Induction of Apoptosis after Expression of PYK2, a Tyrosine Kinase Structurally Related to Focal Adhesion Kinase (original) (raw)
Abstract
Many cells (e.g., epithelial cells) require attachment to the extracellular matrix (ECM) to survive, a phenomenon known as anchorage-dependent cell survival. Disruption of the cell–ECM interactions mediated by the integrin receptors results in apoptosis. Focal adhesion kinase (FAK), a 125-kD protein tyrosine kinase activated by integrin engagement, appears to be involved in mediating cell attachment and survival. Proline-rich tyrosine kinase 2 (PYK2), also known as cellular adhesion kinase β (CAKβ) and related adhesion focal tyrosine kinase, is a second member of the FAK subfamily and is activated by an increase in intracellular calcium levels, or treatment with TNFα and UV light. However, the function of PYK2 remains largely unknown. In this study, we show that over-expression of PYK2, but not FAK, in rat and mouse fibroblasts leads to apoptotic cell death. Using a series of deletion mutants and chimeric fusion proteins of PYK2/FAK, we determined that the NH2-terminal domain and tyrosine kinase activity of PYK2 were required for the efficient induction of apoptosis. Furthermore, the apoptosis mediated by PYK2 could be suppressed by over-expressing catalytically active v-Src, c-Src, phosphatidylinositol-3-kinase, or Akt/protein kinase B. In addition, it could also be suppressed by overexpressing an ICE or ICE-like proteinase inhibitor, crmA, but not Bcl2. Collectively, our results suggest that PYK2 and FAK, albeit highly homologous in primary structure, appear to have different functions; FAK is required for cell survival, whereas PYK2 induces apoptosis in fibroblasts.
Homeostasis of multicellular organisms is controlled not only by the proliferation and differentiation of cells but also by cell death (Raff, 1992). Programmed cell death, or apoptosis, is characterized by the presence of nuclear and cytoplasmic condensation and segmentation and is an important regulatory event in embryogenesis, metamorphosis, endocrine-dependent tissue atrophy, and normal tissue turnover (Raff, 1992; Nagata and Golstein, 1995; Steller, 1995; Muzio et al., 1996). Disregulation of apoptosis contributes to the pathogenesis of several diseases, including cancers, neurodegenerative disorders, immunodeficiency, and autoimmune diseases (Thompson, 1995). Although the intracellular mediators that induce apoptosis are beginning to be defined, relatively little is known about the mechanisms by which cell death programs are executed.
Apoptosis can be triggered by a variety of extrinsic and intrinsic signals. Extrinsic inducers of apoptosis include TNF family proteins (e.g., Fas ligand and TNFα), calcium, growth factor withdrawal, and loss of extracellular matrix (ECM)1 attachment (Nagata and Golstein, 1995; Thompson, 1995). These extrinsic signals induce apoptosis in a wide variety of cell types. Intrinsic inducers of apoptosis comprise a number of genes conserved throughout evolution, including members of interleukin-1β converting enzyme (ICE) proteinase family, Bcl2 family (e.g., Bcl2s, Bad, and Bax), and p53 (Ellis et al., 1991; Vaux et al., 1994). Apoptosis can be suppressed by a variety of extrinsic and intrinsic signals, including growth factors (e.g., IGF1, NGF, and CNTF), signaling molecules activated by these growth factors (e.g., phosphatidylinositol-3-kinase [PI3 kinase]), Bcl2 family proteins (e.g., Bcl2 and Bclxl), and proteinase inhibitors (e.g., crmA; Kapeller and Cantly, 1994; Nagata and Golstein, 1995; Steller, 1995; Thompson, 1995; Yao and Cooper, 1995).
Integrin engagement and activation of FAK are implicated in a number of signaling pathways, including ones that lead to anchorage-dependent cell survival (Burridge and Chrzanowska-Wodnicka, 1996; Frisch et al., 1996; Hungerford et al., 1996; Parsons, 1996; Xu et al., 1996). FAK is a prototypic member of a family of nonreceptor protein tyrosine kinases, containing a central catalytic domain and large NH2- and COOH-terminal noncatalytic regions that are devoid of SH2 and SH3 domains (Hanks et al., 1992; Schaller et al., 1992). FAK is enriched in the brain and expressed in most cell lines and tissues examined (Andre and Becker-Andre, 1993; Grant et al., 1995). In some cell types, the COOH-terminal domain of pp125FAK is expressed autonomously as a 41-kD protein termed focal adhesion kinase (FAK)-related nonkinase (FRNK) (Schaller et al., 1993). FAK is activated by many diverse stimuli, including v-Src transformation (Schaller et al., 1992), attachment to the ECM (Guan and Shalloway, 1992; Schaller et al., 1992), and exposure to growth factors (e.g., PDGF; Rankin and Rozengurt, 1994), neuropeptides (e.g., bombesin; Rozengurt, 1991; Zachary et al., 1992), and lysophosphatidic acid (Moolenaar, 1991). Clustering of integrins through binding to the ECM leads to the tyrosine phosphorylation of FAK (Guan and Shalloway, 1992; Schaller et al., 1992). Phosphorylation of FAK on tyrosine 397 creates a high affinity binding site for the SH2 domains of Src and Fyn, both of which are associated with activated FAK (Cobb et al., 1994; Schaller et al., 1994). Phosphorylation of FAK on tyrosine residues present in the COOH-terminal domain leads to the association of various other SH2 domain–containing signaling proteins, including Grb2 (Schlaepfer et al., 1994) and the p85 subunit of PI3 kinase (Chen and Guan, 1994_b_ ; Guinebault et al., 1995). In addition, the proline-rich regions in the COOH-terminal domain of FAK direct the binding to p130cas (Crk-associated substrate) and Graf (GTPase regulator associated with FAK) in an SH3 domain-dependent manner (Polte and Hanks, 1995; Harte et al., 1996; Hildebrand et al., 1996). The COOH-terminal domain of FAK can also associate with the cytoskeletal proteins paxillin and talin (Bellis et al., 1995; Chen et al., 1995; Hildebrand et al., 1995; Tachibana et al., 1995). Inclusive within the COOH-terminal domain is a 140-amino acid sequence that is both necessary and sufficient for the targeting of FAK to focal adhesions, called the focal adhesion targeting domain (FAT; Hildebrand et al., 1993). FAK is believed to play an important role in regulating signaling events initiated by the activation of various membrane receptors that induce cytoskeletal rearrangement (Parsons, 1996). Cells microinjected with reagents that attenuate the activity of FAK (e.g., anti-sense oligonucleotides and anti-FAK antibodies) undergo apoptosis (Hungerford et al., 1996; Xu et al., 1996). Cells expressing constitutively active CD2-FAK are resistant to apoptosis when detached from the ECM (Frisch et al., 1996), suggesting that FAK may be involved in mediating the anchorage-dependent cell survival.
Recently, a second member of the FAK subfamily, proline-rich tyrosine kinase 2 (PYK2), also known as cellular adhesion kinase β (CAKβ) and related adhesion focal tyrosine kinase (RAFTK), has been identified (Avraham et al., 1995; Lev et al., 1995; Sasaki et al., 1995). PYK2, a 116-kD cytoplasmic protein tyrosine kinase, is rapidly phosphorylated on tyrosine residues in response to various stimuli, including elevation of the intracellular calcium levels, activation of protein kinase C, and exposure to stress factors (e.g., UV light, TNFα; Lev et al., 1995; Tokiwa et al., 1996). PYK2 is highly enriched in the brain and is expressed in fewer tissues and cell lines (e.g., PC12 cells, and many hematopoietic cell lines; Avraham et al., 1995; Lev et al., 1995; Sasaki et al., 1995; Salgia et al., 1996). The restricted expression of PYK2, as compared to FAK, suggests that they may mediate distinct functions. FAK and PYK2 are highly homologous to each other, sharing 45% overall sequence identity and 60% identity in the catalytic domain. Several tyrosine residues are conserved between FAK and PYK2, including the binding site for the SH2 domains of Src and Fyn (Y397 in FAK, Y402 in PYK2) and the putative binding site for the SH2 domain of Grb2 (Y925 in FAK, Y881 in PYK2; Cobb et al., 1994; Schaller et al., 1994; Schlaepfer et al., 1994; Avraham et al., 1995; Lev et al., 1995; Sasaki et al., 1995; Dikic et al., 1996). In addition, PYK2 also contains the putative “FAT” domain and the proline-rich sequences responsible for mediating the binding of p130cas and Graf. Given the high degree of sequence similarity between PYK2 and FAK, it is possible that PYK2 interacts with some or many of the FAK-binding partners. Indeed, tyrosine-phosphorylated PYK2 is able to bind to the SH2 domain of Grb2 in a similar manner to FAK, which is thought to lead to the activation of MAPK pathway in PC12 cells (Lev et al., 1995). While PYK2 is believed to play a role in regulating neurotransmission or neuroplasticity by phosphorylating potassium channels (Lev et al., 1995), other functions of PYK2 are still unidentified.
In this report we demonstrate that over-expression of PYK2 in both fibroblastic and epithelial cell lines (e.g., rat-1, mouse 10T1/2, swiss 3T3, quail QT6, and human embryonic kidney 293 cells [HEK 293]) leads to apoptosis. The NH2-terminal domain and tyrosine kinase activity of PYK2 are required for its full apoptotic activity. Furthermore, we show that the PYK2-mediated apoptosis can be suppressed by over-expression of catalytically active c-Src, v-Src, PI3 kinase, and Akt/PKB. It could also be suppressed by over-expressing crmA, but not Bcl2. These data demonstrate that while PYK2 and FAK are structurally similar, each has the capacity to mediate distinctly different signaling responses.
Materials and Methods
Reagents and Cell Lines
Rabbit polyclonal antisera recognizing PYK2 were raised using a glutathione-S-transferase (GST) fusion protein containing the COOH-terminal 400 amino acids of rat PYK2 (amino acid 587 to 988). Monoclonal antibodies and goat polyclonal antibody recognizing NH2-terminal PYK2 domain were purchased from Santa Cruz Biotechnology (Santa Cruz, CA; anti-myc and anti-PYK2) or Transduction Laboratories (Lexington, KY; anti-phosphotyrosine). Propidium iodide was purchased from Sigma Chemical Co. (St. Louis, MO). TUNEL kit (Apoptag kit) was purchased from Boehringer Mannheim (Indianapolis, IN). GST-c-Jun was purchased from Clonetech (Palo Alto, CA). LA29, a v-Src temperature-sensitive cell line, was provided by M. Weber (University of Virginia, Charlottesville, VA). Mouse fibroblastic 10T1/2, 5HD47 (c-Src), pm430 (kinase inactive c-Src), and dl155 (SH2 defective c-Src) cell lines were provided by S. Parsons (University of Virginia, Charlottesville, VA). HEK 293 cells were provided by Q.-H. Song (Columbia University, New York, NY).
Expression Vectors
The cDNAs of PYK2 (CAKβ, kindly provided by T. Sasaki, Sapporo Medical University, Sapporo, Japan), FAK, and FRNK were subcloned into expression vectors, either downstream of a c-myc epitope tag (MEQKLISEEDL) under the control of the cytomegalovirus (CMV) promoter (pCMV-c-Myc; Evan et al., 1985) or downstream of GST under the control of the elongation factor 1α (EF1α) promoter (pEBG). The “FAT” domain deletion mutant (PYK2Δ936-1009) was generated by inserting an in-frame stop codon between XbaI site in PYK2 and the NheI site in pCMV-c-Myc. The kinase and COOH-terminal domain deletion mutant (PYK2Δ250-1009) was generated by inserting an in-frame stop codon between the Nsi site in PYK2 and the Nsi site in pCMV-c-Myc. NH2-terminal deletion mutants, the kinase inactive (lysine [K] 457 to alanine [A]), and autophosphorylation site (tyrosine [Y] 402 to phenylalanine [F]) mutants were generated by PCR (Ho et al., 1989). The chimeric constructs of PYK2/FAK1 and PYK2/FAK2 were generated by in-frame ligation of individual PCR fragments. In construct PYK2/FAK1, the NH2-terminal domain of PYK2 (amino acids 2 to 385) was amplified by PCR and fused with the PCR fragments of the kinase and COOH-terminal domains of FAK (amino acids 380 to 1052). In construct PYK2/FAK2, the NH2-terminal and kinase domain of PYK2 (amino acids 2 to 695) was ligated with the COOH-terminal domain of FAK (amino acids 692 to 1052). The authenticity of all mutants was verified by DNA sequencing. The constructs encoding constitutively active and inactive PI3 kinase and Akt were gifts from Anke Klippel (Chiron Corporation, Emeryville, CA; Klippel et al., 1996).
Cell Culture and Transfections
Rat-1, mouse 10T1/2, and Cos-1 cells were maintained in DME containing 10% fetal calf serum, 100 μg/ml penicillin G, and 100 mg/ml Streptomycin (GIBCO BRL, Gaithersburg, MD). LA29, 5HD47, pm430, and dl155 cell lines were maintained in the above medium containing G418 (Sigma Chemical Co.). HEK 293 cells were maintained in DME/F12 (1:1) medium with the same additives. Cells were plated 12 h before transfection at a density of 104 cells/22 mm2 coverslip (VWR Scientific, West Chester, PA). Cells were transfected with 30–40 μl of lipofectamine (GIBCO BRL) with various constructs (3 μg DNA) with or without 1 μg of pCMV β-galactosidase in 1.6 ml of DME-serum free medium (GIBCO BRL). After incubation for 5 h, 1 ml of serum-containing DME media was added. 30 h later, cells were fixed with 4% paraformaldehyde for immunostaining or fixed with 0.5% glutaraldehyde for β-galactosidase staining as described below.
Apoptosis Assays
The morphology of transfected cells was examined by β-galactosidase staining or immunostaining of PYK2 using antibodies against PYK2 or the c-Myc epitope tag. For β-galactosidase staining, fixed cells were washed with PBS three times, each for 5 min, and incubated in PBS containing 20 mM K2Fe(CN)6, 20 mM K2Fe(CN)6.3H2O, 1 mM MgCl2, and 0.5 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) until a suitable color developed (∼1–2 h). For immunostaining, fixed cells were incubated with the primary antibody against PYK2 (1:500 dilution) at 37°C for 1 h and visualized with a fluorescence-conjugated anti–rabbit or anti–mouse secondary antibody (1:300 dilution). Condensed or fragmented DNA was detected in situ with propidium iodide or fluorescence-dUTP using terminal deoxynucleotidyl transferase (Apoptag) as described previously (Gabrieli et al., 1992; Xiong and Montell, 1995). These cells were double labeled with anti-PYK2 antibodies to monitor the protein expression. Apoptotic index was determined by counting the number of apoptotic cells, which expressed either β-galactosidase or PYK2, divided by the total number of β-galactosidase or PYK2-expressing cells. For each experiment, a minimum of 200 cells that expressed β-galactosidase or PYK2 was counted. Each construct was examined at least three times.
Immunoprecipitation
For immunoprecipitation, 500 μg of cell lysates was incubated with anti-PYK2 antibodies at 4°C for 1 h in a final volume of 1 ml RIPA buffer. After the addition of protein A-agarose beads, the reaction was incubated at 4°C for another hour. Immunocomplexes were purified and subjected to immunoblotting using antiphosphotyrosine and anti-PYK2 antibodies.
Results
Expression of PYK2 Induced Morphological Changes in rat-1 Cells
To study the function of PYK2, we attempted to generate rat-1 fibroblastic cell lines stably expressing PYK2. After a number of failures, we examined the phenotype of rat-1 cells after transient transfection using a vector carrying cDNA encoding full length PYK2 with an NH2-terminal c-Myc epitope tag under the control of the CMV promoter (PYK2-WT). The expression of PYK2 as well as cell morphology were monitored by immunostaining of the cells using anti-PYK2 antibodies, anti-c-Myc (9E10) monoclonal antibodies, or by β-galactosidase staining of cells that were co-transfected with a reporter plasmid containing the cDNA encoding β-galactosidase (pCMV-β-gal). As shown in Fig. 1, the transfection with a control pCMV vector with or without pCMV-β-gal had no effect on cell morphology (Fig. 1, A and B). However, rat-1 cells expressing the PYK2 protein, identified by both PYK2 or Myc immunostaining or β-galactosidase staining, appeared to be round with condensed cytoplasm, blebbed membrane, and poorly attached to the dish (Fig. 1, C and D). These morphological changes were specific for PYK2- expressing rat-1 cells, since the rat-1 cells overexpressing FAK appeared similar to control cells (Fig. 1, E and F).
Figure 1.
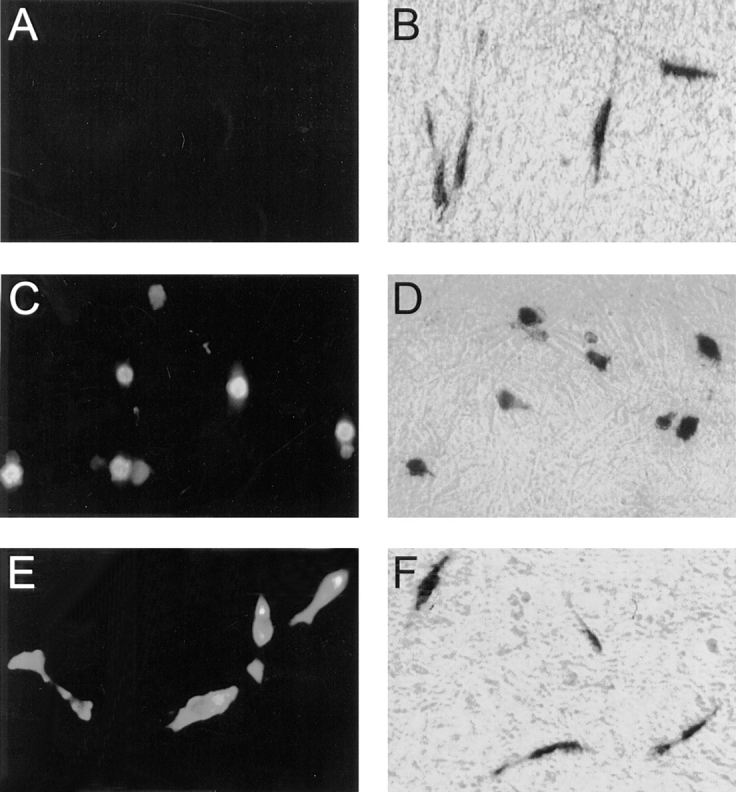
Morphological changes of rat-1 cells expressing PYK2. Rat-1 cells were transiently transfected with pCMV vector (A and B), pCMV-PYK2-WT (C and D), and pCMV-FAK (E and F) plasmids without (A, C, and E) and with pCMV-β-galactosidase (B, D, and F). 30 h after transfection, the cells were fixed with 4% paraformaldehyde and stained with antibodies against c-Myc (9E10) epitope (A, C, and E), or fixed by 0.5% glutaraldehyde and stained for the β-galactosidase activity of the cells cotransfected with pCMV-β-galactosidase (B, D, and F).
Morphologically Altered rat-1 Cells Exhibit Features of Apoptosis
The morphological changes in rat-1 fibroblasts induced by PYK2 expression was indicative of apoptosis. We used both propidium iodide staining and TdT-mediated dUTP-fluorescence nick end labeling (TUNEL) to determine whether DNA condensation and fragmentation, a hallmark of apoptosis, occurred in the cells expressing PYK2 (Fisher, 1994; Muzio et al., 1996). In parallel, the transfected cells were also immunostained with either anti-PYK2 or anti-Myc (9E10) antibodies to monitor the protein expression. Propidium iodide staining of cells expressing wild-type PYK2 demonstrated condensed nuclei (Fig. 2, B and D). This effect was not observed in cells transfected with vector alone (data not shown) or constructs encoding COOH-terminal PYK2 protein (PYK2Δ1-680; Fig. 3, A and C). In addition, PYK2-expressing cells exhibited DNA fragmentation, detected using modified TUNEL (Fig. 2, F and H), whereas no fragmentation was observed in cells transfected with the COOH-terminal PYK2, PYK2Δ1-680 (Fig. 2, E and G). The concomitant induction of cell death with expression of wild-type PYK2 suggested that the expression of PYK2 may be directly responsible for the induction of cell death. Similar morphological changes and apoptosis were observed after transfection of PYK2 into mouse 10T1/2, swiss 3T3, quail QT6, and HEK 293 cells (data not shown). In PC12 cells that express significant levels of endogenous PYK2, no detectable cell death was observed when PYK2 was over-expressed (data not shown).
Figure 2.
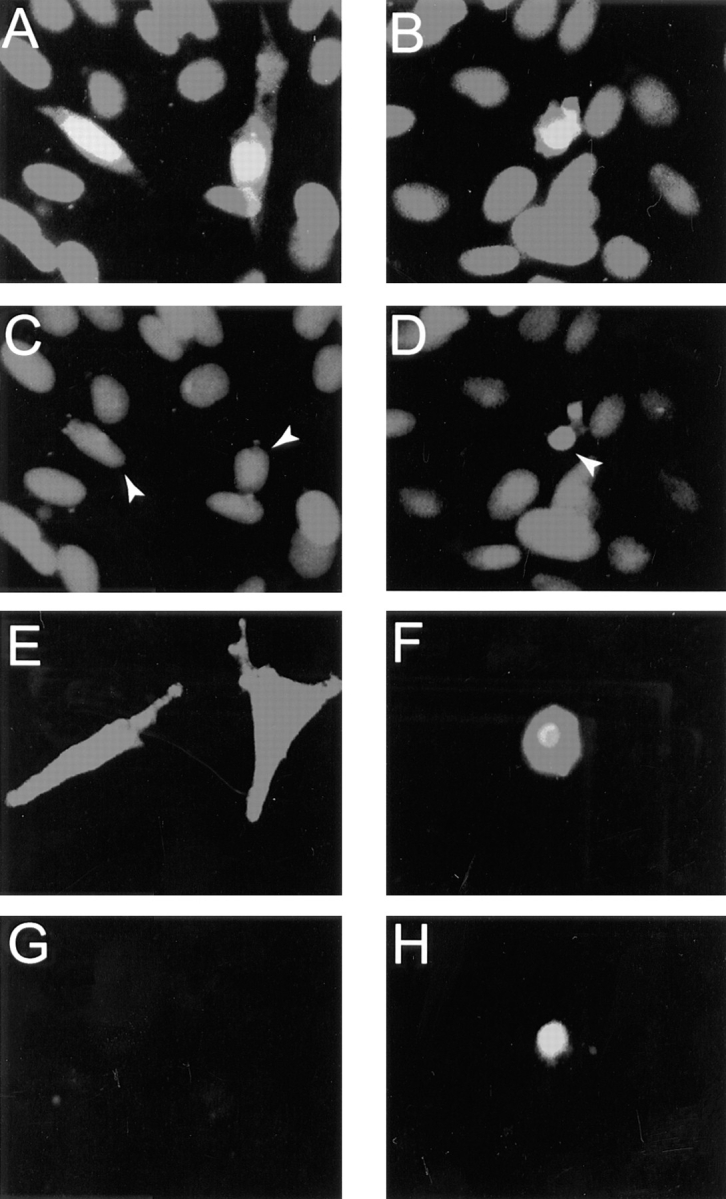
Apoptosis of rat-1 cells expressing PYK2. Rat-1 cells were transiently transfected with PYK2 COOH-terminal (PYK2Δ1-680; A, C, E, and G) and wild-type PYK2 (PYK2-WT; B, D, F, and H) plasmids. 24–30 h after transfection, the cells were fixed and immunostained with anti-PYK2 antibodies and propidium iodide (A–D). In a separate experiment, the transfected cells were immunostained using antibodies against PYK2 and labeled with fluorescence-conjugated dUTP using terminal transferase (E–H). Nuclei of rat-1 cells overexpressing PYK2Δ1-680 with normal morphology stained uniformly with propidium iodide, indicating intact nuclei (C, arrows). Propidium iodide staining of rat-1 cells overexpressing PYK2-WT showed apoptotic nuclei (condensed, indicated by arrows) in a fraction of cells (D). While there was no labeling of fluorescence-conjugated dUTP in cells expressing PYK2Δ1-680 with normal morphology (G), there was labeling of dUTP in cells expressing PYK2-WT (H).
Figure 3.

Schematic diagrams of PYK2, PYK2 mutants, FAK, and FAK mutants. The numbers represent the amino acid residues deleted from wild-type PYK2. The shaded block represents the kinase domain of PYK2. The putative FAT domain and proline-rich sequences are indicated. The constructs tagged with either c-Myc or GST are also indicated. Apoptotic index (mean ± SEM) was determined by counting the apoptotic PYK2 or FAK-expressing cells (or PYK2 positive) divided by total number of PYK2 or FAK expression cells, and listed in the right-hand column.
The Full Apoptotic Activity of PYK2 Requires NH2-terminal and Active Kinase Domains
PYK2 contains a central catalytic kinase domain (from amino acid 419 to 679), flanked by noncatalytic NH2-terminal (amino acids 1 to 418) and COOH-terminal (amino acids 680 to 1009) domains (Fig. 3). Within the COOH-terminal region, there are two proline-rich sequences and a putative FAT domain (Fig. 3). To determine which domains of PYK2 were required for its apoptotic activity, we generated a series of PYK2 deletion mutants, whose structures are summarized in Fig. 3. Mutant proteins were tagged either with GST or a c-Myc epitope at the NH2 terminus. Transfection of individual mutant constructs into HEK 293 cells followed by Western blotting of cell extracts using anti-PYK2, antibodies to the c-Myc epitope or GST, confirmed the expression of mutant proteins of the appropriate molecular sizes (Fig. 4 A). The apoptotic activity of individual mutant proteins was tested by transient transfection of rat-1 cells with each mutant construct in the presence or absence of pCMV β-galactosidase reporter. The “apoptotic index” for each mutant was determined by counting the apoptotic blue cells or apoptotic PYK2-expressing cells and dividing by the total number of transfected cells (total number of blue or PYK2-expressing cells).
Figure 4.


Apoptotic activities and Western blotting of PYK2, PYK2 mutants, FAK, and FAK mutants. (A) Western blotting of HEK 293 cells expressing proteins of PYK2 and PYK2 mutants using antibodies against PYK2 and c-Myc epitope. The position of protein molecular weight markers is indicated at the right. (B) Western blotting of HEK 293 cells expressing proteins of PYK2, FAK, and FAK mutants using antibodies against c-Myc epitope. The position of protein molecular weight markers is also indicated at the right. (C) Histograms of the apoptotic index of cells transfected with constructs encoding PYK2, PYK2 mutants, FAK, and FAK mutants. Apoptotic index (mean ± SEM) was determined by counting the apoptotic PYK2 or FAK-expressing cells (or PYK2-positive) divided by total number of PYK2 or FAK expression cells.
The PYK2-WT demonstrated the highest apoptotic activity or apoptotic index (0.97), while the basal apoptotic index in rat-1 cells transfected with control vector alone was 0.09–0.13. A significant reduction of the apoptotic index (0.3–0.5) was observed after transfection with the NH2-terminal domain deletion mutants (e.g., PYK2Δ1-88 and PYK2Δ1-416; Figs. 3 and 4, A and C), suggesting that the NH2-terminal domain of PYK2 was required for maximal apoptotic activity. This conclusion was further supported by the observation that transfection of cells with the NH2-terminal domain alone (PYK2Δ250-1009) was able to induce significant cell death (0.55; Figs. 3 and 4 C). We next determined if kinase activity was required for PYK2-induced apoptosis. The kinase-inactive PYK2 (K457A [PYK2-KD]), containing a lysine (K) to alanine (A) mutation in the ATP-binding site (Fig. 5 A), was transfected into rat-1 cells. The apoptotic index of rat-1 cells expressing the kinase-inactive mutant was 0.6, a significant decrease from that of the wild-type, suggesting that PYK2 catalytic activity was required for the maximal apoptotic activity (Fig. 5 B). This conclusion was further supported by the observation that the apoptotic index of rat-1 cells expressing the autophosphorylation mutant PYK2-Y402F, containing a tyrosine (Y) 402 to (F) mutant (Fig. 5 A), was also significantly reduced to 0.41 (Fig. 5 B). In contrast to the cells expressing PYK2, cells expressing wild-type FAK, an autophosphorylation mutant (Y397F), or the COOH-terminal domain of FAK (FRNK) exhibited a low level of apoptosis (apoptotic index of 0.1–0.3; Figs. 3 and 4 C). The expression levels of PYK2, FAK, FAK-Y397F, and FRNK were similar based on Western blot analysis of extracts from the transfected cells (Fig. 4 B). The transfection efficiencies using these constructs were similar (data not shown).
Figure 5.

Catalytic and apoptotic activity of wild-type and mutant PYK2. (A) Schematic representation of PYK2 and its mutants. The numbers represent the amino acid residues mutated from wild-type PYK2. The shaded block represents the kinase domain of PYK2. The putative FAT domain and proline-rich sequences are indicated. The constructs tagged with c-Myc are also indicated. The tyrosine phosphorylation and the apoptotic index of these mutants are listed in the right-hand column. (B) Tyrosine phosphorylation of wild-type PYK2 (PYK2-WT), kinase-inactive PYK2 (PYK2-KD), the putative FAT domain deletion mutant (PYK2Δ936-1009), the NH2-terminal domain deletion mutants (PYK2Δ1-88 and PYK2Δ1-416), and the autophosphorylation site mutant (PYK2-Y402F). Cell lysates from 293 cells overexpressing PYK2-WT, PYK2-KD, PYK2Δ936-1009, PYK2Δ1-88, PYK2Δ1-416, and PYK2-Y402F proteins were immunoprecipitated by antibodies against PYK2. The immunoprecipitatied proteins were then subjected to immunoblotting with the antibodies against phosphotyrosine (anti-P-tyr) or PYK2 (anti-PYK2). (C) Histograms of apoptotic index of PYK2Δ936- 1009, PYK2Δ1-88, PYK2-WT, PYK2-KD, PYK2Δ1-416, and PYK2-Y402F.
The putative FAT domain of PYK2 did not appear to be critical for the apoptotic activity. Partial deletion of the FAT domain, PYK2Δ936-1009, did not inhibit induction of apoptosis (apoptotic index of 0.99; Figs. 3 and 4), yet this mutant protein failed to bind to paxillin and localize to focal contacts (data not shown). Paradoxically, transfection of mutant cDNAs encoding the entire COOH-terminal domain (PYK2Δ1-680) or cDNAs encoding only the COOH-terminal FAT domain (PYK2Δ1-781 and PYK2Δ1-868) still retained low but reproducible apoptotic activity (Figs. 3 and 4). These data suggested that while expression of either the NH2 or COOH terminus alone could induce limited apoptosis, the full apoptotic activity required both an intact NH2 terminus and active kinase domain.
To confirm that PYK2-KD was catalytically inactive, we studied autophosphorylation of wild-type PYK2-WT and mutants (PYK2-KD, PYK2Δ1-88, PYK2Δ1-416, PYK2Δ936-1009, and PYK2-Y402F) by immunoblotting using antiphosphotyrosine antibodies. While the PYK2-WT was highly tyrosine phosphorylated, the PYK2-KD failed to react with antiphosphotyrosine antibodies (Fig. 5 B), indicating that PYK2-KD was kinase inactive. The autophosphorylation site of PYK2 may be tyrosine 402 (Y402), a residue also conserved in FAK (Y397) that became phosphorylated upon FAK activation. The Y402 mutant (PYK2-Y402F) and the NH2-terminal deletion mutant PYK2Δ1-416, in which the autophosphorylation site (Y402) was deleted, also did not react with antiphosphotyrosine antibodies (Fig. 5 B). Interestingly, the partial NH2-terminal deletion mutant (PYK2Δ1-88) with reduced apoptotic index also exhibited reduced tyrosine phosphorylation (Fig. 5, B and C).
To further examine the mechanisms by which PYK2, but not FAK, induces apoptosis, we constructed a chimeric protein (PYK2/FAK1) containing the NH2-terminal domain of PYK2 (amino acids 2 to 385) and the catalytic and COOH-terminal domains of FAK (amino acids 380 to 1,052) and a second chimeric protein (PYK2/FAK2) containing the NH2-terminal and kinase domains of PYK2 (amino acids 2 to 695) and the COOH-terminal domain of FAK (amino acids 692 to 1,052; Fig. 6 A). The structure of these chimeric constructs (PYK2/FAK1 and PYK2/FAK2) was confirmed by Western blotting analysis of HEK 293 cell lysates overexpressing PYK2, PYK2/FAK1, PYK2/ FAK2, and FAK using antibodies against c-Myc epitope, PYK2 NH2-terminal domain (anti-PYK2 from Santa Cruz Biotechnology), and FAK COOH-terminal domain (mAb 2A7; Fig. 6 B). Apoptotic activities of these chimera were examined in rat-1 cells. Both PYK2/FAK1 and PYK2/ FAK2 demonstrated similar apoptotic activities (apoptotic index of 0.8 to 0.84) as wild-type PYK2, while the apoptotic activity of wild-type FAK was close to the basal level (0.13; Fig. 6 C). These data indicated that the NH2-terminal domain of PYK2 was sufficient to render FAK indistinguishable from PYK2 in its ability to induce apoptosis.
Figure 6.

Apoptotic activity of PYK2/FAK chimeric fusion proteins. (A) Schematic representation of PYK2, PYK2/FAK1, PYK2/FAK2, and FAK. The shaded block represents the regions of PYK2. The open block represents the regions of FAK. The numbers represent the number of amino acids in FAK or PYK2. The kinase domain sequence is indicated. The constructs tagged with c-Myc are also indicated. NH2-terminal domain of PYK2 (amino acid 2 to 385) was fused with the kinase and COOH-terminal domains of FAK (amino acids 380 to 1,052) in construct PYK2/FAK1. In construct PYK2/FAK2, PYK2 (amino acids 2 to 695) was fused with FAK (amino acids 692 to 1,052). The apoptotic index of these PYK2/FAK chimeric proteins was listed in the right-hand column. (B) Western blotting of HEK 293 cells expressing PYK2, PYK2/FAK1, PYK2/FAK2, and FAK proteins using antibodies against cMyc epitope, goat anti-PYK2 (from Santa Cruz Biotechnology; recognizing epitope in PYK2 NH2-terminal domain), or FAK (2A7, recognizing epitope in FAK COOH-terminal domain). (C) Histograms of the apoptotic index of rat-1 cells transfected with FAK-WT, PYK2/FAK1, PYK2/FAK2, and PYK2-WT.
PYK2-mediated Apoptosis Can Be Suppressed by Over-Expression of Catalytically Active Src, PI3 Kinase, and Akt
To examine the possible effects of Src on PYK2-mediated apoptosis, we expressed PYK2 in LA29 cells, a temperature-sensitive v-Src rat-1 cell line. When PYK2-WT was transiently expressed in LA29 cells grown at permissive temperature (35°C), the apoptotic index was significantly reduced from 0.97 to 0.25 (Fig. 7). No significant inhibition of apoptosis was observed when LA29 cells expressing PYK2 were grown at nonpermissive temperature (39°C) (data not shown), indicating that the tyrosine kinase activity of v-Src was required for this event. To examine whether suppression of apoptosis could be mimicked by c-Src, PYK2 was transiently expressed in mouse 10T1/2 cells stably over-expressing c-Src (5H), as well as its parental 10T1/2 cells. 10T1/2 cells expressing full length PYK2 underwent apoptosis (apoptotic index of 0.95). The apoptotic index was significantly reduced to 0.45 when PYK2 was expressed in the c-Src (5H) cell line (Fig. 7). To determine if the reduced apoptotic index of c-Src was dependent on kinase activity, PYK2 was transiently expressed in cells stably over-expressing either catalytically inactive c-Src (pm430 mutant) or an SH2-defective variant of c-Src (dl155; Wilson et al., 1989). The extent of cell death (apoptotic index of 0.98) mediated by PYK2 in cells expressing catalytically inactive Src, was not appreciably different from the parental 10T1/2 cells (Fig. 5). However, in the SH2-defective c-Src cell line (dl155), the apoptotic index was significantly reduced to 0.55 (Fig. 7). These results suggested that the catalytic activity, but not the SH2 domain of c-Src, appeared to be required for the suppression of PYK2-mediated apoptosis in fibroblasts.
Figure 7.


Suppression of the PYK2-induced apoptosis by overexpression of catalytically active Src. Wild-type PYK2 was transiently transfected into different cell lines stably expressing wild-type Src and various Src mutants. (A) Immunostaining of PYK2 with anti–c-Myc antibodies (9E10 mAB) in rat-1, temperature-sensitive v-Src (ts v-Src, LA29), 10T1/2, c-Src (5HD47), kinase-dead c-Src (430), and SH2-defective c-Src (c-Src dSH2, dl155) cell lines expressing wild-type c-Myc–tagged PYK2. (B) Histograms of apoptotic index mediated by wild-type PYK2 in different Src cell lines.
We next examined the effects of PI3 kinase on PYK2-mediated apoptosis. The plasmids encoding c-Myc–tagged constitutively active or catalytically inactive P110/PI3 kinases, in which a truncated p85 subunit was linked to the catalytic subunit (Hu et al., 1995), were cotransfected in rat-1 cells with wild-type PYK2. Co-expression of the constitutively active PI3 kinase (PI3K) significantly reduced the apoptotic effects of PYK2 (apoptotic index of 0.59; Fig. 8). The reduction in PYK2-induced apoptosis was not observed when the catalytically inactive PI3 kinase (PI3KΔ) was cotransfected into rat-1 cells (Fig. 8), indicating that the catalytic activity of PI3 kinase was required for suppression of apoptosis. The expression of catalytically active (PI3K) and inactive (PI3KΔ) PI3 kinases was examined by immunostaining using antibodies against the c-Myc epitope and p85 subunit of PI3 kinase.
Figure 8.


Suppression of PYK2-induced apoptosis by overexpression of active PI3 kinase and Akt. Full length PYK2 was cotransfected into rat-1 cells with constitutively active and inactive PI3 kinase (PI3K, PI3KΔ) and active and inactive Akt (Akt, AktΔ; at 1:1 molar ratio of DNA). (A) Cotransfected cells were doubly immunostained using antibodies against PYK2 (a, c, e, and g), p85 subunit for PI3 kinase (b and d), and HA-epitope for Akt proteins (f and h). (B) Histograms of the apoptotic index mediated by cotransfection of PYK2-WT with vector alone (1), active PI3 kinase (2), inactive PI3 kinase (3), active Akt (4), inactive Akt (5), crmA (6), and Bcl2 (7).
The serine/threonine protein kinase Akt (also called protein kinase B, PKB) is believed to be one of the effectors in the PI3 kinase signaling pathway (Burgering and Coffer, 1995; Franke et al., 1995). Activation of Akt can protect many cells against apoptosis, whereas inhibition of Akt does not (Dudek et al., 1997; Kauffmann-Zeh et al., 1997; Kulik et al., 1997). To further confirm the suppressive effects of PI3 kinase in PYK2-induced apoptosis, we determined if activated Akt was able to protect cells against PYK2-induced apoptosis. The plasmids encoding HA-tagged constitutively active Akt/PKB, in which Akt was targeted to the membrane by myristoylation, or catalytically inactive Akt were cotransfected in rat-1 cells with wild-type PYK2. Co-expression of the active Akt significantly reduced the apoptotic effects of PYK2 (apoptotic index of 0.58; Fig. 8), whereas catalytically inactive Akt did not (Fig. 8). These data demonstrated that active Akt was able to at least partially block PYK2-induced apoptosis. In addition, we also examined the effects of other cell death suppressors on PYK2-induced apoptosis. CrmA, an ICE-like proteinase inhibitor, was able to efficiently suppress PYK2-induced cell death (apoptotic index of PYK2 was reduced to 0.4, when crmA was cotransfected with wild-type PYK2; Fig. 8 B). In contrast, overexpression of Bcl2, another cell death suppressor, did not block the PYK2-induced cell death (Fig. 8 B).
Discussion
In this study, we have investigated the expression of the FAK-related protein tyrosine kinase, PYK2, in several different fibroblastic and epithelial cell lines including rat-1, mouse 10T1/2, and HEK 293 cells. In each cell line the forced expression of PYK2, but not the related tyrosine kinase, FAK, rapidly (within 24 to 30 h) induced apoptosis. Apoptosis induced by expression of full length PYK2 was very efficient; >90% of the cells expressing PYK2 undergo programed cell death. In contrast, mutant forms of PYK2 deficient in kinase activity (PYK2-KD) or lacking the major site of autophosphorylation (PYK2-Y402F) were significantly less efficient in inducing apoptosis, reducing the overall level of cell death from >90% to 40–58%. Chimeric proteins containing the NH2-terminal domain of PYK2 and the kinase and COOH-terminal domains of FAK were as efficient as wild-type PYK2 in the induction of apoptosis. Co-expression of PYK2 with either active Src, PI3 kinase, or the serine/threonine kinase, Akt, significantly reduced apoptosis. In addition, co-expression of PYK2 with crmA, an inhibitor of the ICE or ICE-like proteinases, but not Bcl2, also significantly inhibited apoptosis. We conclude from these data that the efficient induction of cell death by PYK2 under our assay conditions, requires specific interactions via sequences in the NH2-terminal domain of PYK2, PYK2 kinase activity, and the ability to autophosphorylate at tyrosine residue 402. In addition, efficient induction of cell death requires that PYK2 functionally interacts with a downstream signaling pathway(s) (may be mediated in part by inhibition of the PI3 kinase pathway) to promote apoptosis.
The high apoptotic activity exhibited by the chimeric constructs PYK2/FAK1 and PYK2/FAK2 point to the importance of the NH2-terminal domain of PYK2 in inducing apoptosis. Furthermore, these observations argue that the catalytic and COOH-terminal domains of PYK2 and FAK, while necessary for the apoptotic process, are interchangable. Expression of the PYK2 NH2- and COOH-terminal domains also induces apoptosis, albeit with an efficiency significantly less than full length PYK2, but comparable to catalytically inactive PYK2 (Fig. 3). While it is unclear how these isolated domains induce apoptosis, studies of FAK demonstrate that the NH2- and COOH-terminal domains interact with several well characterized binding partners, linking FAK to both upstream and downstream signaling pathways (Schaller and Parsons, 1994). Thus we speculate that apoptosis induced by forced expression of the chimeric proteins, the NH2-terminal, and COOH-terminal domains of PYK2 may result from the activation of signaling pathways that lead to the apoptotic process, or/ and the inhibition of signaling pathways whose continuity is required to maintain cell viability. What these pathways might be remains unclear, particularly in light of the paucity of data regarding the function of PYK2 in different cell types.
Co-expression of full length PYK2 with either constitutively active PI3 kinase, Akt, or Src results in the partial rescue of PYK2-induced apoptosis (Fig. 6). These data implicate the PI3 kinase pathway in the protection of apoptosis mediated by wild-type PYK2. Akt is a serine/threonine protein kinase (also called protein kinase B or PKB), identified first as an oncogene; however, recent studies indicate that it is one of the major targets of PI3 kinase (Burgering and Coffer, 1995; Franke et al., 1995). The fact that Akt can substitute for activated PI3 kinase in protection of PYK2-induced apoptosis lends further support for the role of PI3 kinase signaling in protection of PYK2-induced apoptosis. Finally, v- and c-Src also partially suppress PYK2-mediated apoptosis. The fact that PI3 kinase is activated by Src (Fukai and Hanafusa, 1989) and that catalytically active Src is required for rescue of PYK2-induced apoptosis, leads us to speculate that activation of PI3 kinase by Src may be a possible mechanism for Src-mediated rescue. Thus one possible mechanism by which PYK2 might induce apoptosis is by inhibiting PI3 kinase either directly (perhaps by a phosphorylation-dependent mechanism) or indirectly by negatively regulating an upstream effector of PI3 kinase activation. This suggestion is supported by recent observations that PI3 kinase activity is required for PC12 cell survival mediated by growth factors including NGF and PDGF (Valius and Kazlauskas, 1993; Yao and Cooper, 1995) and that active Akt is able to rescue the apoptosis induced by c-Myc over-expression in fibroblasts (Kauffmann-Zeh et al., 1997), serum withdrawal in neuronal cells (Dudek et al., 1997), and UV light in Cos-1 cells (Kulik et al., 1997).
The results presented herein are the first report of a role for PYK2 in the induction of apoptosis. PYK2 is present at relatively high levels in brain and is expressed in kidney, liver, spleen, and a variety of cell lines (e.g., PC12 cells and many hematopoietic cell lines; Avraham et al., 1995; Salgia et al., 1996; Sasaki et al., 1995). Elevated expression of PYK2 has been reported in GN4, a transformed rat liver epithelial cell line (Yu et al., 1996). In PC12 and GN4 cells, PYK2 activation appears to be linked to neurotransmitters, growth factors, and hormones that induce alterations in calcium-dependent signaling pathways (Lev et al., 1995; Yu et al., 1996). The cell lines used in this study (rat-1, mouse 10T1/2, and HEK 293 cells) exhibit low levels of endogenous PYK2 expression. Therefore it is unlikely that PYK2 plays a role in regulating apoptotic pathways in these cells. More likely is the possibility that PYK2 expression in these cells is mediating an inappropriate regulation of an existing signaling pathway (such as PI3 kinase), leading to the perturbation of a signaling pathway required for normal cell viability. Interestingly, no apoptosis was observed when PYK2 was overexpressed in PC12 cells where there is significant level of endogenous PYK2. Whether PYK2 can function to trigger apoptosis in cells in which it is normally expressed remains to be examined.
In summary, PYK2 appears to mediate different functions from those of FAK, a related protein tyrosine kinase. Ectopic expression of PYK2 led to apoptosis in several fibroblastic and epithelial cell lines, whereas over-expression of FAK did not result in cell death in any of these cell lines tested. The different effects of PYK2 and FAK on cell growth suggest that these two protein tyrosine kinases, in spite of similarity in overall structure, may mediate signaling via different pathways. Experiments to understand the mechanisms for the opposite effects mediated by PYK2 and FAK are in progress.
Acknowledgments
We are grateful to Dr. T. Sasaki for providing cDNA of CAKβ, to Dr. A. Klippel for providing constructs of PI3 kinase and Akt, to Dr. S. Parsons for providing cell lines transfected with c-Src and c-Src variants, to Dr. M. Weber for providing ts v-Src rat-1 cell line (LA29), and to Drs. Z.J. Luo and J.Y. Wu for suggestions and constructs of Bcl2. We thank Z.-H. Wang, M. Macklem, and C. Borgman for excellent technical assistance. We also thank Drs. L. Mei, S. Weed, R. Malink, M. Hart, and G. Kulik for helpful comments on the manuscript and discussions.
These studies were supported by Department of Health and Human Services grants CA 40042 and CA 29243 to J.T. Parsons; W.C. Xiong is supported by National Institutes of Health National Research Service Award fellowship NS 09918.
Abbreviations used in this paper
ECM
extracellular matrix
FAK
focal adhesion kinase
FRNK
FAK-related nonkinase
FAT
focal adhesion targeting domain
GST
glutathione-S-transferase
PI3 kinase
phosphatidylinositol-3-kinase
Footnotes
Address all correspondence to J. Thomas Parsons, Department of Microbiology, Box 441, Health Science Center, University of Virginia, Charlottesville, VA 22908. Tel.: (804) 924-5395. Fax: (804) 982-1071. E-mail: jtp@virginia.edu
References
- Andre E, Becker-Andre M. Expression of an N-terminally truncated form of human focal adhesion kinase in brain. Biochem Biophys Res Commun. 1993;190:140–147. doi: 10.1006/bbrc.1993.1022. [DOI] [PubMed] [Google Scholar]
- Avraham S, London R, Fu Y, Ota S, Hiregowdara O, Li J, Jiang S, Pasztor JM, White RA, Groopman JE, Avraham H. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J Biol Chem. 1995;270:27742–27751. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- Bellis SL, Miller JT, Turner CE. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J Biol Chem. 1995;270:17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature (Lond) 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–519. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Chen H-C, Guan J-L. Stimulation of phosphatidylinositol 3′-kinase association with focal adhesion kinase by platelet-derived growth factor. J Biol Chem. 1994a;269:31229–31233. [PubMed] [Google Scholar]
- Chen H-C, Guan J-L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1994b;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-C, Appedda PA, Parsons JT, Hidebrand JO, Schaller MD, Guan J-L. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995;270:16995–16999. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein tyrosine kinase, pp125FAK . Mol Cell Biol. 1994;14:147–155. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. MAPKs: new JNK expands the group. TIBS (Trends Biochem Sci) 1994;19:470–474. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for PYK2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature (Lond) 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- Dudek H, Data SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science (Wash DC) 1997;275:661–664. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher O. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai Y, Hanafusa H. Phosphatidylinositol kinase activity associates with viral pp60srcprotein. Mol Cell Biol. 1989;9:1651–1658. doi: 10.1128/mcb.9.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SG, Karl KA, Kiebler MA, Kandel ER. Focal adhesion kinase in the brain: novel subcellular localization and specific regulation by Fyn tyrosine kinase in mutant mice. Genes Dev. 1995;9:1909–1921. doi: 10.1101/gad.9.15.1909. [DOI] [PubMed] [Google Scholar]
- Guan JL, Shalloway D. Regulation of pp125FAKboth by cellular adhesion and by oncogenic transformation. Nature (Lond) 1992;358:690–692. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
- Guinebault C, Payratre B, Racaud-Sultan C, Mazarguil H, Breton M, Mauco G, Plantavid M, Chap H. Integrin-dependent translocation of phosphoinositide 3-kinase to the cytoskeleton of thrombin-activated platelets involves specific interactions of p85α with actin filaments and focal adhesion kinase. J Cell Biol. 1995;129:831–842. doi: 10.1083/jcb.129.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci USA. 1992;89:8487–8491. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte MT, Hildebrand JD, Burnham MR, Bouton AH, Parsons JT. p130cas, a substrate associated with v-Src and c-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996;271:13649–13655. doi: 10.1074/jbc.271.23.13649. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Schaller MD, Parsons JT. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAKto cellular focal adhesions. J Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JD, Schaller MD, Parsons JT. Paxillin, a tyrosine phosphorylated focal adhesion-associated protein binds to the carboxyl terminal domain of focal adhesion kinase. Mol Biol Cell. 1995;6:637–647. doi: 10.1091/mbc.6.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol. 1996;16:3169–3178. doi: 10.1128/mcb.16.6.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site- directed mutagenesis by overlap extension using polymerase chain reaction. Gene (Amst) 1989;7:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hu Q, Klippel A, Muslin J, Fantl WJ, Williams LT. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science (Wash DC) 1995;268:100–102. doi: 10.1126/science.7701328. [DOI] [PubMed] [Google Scholar]
- Hungerford JE, Campton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAKin cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeller R, Cantley LC. Phosphatidylinositol 3-kinase. Bioessays. 1994;16:565–576. doi: 10.1002/bies.950160810. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Cofer P, Downward J, Even G. Suppression of c-Myc-induced apoptosis by Ras signaling through PI(3)K and PKB. Nature (Lond) 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Klippel A, Reinhard C, Kavanaugh WM, Apell G, Escobedo MA, Williams LT. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik, G., A. Klippel, and M.J. Weber. 1997. Anti-apoptotic signaling by the IGF-I receptor, PI3 kinase and Akt. Mol. Cell. Biol. In press. [DOI] [PMC free article] [PubMed]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GO, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature (Lond) 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Moolenaar WH. Mitogenic action of lysophosphatidic acid. Adv Cancer Res. 1991;57:87–102. doi: 10.1016/s0065-230x(08)60996-3. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scattidi C, Bretz JO, Zhang M, Gentz R, et al. Flice, A novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Nagata S, Golstein P. The Fas death factor. Science (Wash DC) 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Parsons, J.T. 1996. Integrin-mediated signaling: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr. Opin. Cell Biol. 146–152. [DOI] [PubMed]
- Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130cas. Proc Natl Acad Sci USA. 1995;92:10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff M. Social controls on cell survival and cell death. Nature (Lond) 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Rankin S, Rozengurt E. Platelet-derived growth factor modulation of focal adhesion kinase and paxillin tyrosine phosphorylation in Swiss 3T3 cells. Bell-shaped dose response and cross-talk with bombesin. J Biol Chem. 1994;269:704–710. [PubMed] [Google Scholar]
- Rozengurt E. Neuropeptides as cellular growth factors: role of multiple signaling pathways. Eur J Clin Invest. 1991;21:123–134. doi: 10.1111/j.1365-2362.1991.tb01801.x. [DOI] [PubMed] [Google Scholar]
- Salgia R, Avraham S, Pisick E, Li J-L, Raja S, Greenfield EA, Sattler M, Avraham H, Griffin JD. The related adhesion focal tyrosine kinase forms a complex with paxillin in hematopoietic cells. J Biol Chem. 1996;271:31222–31226. doi: 10.1074/jbc.271.49.31222. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T. Cloning and characterization of cell adhesion kinase β, a novel protein- tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem. 1995;270:21206–21219. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. Focal adhesion kinase and associated proteins. Curr Opin Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK, a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Parsons JT. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK . Mol Cell Biol. 1993;13:785–791. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src . Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by Grb2 binding to focal adhesion kinase. Nature (Lond) 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Steller H. Mechanisms and genes of cellular suicide. Science (Wash DC) 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- Tachibana K, Sato T, D'Avirro N, Morimoto C. Direct association of pp125FAK with paxillin, the focal adhesion targeting mechanism of pp125FAK . J Exp Med. 1995;182:1089–1099. doi: 10.1084/jem.182.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science (Wash DC) 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of PYK2 by stress signals and coupling with JNK signaling pathway. Science (Wash DC) 1996;273:792–793. doi: 10.1126/science.273.5276.792. [DOI] [PubMed] [Google Scholar]
- Valius M, Kazlauskas A. Phospholipase C-γ1 and phosphatidylinositol-3 kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell. 1993;73:321–334. doi: 10.1016/0092-8674(93)90232-f. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Haecker G, Strasser A. An evolutionary perspective on apoptosis. Cell. 1994;76:777–779. doi: 10.1016/0092-8674(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Wilson LK, Luttrell DK, Parsons JT, Parsons SJ. pp60c-srctyrosine kinase, myristylation, and modulatory domains are required for enhanced mitogenic responsiveness to epidermal growth factor seen in cells overexpressing c-src. Mol Cell Biol. 1989;9:1536–1544. doi: 10.1128/mcb.9.4.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong WC, Montell C. Defective glia induce neuronal apoptosis in the repo visual system of Drosophila. . Neuron. 1995;14:581–590. doi: 10.1016/0896-6273(95)90314-3. [DOI] [PubMed] [Google Scholar]
- Xu LH, Iwens LV, Sturge GC, Yang XY, Liu ET, Craven RJ, Cance WG. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7:413–418. [PubMed] [Google Scholar]
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science (Wash DC) 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- Yu H, Li X, Marchetto GS, Dy R, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ, Graves LM, et al. Activation of a novel calcium-dependent protein-tyrosine kinase. J Biol Chem. 1996;271:29993–29998. doi: 10.1074/jbc.271.47.29993. [DOI] [PubMed] [Google Scholar]
- Zachary I, Sinnett-Smith J, Rozengurt E. Bombesin, vasopressin, and endothelin rapid stimulation of tyrosine phosphorylation in Swiss 3T3 cells. J Biol Chem. 1992;267:19031–19034. [PubMed] [Google Scholar]