoglycosidases, inverting or retaining, the tailspike protein belongs. Both classes share two acidic residues as a common feature. The distance between both is assumed to be a valuable indicator of the enzymatic mechanism, as average distances are about 4.5 to 5.5 Å and 9.0 to 10.0 Å for retaining and inverting endoglycosidases, respectively. The larger distance for inverting enzymes originates from the necessity to harbour both the substrate and a catalytic water molecule between the general acid and the general base McCarter and Withers 1994, Davies and Henrissat 1995. The active site topology of the tailspike protein shows distances of 5.8 Å (Asp392, Asp395), 8.2 Å (Asp392, Glu359) and 7.1 Å (Asp395, Glu359). The distance of 5.8 Å between Asp392 and Asp395 is comparable to the distance range observed for retaining enzymes. In that case Asp395 would serve as nucleophile and attack the anomeric C-atom. The distance of 4.0 Å between the anomeric C-atom of Rha I and Asp395 would make a different binding mode for the substrate necessary, with a disruption of the hydrophobic contact of the 6-methyl group to Trp391. Taking into account the precise location of the extended product in the binding cleft, it is likely that substrate and product bind similarly. However, even if Asp395 would serve as a nucleophile, no acidic residue is present on the opposite side of the carbohydrate to activate a potential water molecule as nucleophile in a double displacement mechanism. On the other hand, the steric situation is ideal for a direct attack on the anomeric C-atom of rhamnose by the bound water molecule, activated by Glu359 and Asp395 as general bases. Therefore, an inverting mechanism with Glu359 and Asp395 as general bases and Asp392 as general acid is proposed.
Multiple host specificity and O-antigen form variation
In O-antigenic repeating unit I, which is proximal to the active site, the 3,6-dideoxyhexose points to the solvent region and makes only van der Waals contacts to Val240 and Ser237 of the dorsal fin domain by its 6-methyl group. The 6-methyl is common to the recognised 3,6-dideoxyhexoses paratose, abequose and tyvelose. Therefore, the position of repeating unit I is almost identical in the three complex structures determined by X-ray crystallography. In contrast, the 3,6-dideoxyhexose of repeating unit II points into a shallow depression. The stereochemistry of the 3,6-dideoxyhexoses abequose and tyvelose is clearly seen in the electron density, as shown for repeating unit II (Figure 12). The accommodation of abequose or tyvelose of serotypes B and D1 is achieved by positional wobbling between two acidic residues, Asp303 and Glu309 (Figure 13). Alternative interactions achieved by positional wobbling combined with water-mediated protein carbohydrate contacts creates a binding subsite with multiple specificities.
Figure 12.
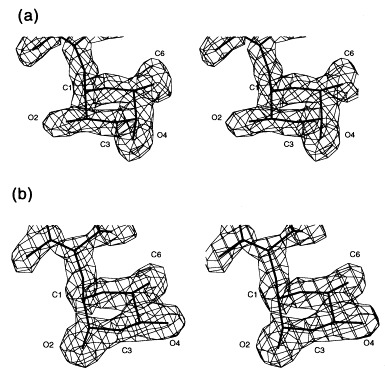
Stereoview of the 2_F_o − _F_c electron density of the 3,6-dideoxyhexoses in repeating unit II pointing towards the protein surface. (a) Abequose (serotype B) and (b) tyvelose (serotype D1) at 1.8 Å resolution contoured at 1σ.
Figure 13.
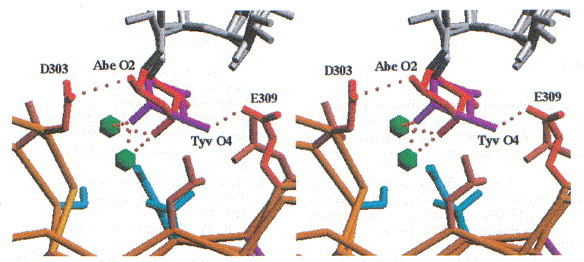
Stereoview of the binding site for 3,6-dideoxyhexoses in repeating unit II. Multiple host specificity is achieved by alternative contacts between the 3,6-dideoxyhexose and the protein, which are partly mediated by two water molecules. The distances between Abe O2 and Asp303 and Tyv O4 and Glu309 are 2.7 and 3.2 Å, respectively.
O-antigen form variation introduces d-glucose residues α-1,4 linked to d-galactose of the trisaccharide backbone. There is only electron density for the glucose of repeating unit I (Figure 14), although it is also present in 80% of repeating unit II of the S. typhi253Ty O-antigen fragments (Weintraub et al., 1988) used for soaking experiments. The electron density for Gal II is much weaker in the case of the S. typhi253Ty O-antigen complex then in the complexes with the homogeneous S. typhimurium and S. enteritidis octasaccharides. No density is visible for Glc II of the S. typhi253Ty decasaccharide, which might be explained by the lack of a well defined subsite for the additional d-glucose at the upper entrance of the binding cleft.
Figure 14.
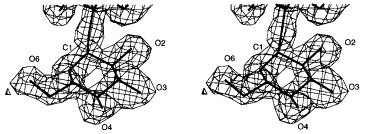
Stereoview of the 2_F_o − _F_c electron density of glucose in repeating unit I of S. typhi253Ty O-antigen introduced by form variation at 1.8 Å resolution contoured at 1σ.
The d-glucose of repeating unit I in S. typhi253Ty O-antigens is accommodated in a well defined additional subsite (Figure 15). The occupation of this subsite requires Lys302 to change its conformation. This is the only major side-chain rearrangement observed upon O-antigen binding.
Figure 15.
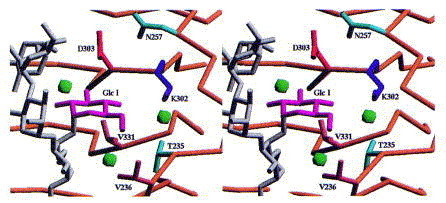
Stereoview of the binding site for glucose I of O-antigen 122 introduced by form variation. Lys302 has to reorient its side-chain to open the binding site. This is the only rearrangement of side-chains observed upon receptor binding.
Comparison to influenza virus hemagglutinin
The overall dimensions of the tailspike protein and influenza virus haemagglutinin (Wilson et al., 1981) are rather similar. Both are trimeric, elongated molecules of 150 and 135 Å length, respectively, either bound to the neck structure of a bacteriophage via a specialised domain by strong but non-covalent protein-protein interactions or fixed in the outer membrane of an enveloped virus via a transmembrane anchor. Nature has solved the requirements for both proteins by a very different architecture. The molecular axis of haemagglutinin comprises a long three-α-helix bundle as a characteristic feature, whereas the central structural element of the tailspike protein is a three-β-helix bundle. The receptor binding site of haemagglutinin is located in a carbohydrate binding domain at its tip, comprising a jelly-roll fold as often observed in viral coat proteins (Weis et al., 1988). The receptor binding site of the tailspike protein is located at the centre of the molecule. Its cleft-structure is ideal to recognise the elongated O-antigen polymer of LPS. The different locations and architectures of the binding sites reflects different substrate types.
Role of receptor cleavage in the phage life cycle
For the tailspike protein, the kinetics of receptor binding, which occurs rapidly and reversibly, suggest that the interaction with the receptor still allows the phage to browse over the surface of the host cell by rapid release and rebinding of the receptor (Baxa et al., 1996). Therefore, the low enzymatic activity will not contribute much to lateral mobility and it will certainly not give the tailspike protein the function of a drill in order to remove the O-antigen from the LPS and make a potential second receptor accessible. However, the activity is high enough to cleave a few O-antigen chains during the infection process. The location of the active site proximal to the cell surface with respect to the binding site is compatible with a liberating function to release newly assembled phages after cell lysis from the cell debris.
Studies on the role of receptor destroying activities have been reported for influenza A virus (Liu et al., 1995) and human parainfluenza 3 virus (Huberman et al., 1995), where binding and receptor destroying activity are either separated on haemagglutinin and neuraminidase or combined in one protein, the haemagglutinin-neuraminidase, respectively. The loss or reduction of receptor destroying neuraminidase activity does not prevent viral entry but blocks multicycle infections due to aggregation of virus particles or delayed virus release.
Point mutants of the tailspike protein defective in endorhamnosidase activity but not in receptor binding activity will allow further studies on the role of the enzymatic activity during infection and phage detachment in vivo.
Conclusions
The tailspike protein of Salmonella phage P22 is a homotrimer of 72 kDa subunits, which functions as viral adhesion protein with a very low receptor destroying enzymatic activity. The head-binding domain and the C-terminal part are probably linked in a flexible manner. Two O-antigenic repeating units are sufficient for receptor binding. They are recognised in a 20 Å long cleft in the centre of the 150 Å long molecule, which is located between insertions into the right-handed parallel β-helix main body of the molecule. The active-site topology suggests an inverting endoglycosidase mechanism with Asp392 as general acid and a catalytic water molecule activated by Glu359 and Asp395 as general bases. The multiple host specificity is achieved by a wobble mechanism between two acidic residues, Asp303 and Glu309 in a subsite for 3,6-dideoxyhexoses (paratose, abequose and tyvelose), which also involves water-mediated contacts to the protein as an essential feature. The glucose introduced by O-antigen form variation is accommodated in an additional subsite for the O-antigen repeating unit proximal to the active site.
Materials and methods
Crystallization and heavy-atom derivative preparation
The N-terminal domain of the tailspike protein (residues 1 to 124 with a C-terminal hexa-His tag) was overexpressed in E. coli Bl21(DE3) under the control of a T7 promoter system and purified by Ni2+-affinity chromatography (Miller, 1995). l-Met(Te) was incorporated as described by Budisa et al. (1995). Briefly, the methionine auxotrophic strain E. coli B834(DE3)(hsd metB) harbouring the expression plasmid was grown on minimal medium with a limited amount of Met(S) (0.05 mM) to an A 600 of 1.0, harvested, washed and resuspended in minimal medium. After induction with 1 mM IPTG, Ac-dl-Met(Te)-OH lithium salt (Karnbrock et al., 1996) was added to a final concentration of 1 mM. Cells were incubated for another 12 hours under vigorous shaking at 37°C. Purification was done as for the wild-type protein (Miller, 1995). The head-binding domain in 50 mM Hepes/NaOH (pH 6.5) was concentrated to 10 mg/ml using a Centricon 30 device (Amicon).
Initial crystallization experiments were performed with Crystal Screen I and II (Hampton Research). In the final condition 5 μl of protein solution were mixed with 3 to 5 μl of reservoir solution containing 20% (w/v) polyethylene glycol 8000, 0.2 M MgCl2, 0.1 M bis-Tris-HCl (pH 6.6) and equilibrated against 5 ml reservoir solution by vapour diffusion. Crystals of approximately 0.2 mm were obtained by seeding the hanging drop 6 to 12 hours after mixing with a small single crystal. Met(Te) material gave larger crystals compared to wild-type protein. Crystals were harvested in the reservoir solution. Uranium derivatives were prepared by soaking either native or Met(Te) crystals in reservoir solution with 15 mM UO2(SO4) for one day.
Data collection
Measurement of X-ray intensities was carried out using a MARresearch image plate (MARresearch, Hamburg) and CuKα-radiation from a Rigaku RU 200 X-ray generator operated at 5.4 kW at 16°C. Images were processed with MOSFLM (Leslie, 1991) and integrated intensities scaled and merged with AGROVATA, ROTAVATA and TRUNCATE of the CCP4 package (CCP4, 1994). The crystals were of space group _P_2(1) with cell dimensions a = 57.3 Å, b = 82.1 Å, c = 73.8 Å, α = 90°, β = 90.9°, γ = 90° and two trimers in the asymmetric unit, resulting in a Matthews coefficient (Matthews, 1968) of 2.05 Å3 Da−1.
Data to 1.56 Å resolution of the C-terminal fragment (residues 109 to 666) were collected at room temperature using a MARresearch image plate (MARresearch, Hamburg) at the beamline BW6 of the HASYLAB at DESY (Hamburg). Data were processed with MOSFLM (Leslie, 1991) and integrated intensities scaled and merged with AGROVATA, ROTAVATA and TRUNCATE of the CCP4 package (CCP4, 1994). The resolution limit of the images was chosen to have I > 5σ(I) in the outermost resolution shell using the program X-DAMAGE (Löwe, 1995).
Structure determination and refinement
The selfrotation function, calculated with GLRF (Tong & Rossmann, 1990) showed only one peak when searching for triads, consistent with almost parallel molecular axes for both asymmetric trimers. The uranium derivative (UOTE) was identified using Met(Te) crystals as parent native crystals as these were easier to grow and diffracted to higher resolution due to their larger size. Six sites were identified in the Met(Te) derivative (TEME) using difference Patterson and difference Fourier maps with the PROTEIN package (Steigemann, 1991). A double derivative UOTE was subsequently prepared. Phases for difference Fourier maps were calculated with MLPHARE (CCP4, 1994) and improved with DM (CCP4, 1994) by solvent flattening and histogram mapping. The MIR phases, calculated with three derivatives, had an overall figure of merit of 0.57 in the resolution range 20 to 3 Å. The _ncs_-operators were derived from the refined tellurium positions with the program O (Jones et al., 1991). An electron density modified with SOLOMON (Abrahams & Leslie, 1996) was used to construct a molecular envelope for one trimer with MAMA (Kleywegt & Jones, 1993). _ncs_-averaging within and between the trimers was done with AVE (Kleywegt & Jones, 1994). The atomic model was built into the resulting electron density with FRODO (Jones, 1978) on an Evans & Sutherland ESV. Refinement was carried out with X-PLOR (Brünger, 1992) using parameters derived by Engh & Huber (1991). The structure was refined with _ncs_-restraints against native and Met(Te) derivative data at 2.6 and 2.3 Å, respectively.
Acknowledgements
We thank Dr H. D. Bartunik for help in data collection at beamline BW6 at the HASYLAB at DESY, Hamburg and Professor Dr L. Moroder and Dr W. Karnbrock (Max-Planck-Institut für Biochemie, Martinsried) for the grateful gift of telluromethionine compounds. The work was supported by grants from the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie (S.M., U.B. and R.S.).
Footnotes
References
- Abrahams J.P., Leslie A.G.W. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallog. sect. D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- Air G.M., Laver W.G. The neuraminidase of influenza virus. Proteins: Struct. Funct. Genet. 1989;6:341–356. doi: 10.1002/prot.340060402. [DOI] [PubMed] [Google Scholar]
- Baumann U., Wu S., Flaherty K.M., McKay D.B. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: a two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 1993;12:3357–3364. doi: 10.1002/j.1460-2075.1993.tb06009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxa U., Steinbacher S., Miller S., Weintraub A., Huber R., Seckler R. Interactions of phage P22 tails with their cellular receptor, Salmonella O-antigen polysaccharide. Biophys. J. 1996:2040–2048. doi: 10.1016/S0006-3495(96)79402-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazinet C., Benbasat J., King J. Purification and organization of gene 1 portal protein required for phage P22 DNA packaging. Biochemistry. 1988;27:1849–1856. doi: 10.1021/bi00406a009. [DOI] [PubMed] [Google Scholar]
- Berget P.B., Poteete A.R. Structure and function of the bacteriophage P22 tail protein. J. Virol. 1980;34:234–243. doi: 10.1128/jvi.34.1.234-243.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D., Waddell C.H., King J. Mechanism of head assembly and DNA encapsulation in Salmonella phage P22: I. Genes, proteins, structures and DNA maturation. J. Mol. Biol. 1973;80:669–695. doi: 10.1016/0022-2836(73)90204-0. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. X-PLOR, Version 3.1: A System for Crystallography and NMR. Yale University Press; New Haven, CT: 1992. [Google Scholar]
- Budisa N., Steipe B., Demange P., Eckerskorn C., Kellermann J., Huber R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur. J. Biochem. 1995;230:788–796. doi: 10.1111/j.1432-1033.1995.tb20622.x. [DOI] [PubMed] [Google Scholar]
- Casjens S., Hendrix R. Control mechanisms in dsDNA bacteriophage assembly. In: Calendar R., editor. vol. 1. Plenum Press; New York: 1988. pp. 15–91. (The Bacteriophages). [Google Scholar]
- Chen B.-L., King J. Thermal unfolding pathway for the thermostable P22 tailspike endorhamnosidase. Biochemistry. 1991;30:6260–6269. doi: 10.1021/bi00239a026. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallog. sect. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Danner M., Fuchs A., Miller S., Seckler R. Folding and assembly of phage P22 tailspike endorhamnosidase lacking the N-terminal, head-binding domain. Eur. J. Biochem. 1993;215:653–661. doi: 10.1111/j.1432-1033.1993.tb18076.x. [DOI] [PubMed] [Google Scholar]
- Davies G., Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3:853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- Earnshaw W., Casjens S. DNA packaging by double stranded DNA bacteriophages. Cell. 1980;21:319–325. doi: 10.1016/0092-8674(80)90468-7. [DOI] [PubMed] [Google Scholar]
- Engh R.A., Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallog. sect. A. 1991;47:392–400. [Google Scholar]
- Emsley P., Charles I.G., Fairweather N.F., Isaacs N.W. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature. 1996;381:90–92. doi: 10.1038/381090a0. [DOI] [PubMed] [Google Scholar]
- Eriksson U., Lindberg A.A. Adsoption of phage P22 to Salmonella typhimurium. J. Gen. Virol. 1977;34:207–221. doi: 10.1099/0022-1317-34-2-207. [DOI] [PubMed] [Google Scholar]
- Eriksson U., Svenson S.B., Lönngren J., Lindberg A.A. Salmonella phage glycanases: substrate specificity of the phage P22 endo-rhamnosidase. J. Gen. Virol. 1979;43:503–511. doi: 10.1099/0022-1317-43-3-503. [DOI] [PubMed] [Google Scholar]
- Goldman R.C., Leive L. Heterogeneity of antigen-side-chain length in lipopolysaccharide from Escherichia coli 0111 and Salmonella typhimurium LT2. Eur. J. Biochem. 1980;107:145–153. doi: 10.1111/j.1432-1033.1980.tb04635.x. [DOI] [PubMed] [Google Scholar]
- Harrison S.C., Skehel J.J., Wiley D.C. Virus structure. In: Fields N.B., Knipe D.M., Howley P.M., editors. Fields Virology. 3rd edit. Raven Publishers; Philadelphia: 1996. pp. 59–99. [Google Scholar]
- Hendrickson W.A. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science. 1991;254:51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- Herrler G., Rott R., Klenk H.-D., Müller H.-P., Shukla A.K., Schauer R. The receptor-destroying enzyme of influenza C virus is neuraminate-O-acetylesterase. EMBO J. 1985;4:1503–1506. doi: 10.1002/j.1460-2075.1985.tb03809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman K., Peluso R.W., Moscona A. Hemagglutinin-neuraminidase of human parainfluenza 3: role of the neuraminidase in the viral life cycle. Virology. 1995;214:294–300. doi: 10.1006/viro.1995.9925. [DOI] [PubMed] [Google Scholar]
- Israel J.V., Anderson T.F., Levine M. In vitro morphogenesis of phage P22 from heads and base-plate parts. Proc. Natl Acad. Sci. USA. 1967;57:284–291. doi: 10.1073/pnas.57.2.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel V. A model for the adsorption of phage P22 to Salmonella typhimurium. J. Gen. Virol. 1978;40:669–673. doi: 10.1099/0022-1317-40-3-669. [DOI] [PubMed] [Google Scholar]
- Iwashita S., Kanegasaki S. Enzymatic and molecular properties of base-plate parts of bacteriophage P22. Eur. J. Biochem. 1976;65:87–94. doi: 10.1111/j.1432-1033.1976.tb10392.x. [DOI] [PubMed] [Google Scholar]
- Jones T.A. A graphics model building & refinement system for macromolecules. J. Appl. Crystallog. 1978;11:268–272. [Google Scholar]
- Jones T.A., Zou J.-Y., Kjelgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog. sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Karnbrock W., Weyher E., Budisa N., Huber R., Moroder L. A new efficient synthesis of acetytelluro- and acetylselenomethionine and their use in the biosynthesis of heavy-atom protein analogs. J. Am. Chem. Soc. 1996;118:913–914. [Google Scholar]
- Kauffmann F. A typhoid variant and a new serological variation in the Salmonella group. J. Bacteriol. 1941;41:127–140. doi: 10.1128/jb.41.2.127-140.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisker C., Schindelin H., Alber B.E., Ferry J.G., Rees D.C. A left-handed β-helix revealed by the crystal structure of a carbonic anhydrase from the archeon Methanosarcina thermophile. EMBO J. 1996;15:2323–2330. [PMC free article] [PubMed] [Google Scholar]
- Kleywegt G.J., Jones T.A. Masks made easy. ESF/CCP4 Newsletter. 1993;28:56–59. [Google Scholar]
- Kleywegt G.J., Jones T.A. From First Map to Final Model (CCP4) Daresbury Laboratory; Warrington WA4 4AD, UK: 1994. Halloween … masks and bones; pp. 59–66. [Google Scholar]
- Kraulis P.J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 1991;24:946–950. [Google Scholar]
- Leslie A.G.W. CCP4 & ESF-EACMB Newsletters on Protein Crystallography. SERC Laboratory; Daresbury, Warrington WA4 4AD, UK: 1991. Recent changes to the MOSFLM package for processing film and image plate data. [Google Scholar]
- Liljas L. Viruses. Curr. Opin. Struct. Biol. 1996;6:151–156. doi: 10.1016/s0959-440x(96)80068-6. [DOI] [PubMed] [Google Scholar]
- Lindberg A.A. Bacteriophage surface carbohydrates and bacteriophage adsorption. In: Sutherland I., editor. Surface Carbohydrates of the Procaryotic Cell. Academic Press; New York: 1977. pp. 289–356. [Google Scholar]
- Liu C., Eichelberger M.C., Compans R.W., Air G.M. Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J. Virol. 1995;69:1099–1106. doi: 10.1128/jvi.69.2.1099-1106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwe J. Röntgenstrukturanalyse des 20S Proteasoms aus Thermoplasma acidophilum. Technische Universität München; 1995. Thesis. [Google Scholar]
- Lüderitz O., Straub A.M., Westphal O. Bacteriol. Rev. 1966;30:192–255. doi: 10.1128/br.30.1.192-255.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B.W. Solvent content of protein crystals. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- Maurides P.A., Schwarz J.J., Berget P.B. Intragenic suppression of a capsid assembly-defective P22 tailspike mutation. Genetics. 1990;125:673–681. doi: 10.1093/genetics/125.4.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter J.D., Withers S.G. Mechnisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 1994;4:885–892. doi: 10.1016/0959-440x(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Miller S. Deletions- und Punktmutanten des P22 Tailspike-Proteins. Universität Regensburg; 1995. Thesis. [Google Scholar]
- Nicholls A., Bharadwaj R., Honig B. Grasp-graphical representation and analysis of surface properties. Biophys. J. 1993;64:A166. [Google Scholar]
- Palva E.T., Mäkelä H. Lipopolysaccharide Heterogeneity in Salmonella typhimurium analyzed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis. Eur. J. Biochem. 1980;107:137–143. doi: 10.1111/j.1432-1033.1980.tb04634.x. [DOI] [PubMed] [Google Scholar]
- Pickersgill R., Jenkins J., Harris G., Nasser W., Robert-Baudouy J. The structure of Bacillus subtilis pectate lyase in complex with calcium. Nature Struct. Biol. 1994;1:717–723. doi: 10.1038/nsb1094-717. [DOI] [PubMed] [Google Scholar]
- Poteete A.R. P22 bacteriophage. In: Webster R.G., Granoff A., editors. Encyclopedia of Virology. Academic Press; London: 1994. pp. 1009–1013. [Google Scholar]
- Prasad B.V.V., Prevelige P.E., Marietta E., Chen R.O., Thomas D., King J., Chiu W. Three-dimensional transformation of capsids associated with genome packaging in a bacterial virus. J. Mol. Biol. 1993;231:65–74. doi: 10.1006/jmbi.1993.1257. [DOI] [PubMed] [Google Scholar]
- Raetz C.R.H., Roderick L. A left-handed parallel β helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- Ramachandran G.N., Sasisekharan V. Conformation of polypeptides and proteins. Advan. Protein Chem. 1968;23:283–437. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- Rey F.A., Heinz F.X., Mandl C., Kunz C., Harrison S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- Sauer R.T., Krovatin W., Poteete A.R., Berget P.B. Phage P22 tail protein: gene and amino acid sequence. Biochemistry. 1982;21:5811–5815. doi: 10.1021/bi00266a014. [DOI] [PubMed] [Google Scholar]
- Schwarz J.J., Berget P.B. Characterization of bacteriophage P22 tailspike mutant proteins with altered endorhamnosidase and capsid assembly activities. J. Biol. Chem. 1989;264:20112–20119. [PubMed] [Google Scholar]
- Steigemann W. Recent advances in the PROTEIN program system for the X-ray structure analysis of biological macromolecules. In: Moras D., Podjarny A.D., Thiery J.C., editors. Crystallographic Computing V. Oxford Univ. Press; Oxford: 1991. pp. 115–125. [Google Scholar]
- Steinbacher S., Seckler R., Miller S., Steipe B., Huber R., Reinemer P. Crystal structure of P22 tailspike protein: interdigitated subunits in a thermostable trimer. Science. 1994;265:383–386. doi: 10.1126/science.8023158. [DOI] [PubMed] [Google Scholar]
- Steinbacher S., Baxa U., Miller S., Weintraub A., Seckler R., Huber R. Crystal structure of phage P22 tailspike protein complexed with Salmonella sp. O-antigen receptors. Proc. Natl Acad. Sci. USA. 1996;93:10584–10588. doi: 10.1073/pnas.93.20.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenson S.B., Lönngren J., Carlin N., Lindberg A.A. Salmonella bacteriophage glycanases: endorhamnosidases of Salmonella typhimurium bacteriophages. J. Virol. 1979;32:583–592. doi: 10.1128/jvi.32.2.583-592.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuman-Commike P.A., Greene B., Jakana J., Prasad B.V.V., King J., Prevelige P.E., Chiu W. Three-dimensional structure of scaffolding-containing phage P22 procapsids by electron cryo-microscopy. J. Mol. Biol. 1996;260:85–98. doi: 10.1006/jmbi.1996.0383. [DOI] [PubMed] [Google Scholar]
- Tong L., Rossmann M.G. The locked rotation function. Acta Crystallog. sect. A. 1990;46:783–792. doi: 10.1107/s0108767390005530. [DOI] [PubMed] [Google Scholar]
- Varghese J.N., Laver W.G., Colman P.M. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature. 1983;303:35–40. doi: 10.1038/303035a0. [DOI] [PubMed] [Google Scholar]
- Weintraub A., Lindberg A.A., Lipniunas P., Nilsson B. Heterogeneity in oligosaccharides from the O-polysaccharide chain of the lipopolysaccharide from Salmonella typhi253Ty determined by fast atom bombardment mass spectrometry. Glycoconjugate J. 1988;5:207–213. [Google Scholar]
- Weis W., Brown J.H., Cusack S., Paulson J.C., Skehel J.J., Wiley D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature. 1988;333:426–431. doi: 10.1038/333426a0. [DOI] [PubMed] [Google Scholar]
- Wilson I.A., Skehel J.J., Wiley D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Xia D., Henry L.J., Gerard R.D., Deisenhofer J. Crystal structure of the receptor-binding domain of adenovirus type 5 fiber protein at 1.7 Å resolution. Structure. 1994;2:1259–1270. doi: 10.1016/s0969-2126(94)00126-x. [DOI] [PubMed] [Google Scholar]
- Yoder M.D., Jurnak F. The parallel β helix and other coiled folds. FASEB J. 1995;9:335–342. doi: 10.1096/fasebj.9.5.7896002. [DOI] [PubMed] [Google Scholar]
- Yoder M.D., Noel T.K., Jurnak F. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science. 1993;260:1503–1607. doi: 10.1126/science.8502994. [DOI] [PubMed] [Google Scholar]