Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture (original) (raw)
Abstract
Previous studies suggest that plectin, a versatile cytoskeletal linker protein, has an important role in maintaining the structural integrity of diverse cells and tissues. To establish plectin’s function in a living organism, we have disrupted its gene in mice. Plectin (−/−) mice died 2–3 days after birth exhibiting skin blistering caused by degeneration of keratinocytes. Ultrastructurally, hemidesmosomes and desmosomes appeared unaffected. In plectin-deficient mice, however, hemidesmosomes were found to be significantly reduced in number and apparently their mechanical stability was altered. The skin phenotype of these mice was similar to that of patients suffering from epidermolysis bullosa simplex (EBS)-MD, a hereditary skin blistering disease with muscular dystrophy, caused by defects in the plectin gene. In addition, plectin (−/−) mice revealed abnormalities reminiscent of minicore myopathies in skeletal muscle and disintegration of intercalated discs in heart. Our results clearly demonstrate a general role of plectin in the reinforcement of mechanically stressed cells. Plectin (−/−) mice will provide a useful tool for the study of EBS-MD, and possibly other types of plectin-related myopathies involving skeletal and cardiac muscle, in an organism amenable to genetic manipulation.
Keywords: Plectin, gene targeting, cytoskeleton, skin and muscle cytoarchitecture, EBS-MD, hemidesmosome
Plectin, a versatile cytoskeleton-associated protein of extraordinarily large size, is expressed abundantly in a wide variety of mammalian tissues and cell types. It is particularly prominent in stratified and simple epithelia, various types of muscle, and cells forming the blood–brain barrier (Wiche 1989; Errante et al. 1994). At the cellular level, plectin codistributes with different types of intermediate filaments (IFs) and is located at plasma membrane attachment sites of IFs and microfilaments, such as hemidesmosomes (Wiche et al. 1984), desmosomes (Eger et al. 1997), Z-line structures and dense plaques of striated and smooth muscle, intercalated discs of cardiac muscle, and focal contacts (Wiche et al. 1983; Seifert et al. 1992). Furthermore, direct linkages of IFs with actin filaments (Seifert et al. 1992; Foisner et al. 1995) and microtubules (Svitkina et al. 1996) have been demonstrated by whole-mount electron microscopy.
Electron microscopy of single molecules and secondary structure predictions based on the cloning and sequencing of rat plectin (Wiche et al. 1991; Elliott et al. 1997) revealed a multidomain structure comprising an ∼200-nm-long central α-helical coiled–coil rod structure flanked by large globular domains. A similar domain organization has been found in proteins that share partial sequence homology with plectin, such as desmoplakin (Green et al. 1990), the neuronal and epithelial isoforms of bullous pemphigoid antigen (BPAG) 1 (Sawamura et al. 1991; Yang et al. 1996), and envoplakin (Ruhrberg et al. 1996). The amino-terminal domain of plectin contains a highly conserved actin-binding domain of the type found in spectrin, dystrophin, and other related proteins (Elliott et al. 1997), whereas the carboxyl terminus harbors one or more IF-binding sites, including at least one for cytokeratins (Nikolic et al. 1996). Other identified interaction partners of plectin include α-spectrin (fodrin), high molecular weight microtubule-associated proteins, the nuclear IF protein lamin B, and integrin β4 (Herrmann and Wiche 1987; Foisner et al. 1991; G. Rezniczek, unpubl.). A number of different isoforms of plectin, not yet analyzed in full, can be expected to be involved in these various interactions. This notion is supported by the emergence of a complex gene regulatory mechanism involving close to 40 exons (Liu et al. 1996; Elliott et al. 1997; G. Wiche, unpubl.) and differentially spliced transcripts, including some that are derived from alternative first-coding exons (Elliott et al. 1997).
The strategic localization of plectin at the cytoskeleton–plasma membrane interface and its versatile binding capacities suggest that plectin could strengthen cells against mechanical stress, both along their surface and at their internal cytoskeleton anchorage sites. This notion is supported by several recent reports demonstrating defects in the plectin gene in epidermolysis bullosa simplex (EBS)-MD, an autosomal recessive disease characterized by skin blistering combined with muscular dystrophy (Chavanas et al. 1996; Gache et al. 1996; McLean et al. 1996; Pulkkinen et al. 1996; Smith et al. 1996). Preliminary evidence indicates that defects in plectin also have a role in EBS-Ogna, an autosomal dominant severe skin blistering disease (Koss-Harness et al. 1997). The concept of cells being mechanically reinforced through plectin crosslinking activities is fully in line with previous findings showing that perturbations in IF assembly (for review, see Fuchs 1996) and IF-cell membrane connections (Guo et al. 1995) can jeopardize the mechanical integrity of cells.
Despite the wealth of supportive indications, functional in vivo evidence for plectin’s role as a cytoskeletal linking and stabilizing element of cells in mechanically stressed tissues is lacking. In particular, the fundamental question, whether plectin is essential for the formation and/or functional maintenance of the cytoarchitecture of such cells, is not yet clear. Is plectin required for the assembly of junctional complexes prominent in such tissues, such as hemidesmosomes and desmosomes? Is it important for filament anchorage at junctional complexes, and what role does it have in the functional integrity and physiology of cells under constant mechanical stress? To begin to address some of these questions, to define more precisely the role of plectin in tissue development and integrity, and to investigate its relation to EBS-MD, we have generated plectin-deficient mice of two types by targeted disruption of gene regions corresponding to amino-terminal or central rod domains. Data obtained with both types of mice demonstrate that embryonal development can take place in the absence of plectin, but that the protein is essential in maintaining the integrity of skin, skeletal muscle, and heart cytoarchitecture.
Results
Plectin (−/−) mice exhibit skin blistering and die within a few days after birth
To generate plectin null mice, two separate targeting vectors were constructed. In one, exon 31, which encodes the entire central rod domain of plectin, was disrupted (Fig. 1A), in the second, 5′ sequences were deleted that included exon 2, whose 5′ end serves as splice acceptor of all four recently identified alternative first-coding exons of the protein (Elliott et al. 1997), exon 3, and part of exon 4 (Fig. 2). Both targeting vectors were transfected separately into embryonal stem (ES) cells, line R1 (Nagy et al. 1992). Using Southern analysis, four ES cell clones were identified that contained one allele carrying a mutated exon 31 (recombination frequency 1:30) and three clones containing one allele with deletions of exons 2–4 (recombination frequency 1:80). Two ES cell clones bearing a mutated exon 31 allele (p12-88 and p12-61) and one clone heterozygous for deleted exons 2–4 (p2-86) were used to generate chimeric male mice of two types (p12-88 and p12-61; or p2-86). By crossing chimeric founders with wild-type females, offspring heterozygous for each type of mutated plectin allele were obtained, as confirmed by Southern analysis (Figs. 1B and 2B). Heterozygous mice of both types developed and reproduced normally and were indistinguishable from their wild-type littermates. Crossing of heterozygous animals resulted in offspring homozygous for each type of plectin mutation in the expected ratio. Homozygous offspring from all three independently derived lines exhibited a similar phenotype—they died between day one and three after birth showing skin detachment, especially at fore- and hindlimbs, and in some cases around the mouth and nasal cavities. Large blisters reaching the size of up to one centimeter in diameter were found particularly at the upper and lower extremities and those were often accompanied by bleedings, particularly at their extremities (Fig. 3A). Usually not found in an EBS type of skin blistering, this type of bleeding might indicate a possible perturbation of vascular endothelial cells, which normally express plectin at relatively high levels (Wiche et al. 1983; Errante et al. 1994). The extent of phenotypic alterations expressed before death varied among plectin (−/−) mice; in most cases they were smaller than their normal littermates (Fig. 3A), because of the reduced gain in body weight during their short life compared with controls.
Figure 1.
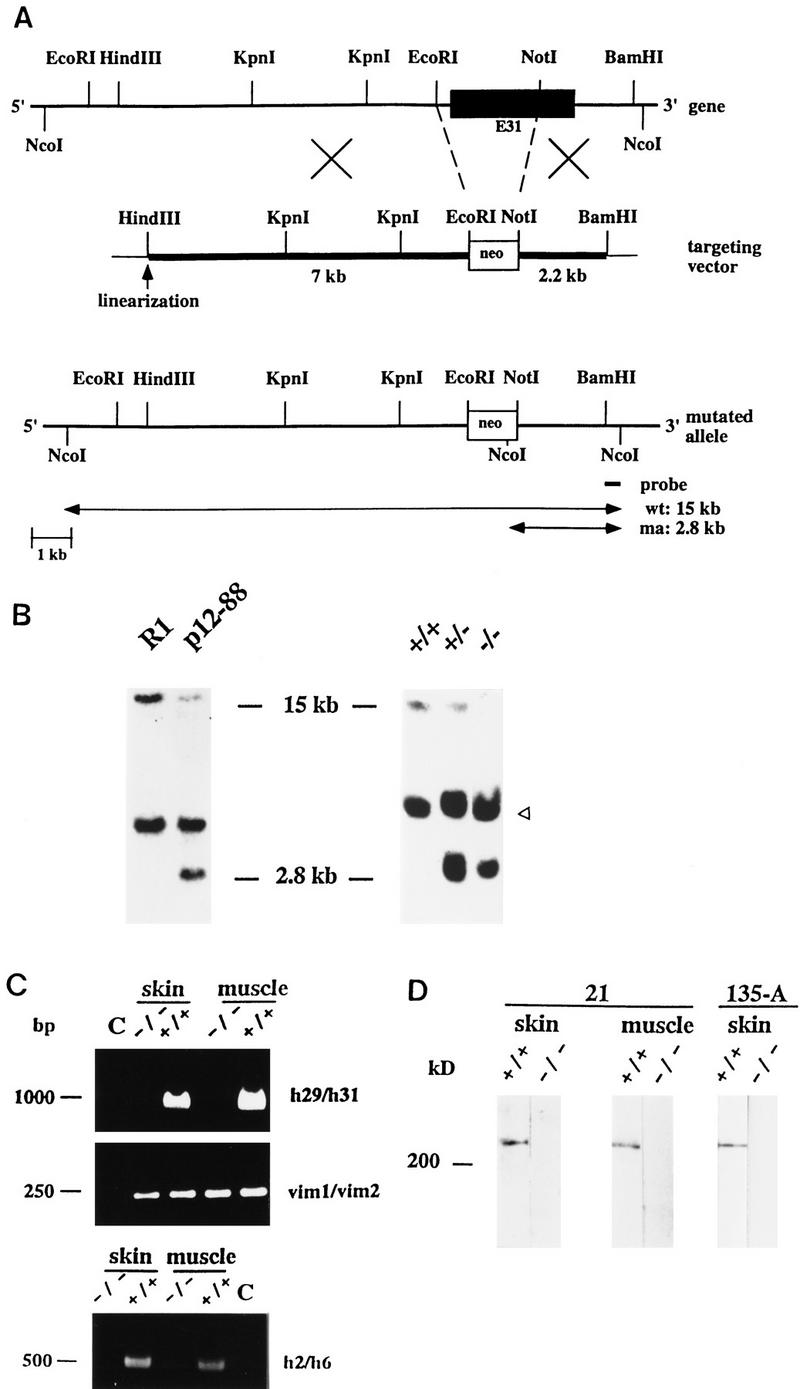
Targeted inactivation of the plectin gene by disruption of rod-encoding exon 31. (A) Schematic representation of the plectin gene locus, the targeting vector, and the disrupted allele. Thick lines in targeting vector diagram represent homology regions. Important restriction sites, exon 31 (solid box), and part of gene replaced by neor-cassette (broken lines) are indicated. Digestion of the genomic DNA with restriction enzyme _Nco_I released a 15-kb fragment from the wild-type (wt) allele and a 2.8-kb fragment from the mutated allele (ma). Both alleles are detectable by a 3′ external probe generated by _Nco_I digestion, as indicated. (B) Southern blot analysis of genomic DNA prepared from parental ES cell clone (R1), targeted clone (p12-88), and from tail biopsies of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice. Presence of wild-type and mutant alleles are indicated by 15- and 2.8-kb fragments, respectively. Note, nonspecific hybridization of probe with a 5-kb fragment (arrowhead) present in all samples. (C) RT–PCR analysis of skin and muscle tissues of plectin wild-type (+/+) and mutant (−/−) mice using plectin-specific or vimentin-specific primer pairs as indicated. (Lanes C) RT–PCRs performed without reverse transcriptase. Size markers (in bp) are indicated. (D) Immunoblotting of skin and muscle tissue homogenates from wild-type (+/+) and plectin (−/−) mice using rabbit antiserum to plectin (21) and mouse antiserum 135-A to the carboxy-terminal domain of plectin (right). Note that no immunoreactive protein bands were found below the ∼300-kD species on the blot portion shown (lower end of lanes, ∼90 kD) or below (data not shown). Comparable amounts of protein were loaded onto gels as confirmed by Ponceau S staining of membranes (data not shown). Molecular mass marker, 200 kD.
Figure 2.
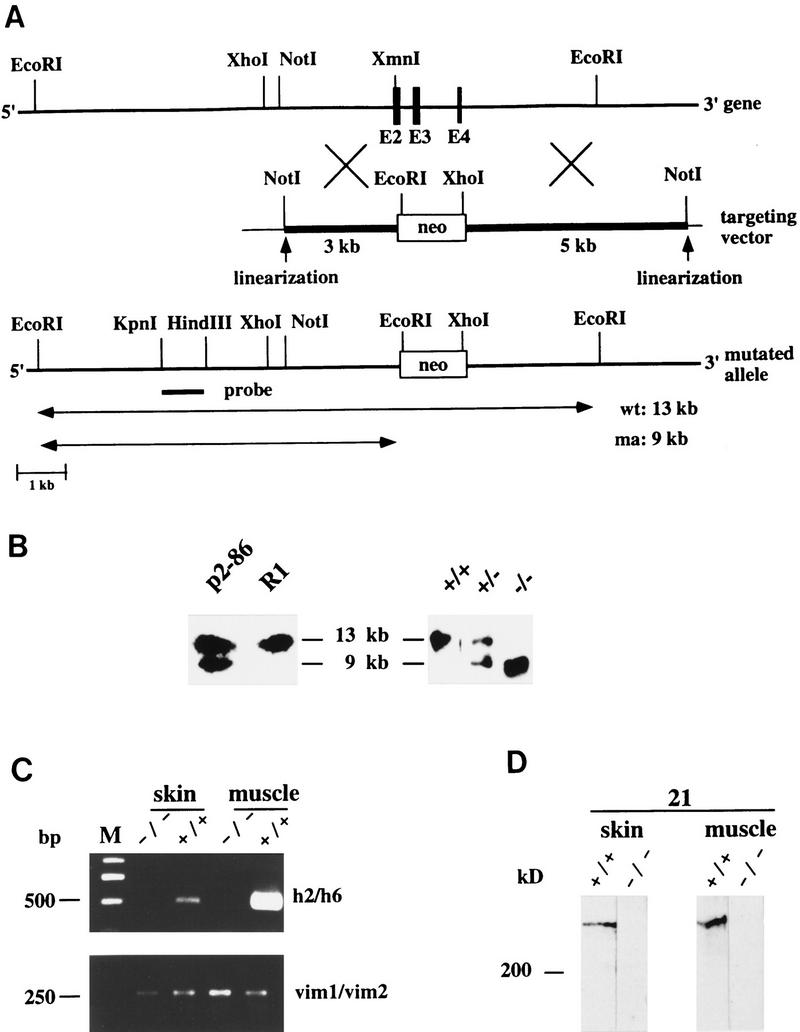
Targeted disruption of the plectin gene by replacement of exons 2–4 with a neor-cassette. (A) Schematic representation of the plectin gene locus, the targeting vector, and the disrupted allele. For specifications, see Fig. 1. Exons 2–4, disrupted by replacement with neor-cassette, are shown as solid boxes. _Eco_RI–DNA fragments of 13 and 9 kb (both detectable with a 5′ external probe generated by _Kpn_I–_Hin_dIII digestion) were indicative of wild-type (wt) and mutant alleles (ma), respectively. (B) Southern blot analysis of genomic DNA prepared from the parental ES cell clone (R1), the targeted clone (p2-86), and tail biopsies of wild-type mice (+/+), and mice heterozygous (+/−), or homozygous (−/−) for the mutation. (C) RT–PCR analysis of skin and muscle tissues of plectin (+/+) and (−/−) mice using plectin-specific (h22/h6; h2/h6) or vimentin-specific (vim1/vim2) primer pairs. Size markers (in bp) are indicated. (D) Immunoblotting of skin and muscle tissue homogenates from wild-type (+/+) and plectin (−/−) mice.
Figure 3.
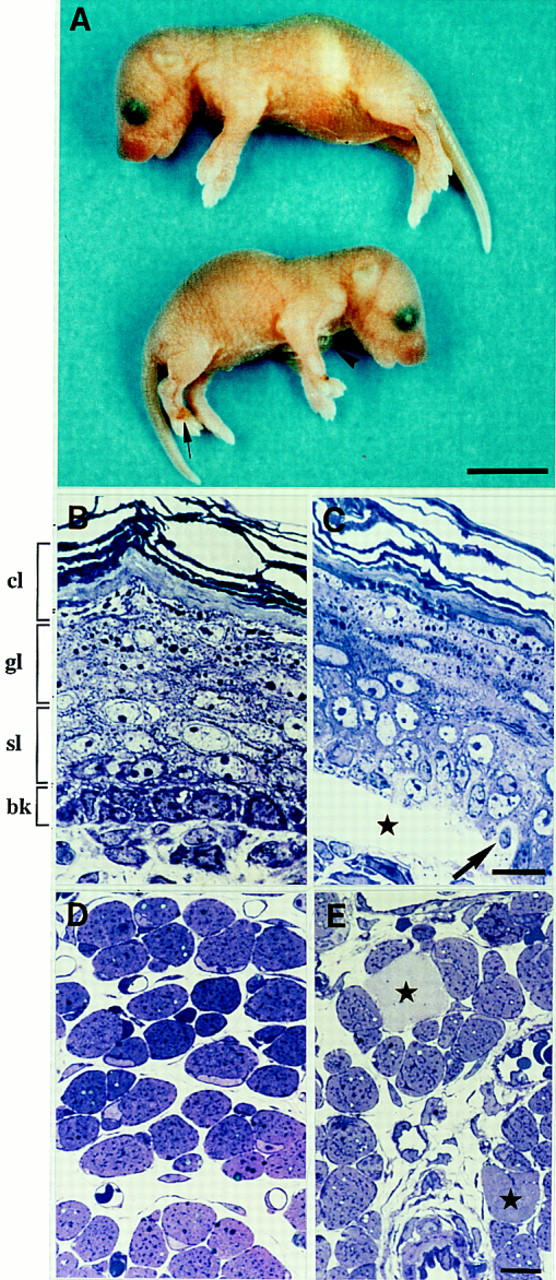
Phenotypic analysis of plectin-deficient mice. (A) Comparison of 2-day-old wild-type (top) and plectin (−/−) (bottom) offsprings. Note large blister on forelimb (arrowhead), bleedings at extremities (arrow) and smaller size of mutant mouse. (B–E) Epoxy resin-embedded and Toluidine blue-stained sections (0.5 μm) of skin (B,C) or muscle biopsies (D,E) taken from 2-day-old animals. Note regular arrangement of epidermal cell layers [(cl) cornified layers; (gl) granular layers; (sl) spinal layers; (bk) basal keratinocytes] in wild-type littermate mouse (B), and blister formation between the dermis and the upper epidermal layers of a plectin (−/−) mouse with EBS (C, asterisk); at the blister margin basal keratinocytes are pale and swollen (arrow). (D) Cross section of skeletal muscle of a heterozygous control littermate animal with normal appearance and arrangement of muscle fibers. (E) Skeletal muscle of a plectin (−/−) mouse, showing scattered degenerating muscle fibers (asterisks). Bars, A, 0.5 cm; B–E, 10 μm.
Plectin mRNA and plectin protein are undetectable in skin and muscle of (−/−) mice
To demonstrate that the mutations generated were plectin null mutations, we conducted reverse transcriptase (RT)–PCR with RNA extracted from skin and muscle tissues of 2- to 3-day-old (−/−) mice. Using primer pairs leading to amplified fragments covering the targeted exon 31 (Fig. 1C, upper panel), exons 2–4 (Fig. 2C, upper panel), and exon 32, which encodes the carboxy-terminal part of plectin (data not shown), no mRNAs could be detected in the tissues of (−/−) mice, whereas positive signals were obtained in control animals. All RNA samples examined were of comparable amount and quality, as verified by amplification of a reversely transcribed cDNA fragment of the unrelated gene vimentin (Figs. 1C, middle panel, and 2C, lower panel). To assess whether truncated transcripts were expressed in the case of the rod/exon 31-deficient mice, an additional RT–PCR was performed using primers (h2/h6) to detect 5′ sequences of the plectin gene (exons 2–6). Contrary to control animals, no amplified fragment of the expected size (0.5 kb) could be detected in these mutant mice, eliminating the possibility that truncated versions of plectin were expressed in exon 31 (−/−) mice.
The major isoform of plectin, a polypeptide of apparent molecular mass of ∼300 kD has been shown previously to be abundantly expressed in a wide variety of different tissues and cell types, including skin and various types of muscle (for review, see Wiche 1989). To confirm the absence of plectin expression in mutant mice on the protein level, skin and muscle tissue homogenates prepared from mutant and wild-type mice were analyzed by immunoblotting using a rabbit anti-plectin antiserum (Figs. 1D and 2D) and monoclonal antibodies to plectin (data not shown), both immunoreactive with the rod domain of the mouse antigen. The ∼300-kD isoform was clearly detectable in both tissue extracts from wild-type mice, but undetectable in those from any of the plectin (−/−) mice. Similar observations were made using a mouse antiserum directed against a carboxy-terminal domain (repeat 4) of plectin (Fig. 1D, lanes 135-A). Because in this case no immunoreactive proteins of molecular mass smaller that the 300 kD species were found (Fig. 1D, 135-A, −/−; data not shown), expression of truncated plectin mutant proteins encoded by plectin mRNA aberrantly expressed from altered genes seemed highly unlikely. The mRNA and protein data combined, convincingly demonstrated that the disruption of the plectin gene by deletion of a central exon (exon 31) or of 5′ exons (exons 2–4) was successful.
Plectin deficiency leads to rupture of basal keratinocytes without preventing hemidesmosome or desmosome formation
Tissue examination at the light microscopy level revealed pathological alterations in homozygous plectin-deficient animals that were restricted to skin and skeletal muscle, with no major phenotypic differences apparent between the two types of null mutations. Heterozygous animals did not show this alteration and were indistinguishable from wild-type animals in overall morphology.
In the skin, large blisters were located between dermis and the superficial epidermal layers (Fig. 3C). Smaller blisters were also present at other sites of the integumentum and in particular in the epithelial layers of the oral mucosa and the tongue (data not shown). At the border of the blisters toward the surrounding intact epidermis, degenerating cells were encountered frequently in the region of the basal cell layer (Fig. 3C, arrow). Furthermore, the basal cell layer was absent completely from the upper epidermal layers that formed the blister roof.
Electron microscopy revealed disruption and degeneration of the basal epidermal cell layer that were particularly evident at the blister margins. Disruption of basal keratinocytes occurred throughout their cytoplasm, in some instances in perinuclear regions (Fig. 4A), in others through basal (Fig. 4B) or apical (data not shown) cytoplasmic regions, without an apparent preference for any of these sites. Other basal keratinocytes showed signs of more advanced degeneration with granular dissolution of cytoplasmic components in spite of preservation of some mitochondria and dense bodies (Fig. 4C). In these cells, keratin filaments were not detectable, although basal hemidesmosomes displaying inner and outer plate structures were preserved (Fig. 4D, arrowhead). At the center of the blisters, basal keratinocytes were no longer detectable and replaced by a fluid-filled space. Parts of the basal lamina overlying the dermis were covered by a partially fragmented cell membrane, which apparently was a remnant of the basal cell surface membrane of keratinocytes (Fig. 4F, arrow).
Figure 4.
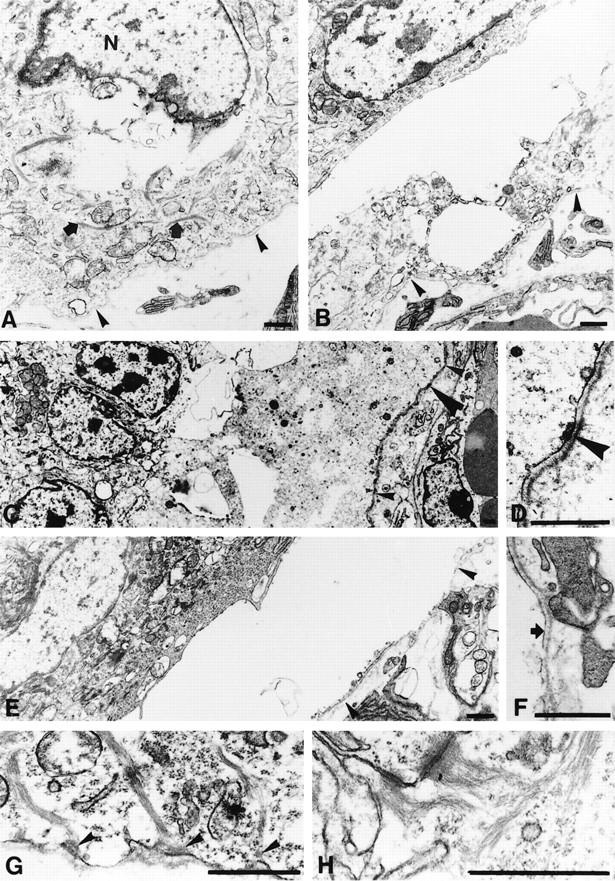
Ultrastructural analysis of plectin (−/−) mouse skin. Specimens shown are from exon 2–4 (A,B,E–H) and exon 31-deficient mice (C,D). (A) Basal keratinocyte at the edge of an epidermal blister with cytoplasmic rupture in perinuclear regions; cytoplasmic components, including keratin filaments, still appear intact (arrows). The epidermal basement membrane is marked with arrowheads. (N) nucleus. (B,C) More advanced degeneration of basal keratinocyte with appearance of empty spaces in the cytoplasm and disorganization of cytoplasmic components; these are replaced by granular, moderate electron-dense material, as particularly evident in C. Arrowheads in B and small arrowheads in C mark epidermal basement membrane; large arrowhead in C, position of insert shown in D. (D) Higher magnification of C; note, inner plate of the hemidesmosome (arrowhead) is still preserved. (E) Complete dissolution of the basal keratinocyte layer, which is replaced by the fluid-filled blister. Arrowheads indicate position of epidermal basement membrane. (F) Higher magnification of the same blister as shown in E; in spite of complete dissolution of the basal keratinocyte, remnants of the cell membrane are still preserved and attached to the epidermal basement membrane (arrow). (G) Hemidesmosomes are present in basal keratinocytes outside blisters. Arrowheads denote intact outer and inner plates as well as insertion of keratin filaments into the inner plate structure. (H) Desmosomes in the upper epidermal layers are regularly structured. Bars, 1 μm.
In the adjacent parts of the epidermis that were not affected by blistering, we observed keratin filaments dispersed throughout the cytoplasm of basal keratinocytes (Fig. 4A,B,G). On a qualitative basis, the distribution of these filaments appeared unaltered compared with that of control animals. This suggested that the formation of the keratin cytoskeleton in these cells was independent of plectin expression. In such nonruptured areas of plectin (−/−) mouse skin, hemidesmosomes were observed along the basal cell surface membrane of basal keratinocytes (Fig. 4G). These structures showed well developed outer and inner plates and exhibited keratin filament association. Compared to basal keratinocytes of control mice (Fig. 5A), however, the keratin filaments appeared looser and less bundled, particularly at their insertion site into the inner plate structure (cf. Figs. 4G and 5A). On a semiquantitative basis, the number of hemidesmosomes in plectin-deficient mice was found to be reduced to ∼50% compared with control mice.
Figure 5.
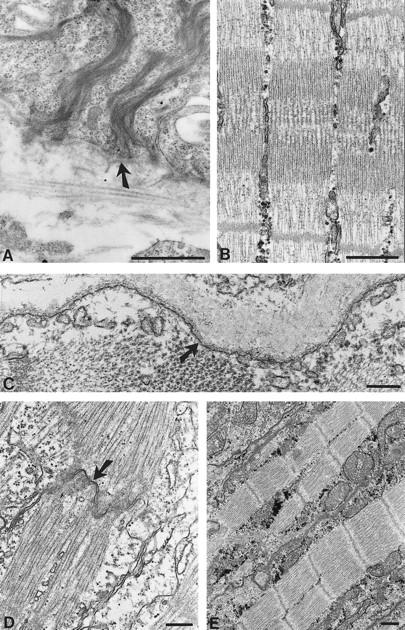
Electron micrographs of skin, skeletal muscle, and heart muscle biopsies taken from 2-day-old wild-type mice. (A) Skin sections showing normal hemidesmosomes with emanating dense keratin filament bundles (arrow). (B,C) Sections through skeletal muscle showing intact sarcomeric structures (B) and the intact plasma membrane (arrow) of a muscle fiber (C). (D,E) Sections through heart muscle exhibiting intact intercalated discs (D, arrow) and normal sarcomeric structures (E). Bars, 1 μm (A,D); 0.5 μm (B); 0.2 μm (C,E).
The upper layers of the epidermis, which were not affected by the blistering process, were ultrastructurally unaltered in comparison with controls. In particular, desmosomes between adjacent epithelial cells appeared unaffected, displaying numerous keratin filaments anchored to their plate structures (Fig. 4H).
Using immunohistochemistry and confocal microscopy, plectin expression was detected in normal mouse skin as a bright outlining of all epidermal keratinocyte membranes, a pronounced staining of the dermo-epidermal borderline along with weak cytoplasmic staining (Fig. 6A). In a side view, generated by optically sectioning the skin sample in a plane perpendicular to the keratinocyte cell layers, plectin was shown clearly to exhibit the strongest signal at the basal membrane of basal keratinocytes and weaker staining in the intracellular space and at the apical membrane of the cell (Fig. 6G,H). In contrast, no plectin signal was obtained in epidermal cells of plectin (−/−) mice (Fig. 6B). To assess whether the absence of plectin affected the expression or localization of other hemidesmosomal proteins, skin sections of plectin (−/−) mice were stained using antibodies to integrin β4 and BPAG1. Double immunfluorescence of skin sections of wild-type mice using an antiserum directed against the cytoplasmic domain of integrin β4 (Giancotti et al. 1992) revealed a prominent integrin β4 signal clearly colocalizing with plectin at the basal side of epidermal basal keratinocytes (Fig. 6A and 6A′). In addition, less-pronounced integrin β4-specific staining could be observed in the cytoplasm of these cells, consistent with the previously reported proteolytic processing of the cytoplasmic domain of this integrin subunit protein (Giancotti et al. 1992). Unexpectedly, however, in plectin (−/−) mice, the integrin β4 signal was substantially reduced in intensity, particularly at the basal pole of basal keratinocytes, suggesting a dependence on plectin expression (Fig. 6B′). To confirm this observation, additional immunofluorescence microscopy experiments were carried out using a monoclonal anti-integrin β4 antibody that was immunoreactive with the extracellular portion of the molecule (Kennel et al. 1989). As expected, the integrin β4 signal was found to be confined to the basal membrane of basal keratinocytes in both wild-type and mutant mice skin sections (Fig. 6C,D). Again, however, in plectin-deficient mice, the detectable signal was much weaker and in some regions of skin even absent (Fig. 6D).
Figure 6.
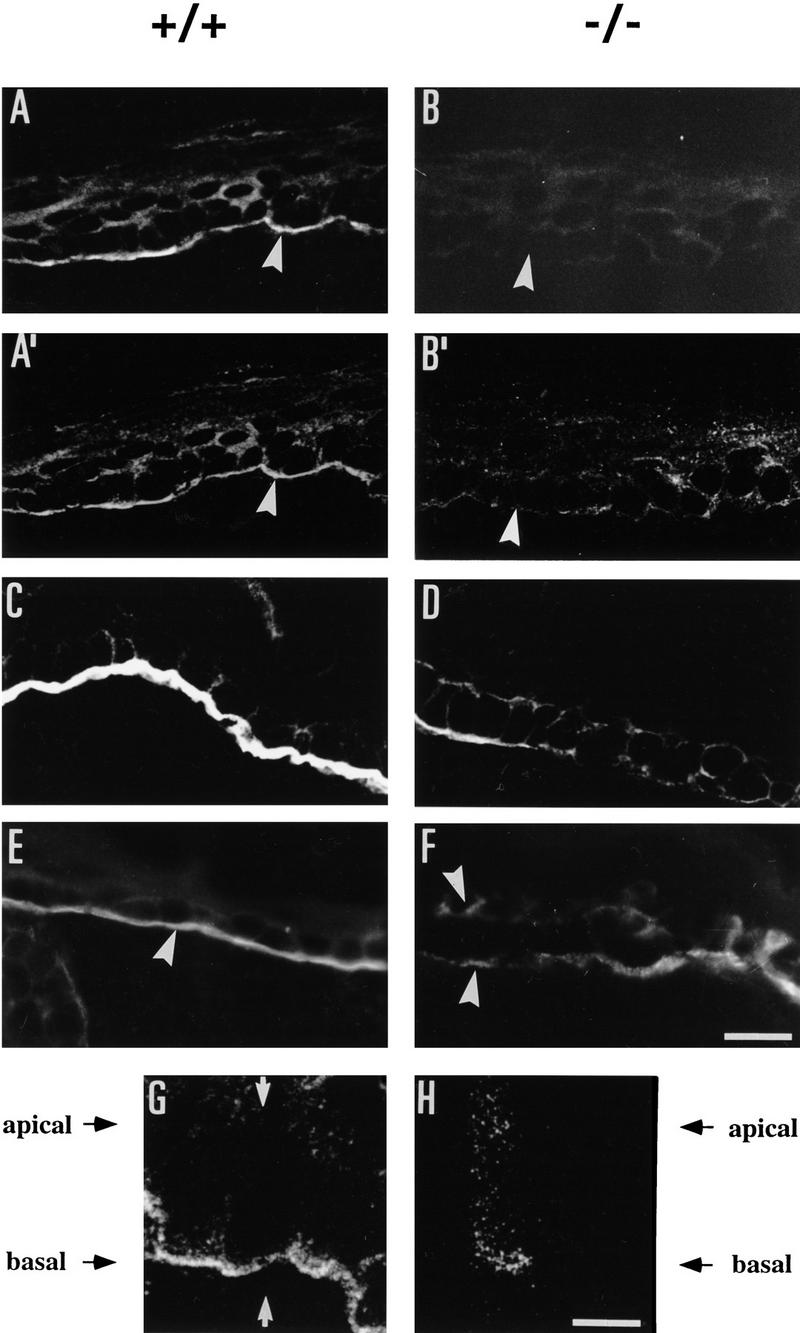
Detection of plectin, integrin β4, and BPAG1 in skin of wild-type (+/+) and plectin-deficient (−/−) mice using immunofluorescence microscopy. Frozen sections were prepared from skin biopsies of 2-day-old mice. (A, A′ and B, B′) Double staining using anti-plectin mAb 10F6 (A,B) and anti-integrin β4 cytoplasmic domain antiserum (A′,B′). (C,D) Anti-integrin β4 extracellular domain mAb 346-11A; (E,F) anti-BPAG1 (mAb-5E). Arrowheads in A, A′, B, B′, and E denote basal membrane of basal keratinocytes, in F, blister floor and blister top. (G,H) Optical confocal microscopy sections in a plane (G, arrows) perpendicular to the layer of the basal keratinocytes. Bars, 20 μm (A–F); 6 μm (G,H).
BPAG1 expression was well preserved at the basal cell surface membrane of basal keratinocytes in dermal/epidermal border areas that showed no blistering (Fig. 6E). In the blisters, the BPAG1 signal was detected mostly at the blister floor, continuing the linear signal from non-blistered areas. In addition, specific, but somewhat weaker and more patchy BPAG1 staining was detected at the roof of the blisters (Fig. 6F), consistent with a mechanism in which cell rupture occurred at the level of the hemidesmosome inner plate structure, which is formed at least in part by BPAG1 (Guo et al. 1995).
Myofibril integrity is impaired severely in skeletal and heart muscle of plectin (−/−) mice
The overall hallmark of the plectin (−/−) phenotype in skeletal muscle observed at the light microscopy level was an increased number of necrotic muscle fibers. Whereas in normal littermates degenerating changes of muscle were found in less than 0.1% of single muscle fibers, in plectin (−/−) mutant mice, necrotic changes of various degrees were found in 0.5%–1% of muscle fibers (Fig. 3E). Occasionally, myophagocytosis was observed in association with the necrotic fibers of mutant mice (data not shown). Although these pathological alterations were noticed in all muscle groups, the paravertebral muscles as well as the distal limb muscles seemed to be affected preferentially. At the ultrastructural level, necrotic changes involving focal loss of myofilaments of various degrees were observed in ∼20% of the muscle fibers. No evidence of apoptotic events in the nuclei of muscle cells was observed at the light and electron microscopy levels. Focal disruptions of sarcomeres affecting Z-lines and adjacent myofibrils were frequently noticeable on longitudinal sections obtained from plectin-deficient mice (Fig. 7A,B), but not on those from wild-type muscle tissue (Fig. 5B). In affected areas, the Z-lines appeared to have lost their tight arrangement, displaying a poorly focused, smearing and streaming appearance. Similar morphological characteristics, including the focal disintegration of myofibrils, focal loss of cross-striation and Z-band streaming are symptomatic of one type of benign nonprogressive congenital myopathy, termed minicore or multicore disease (Penegyres and Kakulas 1991). The overall structure of the sarcolemma seemed to be intact, except for short ruptures of the plasma membrane observed occasionally (Fig. 7C, arrowheads), which were not detectable in control cells (Fig. 5C).
Figure 7.
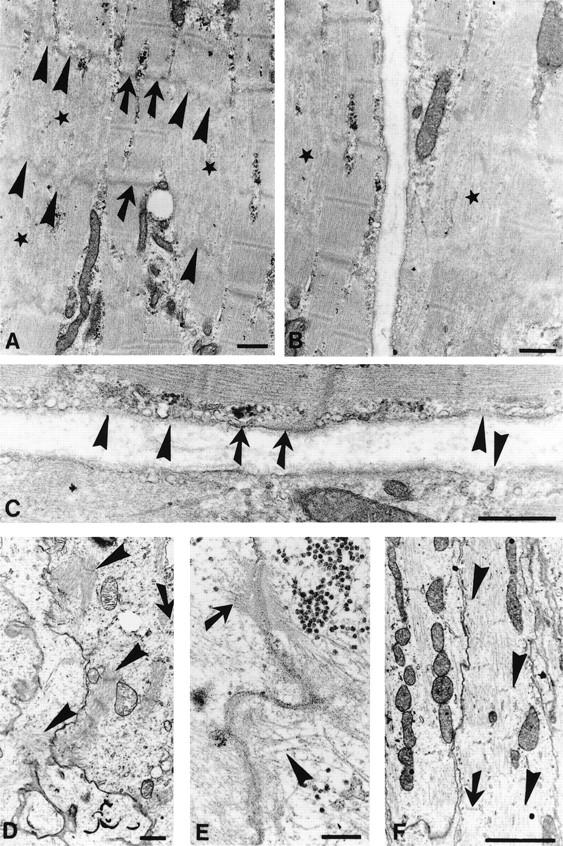
Electron micrographs of skeletal and heart muscle of plectin (−/−) mice. (A,B) Longitudinal sections through skeletal muscle biopsies obtained from 2-day-old plectin (−/−) mice. Corelike focal disruption of myofibrils are indicated by stars, intact Z-bands by arrows, and disrupted Z-bands by arrowheads. (C) Higher magnification of B; intact plasma membrane is indicated by arrows, disrupted membrane regions by arrowheads. (D–F) Heart muscle biopsies of 2-day-old plectin-deficient mice. Note aberrant isolated myofibril bundles (D, arrowheads), dilated intercalated disc (E, arrowhead) alongside normal desmosome exhibiting keratin filament anchorage (E, arrow), and focal loss of Z-bands (F, arrowheads) next to still intact Z-bands (F, arrow). Bars, 0.5 μm (A–C); 1 μm (D); 0.2 μm (E); and 2 μm (F).
Similar to rat and human tissues (Wiche et al. 1983, 1984), immunofluorescence microscopy of skeletal muscle of wild-type mice revealed plectin expression at the periphery of muscle fibers and in the interior of the sarcoplasm (Fig. 8A), where it was particularly prominent at Z-lines (Fig. 9A). On the other hand, no expression of plectin was detectable in skeletal muscle of (−/−) mice by immunohistochemistry (Fig. 8B and 9B). Among other cytoskeletal proteins assumed to have a role in the development and integrity of sarcomeric cytoarchitecture, such as vinculin, β-spectrin, and dystrophin, vinculin expression was found to be the one affected most significantly by plectin deficiency. In mutant mice, the vinculin signal at the sarcolemma of most skeletal muscle fibers was reduced dramatically compared with wild-type tissue, resulting in a patchy, rather than continuous outlining of muscle fiber surfaces (Fig. 8D). The expression levels of β-spectrin (Fig. 8F) and dystrophin (data not shown) were also found to be diminished compared with control mice, indicating a possible functional coupling of plectin and these proteins, all of which are involved in the anchorage of cytoskeletal filament networks to the sarcolemma.
Figure 8.
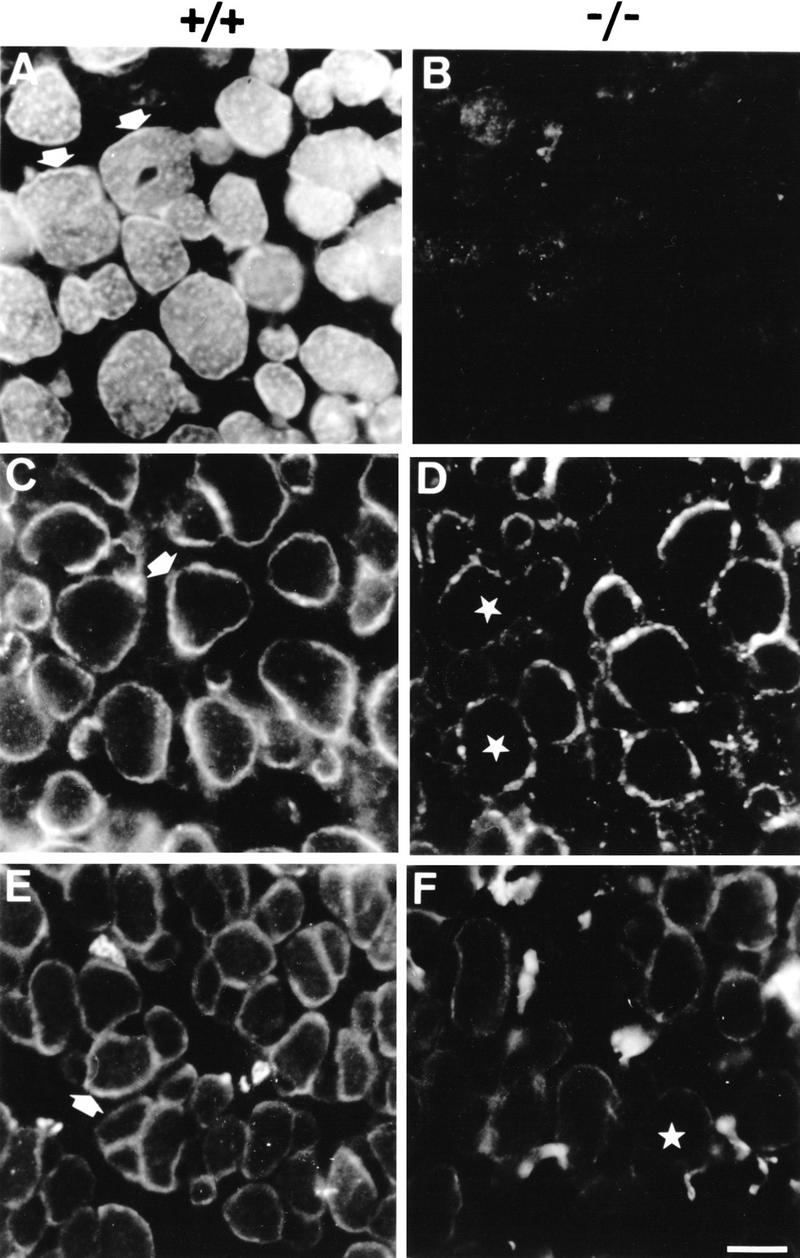
Immunofluorescence microscopy of cross sections through skeletal muscle from 2-day-old wild-type (+/+) and plectin-deficient (−/−) mice. (A,B) Anti-plectin mAb 10F6; arrows in A indicate pronounced staining at the periphery of muscle fibers. (C,D) Anti-vinculin; note prominent staining at the periphery of control muscle fibers (C, arrow) and the irregular, patchy staining pattern of muscle fibers in plectin-deficient mice (D, stars). (E,F) anti-spectrin; the peripheral staining observed in wild-type mice (E, arrow) is substantially diminished in plectin-deficient mice (F, star). Bar, 20 μm.
Figure 9.
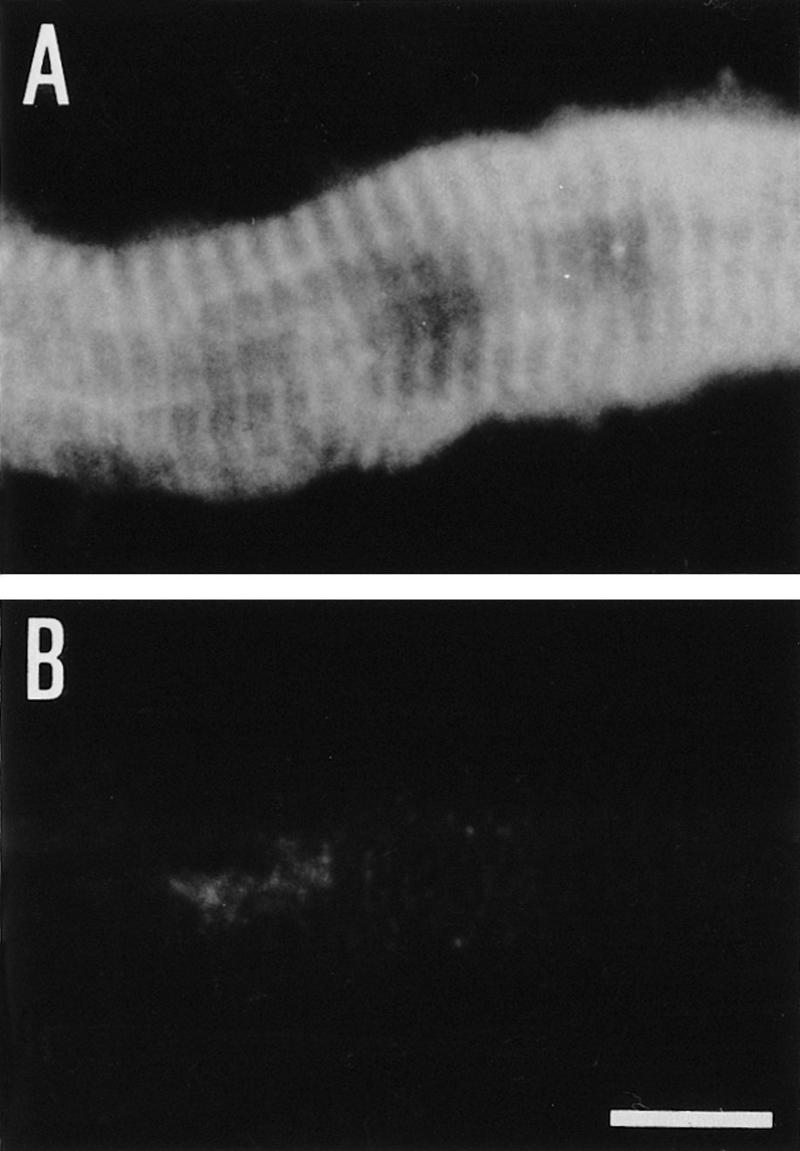
Immunfluorescence microscopy (anti-plectin mAb 10F6) of longitudinal sections through skeletal muscle. Muscle biopsies were obtained from 2-day-old wild type (A) and plectin-deficient (B) mice. Bar, 12 μm.
In the myocardium of plectin (−/−) mice, no overt abnormalities were found at the light microscopy level. The atrial and ventricular walls appeared regularly developed and necrotic cardiomyocytes were not observed. At the ultrastructural level, however, partial disintegration of intercalated discs and adjacent myofibers were a common finding in cardiac muscle cells of plectin-deficient mice (Fig. 7E), contrary to normal myocardium (Fig. 5D,E). In addition, sarcomeres were found frequently to be loosely and irregularly arranged in these cells. The observed focal streaming of Z-lines and the disintegration of myofibril alignment were similar to those characteristic of the corelike alterations observed in skeletal muscle of mutant mice (Fig. 7, cf. E and F with A and B). Furthermore, aberrant isolated myofibril bundles and aberrant accumulation of Z-band material were observed (Fig. 7D).
Discussion
Knockout mice reveal essential function of plectin in mechanical strengthening of cells
Previous studies revealed that plectin molecules interlink IFs with different cytoskeletal filament systems and are involved in the anchoring of IFs at the plasma membrane. Based on this, it has been proposed that plectin has a major role in the structural organization of the cytoskeleton and may mechanically strengthen cells (Wiche 1989). To test this hypothesis in a living organism, we generated two types of homozygous plectin (−/−) mice by independent disruption of a central exon or 5′ exons of the gene. In both types of plectin-deficient mice the lack of plectin resulted in cell degeneration and damage of different severity in a number of tissues.
By far the most severe phenotype was observed in skin where the separation of epidermis from dermis resulted in blistering that might have been responsible, at least partly, for death occurring 2–3 days after birth of the animals. The degeneration of basal keratinocytes could be traced over different stages. It started with the disruption and lysis of internal cytoplasmic structures and organelles and ended with total disruption and loss of basal keratinocytes. The suprabasal keratinocyte layers remained intact. This skin phenotype was very similar to those observed in transgenic mice expressing a truncated version of keratin 14 (Vassar et al. 1991) and in humans exhibiting a homozygous mutation in the keratin 14 gene resulting in a null mutation (Chan et al. 1994; Rugg et al. 1994). In both cases it was postulated that blistering reflected the disruption of the cytoplasm of basal keratinocytes as a consequence of an impaired keratin filament formation. The observation, however, of apparently intact keratin filaments in plectin-deficient, but not yet disrupted, keratinocytes indicated that keratin filament formation per se was independent of plectin expression. Nevertheless, the stability of keratin structures in basal keratinocytes of plectin (−/−) mice appeared severely compromised, probably because of missing crossbridges between filaments themselves and their impaired linkages to other cellular structures, in particular the plasma membrane. Similarly, in skeletal muscle and heart of plectin (−/−) mice, only a fraction of the myofibers were found to be necrotic and focal lesions occurred alongside intact tissue exhibiting a proper muscle cytoarchitecture. Again, this indicated that plectin is not essential for the formation and assembly of myofibrils, but is required for maintaining their structural integrity and stability.
Plectin, a stabilizer of hemidesmosomes and other junctional complexes
Plectin or its putative isoform HD1 (Hieda et al. 1992) have been shown to colocalize with IF-associated as well as actin-based cell junctional complexes, including hemidesmosomes, desmosomes, focal contacts of cells in culture, dense plaques of smooth muscle, and other junctional adhesive complexes (Wiche et al. 1983, 1984; Seifert et al. 1992; Eger et al. 1997; Sanchez-Aparicio et al. 1997). This raises the question of whether the formation and/or functional integrity of such junctions were affected by plectin deficiency. Regarding hemidesmosomes and desmosomes, no impairment of their formation was noticeable in epidermal keratinocytes of plectin (−/−) mice. Both structures were found to be ultrastructurally intact and numerous filaments emanating from their cytoplasmic plate structures were visualized by eletron microscopy, indicating that both structures were still able to serve as filament anchorage sites at least to a certain extent. In contrast to plectin (−/−) mice, BPAG1-deficient mice clearly lacked the inner plate and consequently did not show keratin filament anchorage to the basal cell surface membrane (Guo et al. 1995). Furthermore, because BPAG1 expression in nonblistered tissue areas of plectin-deficient mice appeared to be normal, we conclude that plectin, in contrast to BPAG1, is dispensable for hemidesmosome formation and that BPAG1 expression is not influenced by plectin ablation.
On the other hand, the expression of integrin β4, another well defined constituent of hemidesmosomes, was clearly reduced in plectin-deficient mice, which was consistent with the observed reduction in the number of hemidesmosomes. A reduction of integrin β4 expression also has been reported in mice deficient in integrin α6, another binding partner of integrin β4 (Georges-Labouesse et al. 1996), whereas in integrin β4 (−/−) mice (Dowling et al. 1996; van der Neut et al. 1996) a reduction in the expression of HD1 was noticed (van der Neut et al. 1996). In addition, a recent report suggested that the subcellular distribution of HD1 is determined by the cytoplasmic domain of the β4 subunit (Sanchez-Aparicio et al. 1997), arguing in favor of a direct interaction between plectin and integrin β4 within the hemidesmosomal complex. In this context, it seems reasonable to propose that plectin stabilizes hemidesmosomal structures and thereby promotes their formation.
Additional evidence indicating the importance of plectin for hemidesmosome integrity comes from the peculiar, bipartite distribution of BPAG1 observed in a discontinuous pattern both at the bottom and the top of blisters. This suggests a mechanism of cell breakage in which rupture can occur irregularly on both sides (apical and basal) of the inner plate or within the plate structure itself.
Because there is evidence for integrin β4-binding sites residing in the amino-terminal domain of plectin molecules (G. Rezniczek, unpubl.) and for a highly conserved cytokeratin-binding site located in the globular carboxy-terminal domain of the molecule (Nikolic et al. 1996), it is intriguing to speculate that plectin may act as a stabilizing clamp bridging outer plate components, in particular integrin β4 with the keratin filament network.
Analogous to the stabilization of keratin filament anchoring at hemidesmosomes, plectin molecules may mechanically enforce the attachment of actin filament networks to focal contact sites via the amino-terminal actin-binding domain contained in at least some of its isoforms (Elliott et al. 1997). The apparently reduced mechanical resistance of the sarcolemma observed in some regions of skeletal muscle in plectin (−/−) mice could have been a consequence of unstable actin filament attachment and could explain the observed necrosis of myofibrils. The finding that vinculin expression was reduced significantly and the protein distributed abnormally in muscle tissue of plectin-deficient mice may reflect an impaired formation of focal contact sites.
The destabilization of cellular integrity and cytoarchitecture observed as a consequence of plectin deficiency in tissues exposed to sustained mechanical stress, such as skin and muscle, provides compelling evidence for the hypothesis based on previous findings that plectin molecules act as integration elements and stabilizers of the cytoskeleton. Plectin molecules are likely to achieve this task by linking cytoskeletal filaments to each other and to other organelles in the interior of the cytoplasm, as well as by attachment of cytoskeletal filament networks to the plasma membrane via components of the submembraneous protein skeleton or membrane-associated junctional complexes.
Plectin-deficient mice as a model for EBS-MD
Patients suffering from the hereditary skin blistering disease EBS-MD lack plectin expression in skin and skeletal muscle tissues. In several cases, this defect has been traced to homozygous mutations in the plectin gene leading to premature stop codons either within the rod-encoding exon 31, or within an exon preceding exon 31 (for review, see Uitto et al. 1996). The skin phenotype associated with this disease in general was very similar to that of plectin-deficient mice. In all cases reported, skin blistering arose from the rupture of keratinocytes in the basal cell layer of the epidermis, probably as a consequence of impaired keratin filament anchorage into the plasma membrane. In addition, the presence of necrotic muscle fibers observed in muscle biopsies of EBS-MD patients (Gache et al. 1996; Smith et al. 1996) was confirmed in skeletal muscle of plectin (−/−) mice. In one EBS-MD patient in particular, it was reported that the absence of plectin was accompanied by a disorganization of myofibrils and sarcomeres based on immunohistochemical examination (Gache et al. 1996).
Several phenotypic differences between EBS-MD patients and plectin (−/−) mice were noticed, however. First, the rupture of basal keratinocytes of the patients was reported to occur in a well defined zone, just above the basal plasma membrane or at the level of the inner plate structure of hemidesmosomes, whereas in plectin (−/−) mice, cell rupture occurred throughout the cytoplasm, including the basal zone. Therefore, the phenotype of plectin (−/−) mice resembled more that observed in EBS patients with mutations in keratins 5 and 14. Second, it has been reported that in skin samples taken from EBS-MD patients, a fraction of hemidesmosomes lacked the inner plate structure (Smith et al. 1996), suggesting an involvement of plectin in the formation of hemidesmosomes. In the same study, however, a substantial number of hemidesmosomal structures were found to be morphologically intact and the keratin filament network appeared unaltered, except for its reduced attachment to hemidesmosomes (Smith et al. 1996). This supports our notion that plectin serves as a stabilizing element of keratin filament insertion into the inner plate structure. Third, plectin (−/−) mice died only a few days after birth, most likely from the loss of body fluid and proteins as a consequence of skin blistering, possibly combined with malfunction of the heart and vascular endothelial system, whereas EBS-MD patients apparently have a much higher life expectancy. A possible explanation for these differences could be that in EBS-MD patients, plectin isoforms or truncated versions of the protein are still expressed that are not affected by the mutations found so far. As no truncated plectin molecules could be detected in plectin (−/−) mice, these animals probably exhibited a more severe phenotype, attributable to the abolition of all the plectin isoforms.
Is plectin involved in diseases other than EBS-MD?
Focal lesions similar to those found in skeletal muscle of the plectin (−/−) mice are typical of an autosomal recessive type of myopathy (minicore/multicore disease) characterized by multiple, small randomly distributed areas in the muscle fibers with myofibrillar degenerative changes. Because the genetic basis for this disease is unknown, it would be interesting to establish whether it can be mapped to the plectin gene locus on chromosome 8.
The pathological alterations of heart muscle in plectin (−/−) mice, encompassing focal lesions similar to those observed in skeletal muscle and partial disruption of intercalated discs, demonstrated for the first time an involvement of plectin in the integrity of this type of tissue. A similar condition in EBS-MD patients up to now has not been reported. In plectin-deficient mice, this phenotype is most likely a consequence of cardiomyocyte instability attributable to the lack of plectin in Z-line and intercalated disc structures, both of which have a crucial role in the maintenance of cardiomyocyte cytoarchitecture. The reduction in the number of intact desmosomes, which represent a major adhesive device along the intercalated disc structure of heart, has been shown to dramatically affect myocardium function in mice lacking plakoglobin (Bierkamp et al. 1996; Ruiz et al. 1996). Therefore, the partial disruption of intercalated discs in plectin-deficient mice may reflect an increased fragility of desmosomes, assuming that plectin molecules mechanically strengthen these structures similar to hemidesmosomes. Interestingly, in some studies, minicore/multicore diseases were found to be associated with cardiomyopathy (Magliocco et al. 1989; Bertini et al. 1990), which was at least in one case associated with abnormal accumulation of IFs (Bertini et al. 1990). It is therefore tempting to speculate that a common molecular mechanism involving plectin exists for these myopathies. Whether a cardiomyopathy in conjunction with the severe skin blistering of plectin (−/−) mice is a cause for the early death of these animals awaits a more detailed analysis of their heart function.
Although there is suggestive evidence for a possible functional role of plectin in neuronal tissue (Errante et al. 1994; Smith et al. 1996), no pathological alteration could be observed in the brain of plectin (−/−) mice. Considering that symptoms of neurodegeneration usually arise at older age, the most likely explanation for the absence of any such alterations could be the early death of the animals.
In identifying defined alterations in muscle and heart cytoarchitecture in addition to skin defects, our analysis of the plectin (−/−) phenotype in mice extends considerably the phenotypic analysis of EBS-MD patients reported previously, which was focused primarily on the skin phenotype. Plectin mice could therefore become a useful model in which to investigate on a broad basis the molecular mechanisms underlying EBS-MD and other plectin-related diseases, and to explore possibilities of therapy. Furthermore, plectin-deficient mice and cells derived from these animals should become a useful tool for the characterization of tissue-specific isoforms of the protein, the analyses of their functions and of mechanisms controlling their expression.
Materials and methods
Construction of targeting vectors
Genomic clones were isolated from a mouse genomic library (129 strain, Stratagene), using rat or human plectin cDNA probes for screening. To generate a plectin rod-targeting vector, a 2.8-kb _Eco_RI–_Not_I fragment containing a large part of exon 31 and part of the preceding 1-kb intron was excised from a 13-kb _Hin_dIII–_Bam_HI mouse genomic DNA fragment and replaced with a 1.4-kb _Eco_RI–_Not_I fragment containing a HSV-TK promoter-driven neomycin casette in the same transcriptional orientation. The exon 2–4 targeting vector was constructed by first inserting an _Eco_RI–_Sal_I fragment (containing a PGK promoter-driven neor-cassette) into a 3-kb _Not_I–_Xmn_I mouse genomic DNA fragment, and subsequent insertion of a 5-kb _Kpn_I mouse genomic DNA fragment downstream of the neor-cassette to serve as 3′ homology region.
Electroporation of ES cells, generation and analysis of plectin-deficient mice
R1 ES cells (Nagy et al. 1992) were electroporated independently with the linearized targeting vectors followed by positive selection of ES cell colonies (Hogan et al. 1994) in the presence of G418 (Sigma; 400 μg/ml). Appropriate homologous recombination and unique integration were assayed by Southern blotting of genomic DNA prepared from selected colonies after expansion (see Figs. 1 and 2). To generate chimeric mice, selected ES-cell clones were injected into blastcysts [isolated from C57BL/6 mice at day 3.5 postcoitum (p.c.)], which were transferred into pseudopregnant C57BL/6× DBA F1 or C57BL/6× CBA females (3.5 days p.c.). After breeding, genotyping of offspring was performed by Southern blot analysis of genomic DNA isolated from mouse tails (Hogan et al. 1994).
For RT-PCR analysis, total RNA was isolated from skin and muscle tissues of 2- to 3-day-old plectin (+/+) and (−/−) mice according to the method of Chomzcynski and Sacchi (1987). Total RNA (10 μg) was transcribed reversely using Amplimer Sets for RT-PCR (Clontech), following the manufacturer’s instructions. PCRs were carried out using eLongase Enzyme Mix (GIBCO) and the following primer pairs: h2 (5′-CTTGTGCCGGTGGATGATG) and h6 (5′-CTTGTGCCGGTGGATGATG); h29 (5′-CTCCCCCGCCAAGAAGCCCAAGG) and h31 (5′-CGGCCTCCTCGGCCTGCAGTTTC); r32-1 (5′-TATGAGGCCTACCGCAAGGGC) and r32-2 (5′-ATCCACAGTGCCACGTT); and vim1 (5′-TACCAAGATCTGCTCAAT) and vim2 (5′-TTCAAGGTCATCGTGATGCTG); corresponding to sequences in exons 2 and 6 (h2 and h6), 29 and 31 (h29 and h31), and 32 (r32-1 and r32-2), respectively, and mouse vimentin gene sequences (vim1 and vim 2). Cycle parameters were 94°C for 3 min (1×), followed by 94°C for 30 sec, 60°C for 30 sec, and 68°C for 6 min (35×).
Tissue homogenates of skin and muscle from plectin (+/+) and (−/−) mice were prepared for immunoblot analysis by mechanical disruption of 300 mg of tissue using conditions described by Eger et al. (1997). The efficiency of protein transfer to membranes and equal protein loadings were monitored by staining with Ponceau S (Sigma). Blots were developed using rabbit antiserum to plectin 21 (Wiche et al. 1983) or mouse antiserum 135-A, raised against a recombinant rat plectin mutant protein corresponding to the carboxy-terminal repeat 4 domain of plectin (B. Nikolic, unpubl.), both diluted 1: 500, followed by alkaline phosphatase-conjugated anti-rabbit IgG (Promega; dilution 1:7,500) and BCIP/ NBT.
Histology, immunohistochemistry, and ultrastructural analysis
For general pathological screening, animals were transcardially perfused with 4% formaldehyde in PBS and dissected into 2 mm thick transverse slices that were routinely embedded in paraffin. Paraffin sections (5 μm) were stained with hematoxylin/eosin, periodic acid Schiff reaction, Van Giesson/Elastica stain, and Gomori silver impregnation. Sections that contained parts of the central or peripheral nervous system were stained additionally with Luxol fast blue for myelin and Bielschowski silver impregnation for axons. Special care was taken that the respective transverse sections contained all parenchymal organs.
For immuncytochemistry, selected tissues were snap frozen before fixation, and thin sections (5 μm) cut with a cryomicrotome were incubated with 10% horse serum in PBS for 1 hr. Primary antibodies were used at the following dilutions—mouse anti-plectin mAbs (10F6 and 5B3; Foisner et al. 1994), 1:1 (hybridoma supernatant); mouse anti-dystrophin mAb dys2 (Novocastra), 1:100; mouse anti-β-spectrin mAb (Novocastra), 1:100; mouse anti-vinculin mAb (Novocastra), 1:50; human anti-BPAG1 mAb-5E (Hashimoto et al. 1993), 1:500; rabbit antiserum to the carboxy-terminal domain of integrin β4 (Giancotti et al. 1992), 1:100; rat mAb 346-11A directed against an epitope in the extracellular portion of integrin β4 (Kennel et al. 1989), 1:100. Biotinylated secondary antibodies (donkey anti-rabbit- and sheep anti-human IgGs; both purchased from Dako) and goat anti-mouse IgG (Amersham) were used at dilutions recommended by the manufacturers. Texas-red-conjugated avidin (Amersham) was used for visualization of antibodies. Signal specificity was controlled by omission of primary antibodies or by using normal mouse or rabbit serum in their place.
Small pieces of skin, various muscles, and heart, either fixed by perfusion as described above, or immersed in 2% glutaraldehyde in PBS, were osmicated and embedded in epoxy resin. From these blocks, 0.5 μm thick sections were stained with toluidine blue for light microscopic screening and thin sections were treated with uranyl acetate and lead citrate and analyzed by electron microscopy.
Acknowledgments
Special thanks go to Dr. Carol Ware for expert guidance and practical input in the setting up of ES cell cultures in our laboratory. We also thank Zeljka Stojanovic for dedicated technical assistance in generating knockout mice. Dr. Takashi Hashimoto (Kurume University School of Medicine, Japan), Dr. Filippo Giancotti (NYU Medical Center, New York), and Dr. Stephen Kennel (Oak Ridge National Laboratory, Oak Ridge) are thanked for generous gifts of antibodies to BPAG1 and integrin β4, respectively. We also gratefully acknowledge the gift of R1 ES cells from Dr. Andras Nagy, University of Toronto. Mouse anti-plectin antiserum was provided by B. Nikolic. We thank Dr. Anthony Infante and Dr. Maria J. Castañón for suggestions leading to the improvement of the manuscript. R.F. was supported by the Hermann Lilly Schilling Stiftung. K.A. was the recipient of a short-term fellowship from the European Molecular Biology Organization (EMBO). The major part of this work was supported by grant SFB 006/611 from the Austrian Science Research Fund.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL wiche@abc.univie.ac.at; FAX 43-1-79515-5121.
References
- Bertini E, Bosman C, Bevilacqua M, Ricci E, Gagliardi GM, Parisi F, Servidei S, Dionisi VC, Ballerini L. Cardiomyopathy and multicore myopathy with accumulation of intermediate filaments. Eur J Pediatr. 1990;149:856–858. doi: 10.1007/BF02072073. [DOI] [PubMed] [Google Scholar]
- Bierkamp C, McLaughlin JK, Schwarz H, Huber O, Kemler R. Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol. 1996;180:780–785. doi: 10.1006/dbio.1996.0346. [DOI] [PubMed] [Google Scholar]
- Chan Y, Anton-Lamprecht I, Yu Q-C, Jäckel A, Zabel B, Ernst JP, Fuchs E. A human keratin 14 “knock out”: the absence of K14 leads to severe epidermolyis bullosa simplex and a function for an intermediate filament protein. Genes & Dev. 1994;8:2574–2587. doi: 10.1101/gad.8.21.2574. [DOI] [PubMed] [Google Scholar]
- Chavanas S, Pulkkinen L, Gache Y, Smith FJD, McLean WHI, Uitto J, Ortonne JP, Meneguzzi G. A homozygous nonsense mutation in the PLEC1 gene in patients with epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;98:2196–2200. doi: 10.1172/JCI119028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dowling J, Yu Q-C, Fuchs E. β4 Integrin is required for hemidesmosome formation, cell adhesion and cell survival. J Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Wiche G, Foisner R. Polarization dependent association of plectin with desmoplakin and the lateral submembrane skeleton in MDCK cells. J Cell Sci. 1997;110:1307–1316. doi: 10.1242/jcs.110.11.1307. [DOI] [PubMed] [Google Scholar]
- Elliott CE, Becker B, Oehler S, Castañón MJ, Hauptmann R, Wiche G. Plectin transcript diversity: Identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics. 1997;42:115–125. doi: 10.1006/geno.1997.4724. [DOI] [PubMed] [Google Scholar]
- Errante LD, Wiche G, Shaw G. Distribution of plectin, an intermediate filament-associated protein, in the adult rat central nervous system. J Neurosci Res. 1994;37:515–528. doi: 10.1002/jnr.490370411. [DOI] [PubMed] [Google Scholar]
- Foisner R, Traub P, Wiche G. Protein kinase A- and protein kinase C-regulated interaction of plectin with lamin B and vimentin. Proc Natl Acad Sci. 1991;88:3812–3816. doi: 10.1073/pnas.88.9.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R, Feldman B, Sander L, Seifert G, Artlieb U, Wiche G. A panel of monoclonal antibodies to rat plectin: distinction by epitope mapping and immunoreactivity with different tissues and cell lines. Acta Histochem. 1994;96:421–438. doi: 10.1016/S0065-1281(11)80029-2. [DOI] [PubMed] [Google Scholar]
- Foisner R, Bohn W, Mannweiler K, Wiche G. Distribution and ultrastructure of plectin arrays in subclones of rat glioma C6 cells differing in intermediate filament protein (vimentin) expression. J Struct Biol. 1995;115:304–317. doi: 10.1006/jsbi.1995.1055. [DOI] [PubMed] [Google Scholar]
- Fuchs R. The cytoskeleton and disease: Genetic disorders of intermediate filaments. Annu Rev Genet. 1996;30:197–231. doi: 10.1146/annurev.genet.30.1.197. [DOI] [PubMed] [Google Scholar]
- Gache Y, Chavanas S, Lacour JP, Wiche G, Owaribe K, Meneguzzi G, Ortonne JP. Defective expression of Plectin/HD1 in epidermolysis bullosa simplex with mucular dystrophy. J Clin Invest. 1996;97:2289–2298. doi: 10.1172/JCI118671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges-Labouesse E, Messaddeq N, Ghassan Y, Cadalbert L, Dierich A, LeMeur M. Absence of integrin alpha6 leads to epidermolysis bullosa and neonatal death in mice. Nature Genet. 1996;13:370–373. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Stepp MA, Suzuki S, Engvall E, Ruoslahti E. Proteolytic processing of endogenous and recombinant β4 integrin subunit. J Cell Biol. 1992;118:951–959. doi: 10.1083/jcb.118.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KJ, Parry DAD, Steinert PM, Virata MLA, Wagner RM, Angst BD, Nilles LA. Structure of the human desmoplakins. Implications for function in the desmosomal plaque. J Biol Chem. 1990;265:2603–2612. [PubMed] [Google Scholar]
- Guo L, Degenstein L, Dowling J, Yu QC, Wollmann R, Perman B, Fuchs E. Gene targeting of BPAG1: Abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Amagai M, Ebihara T, Gamou S, Shimizu N, Tsubata T, Hasegawa A, Miki K, Nishikawa T. Further analyses of epitopes for human monoclonal anti-basement membrane zone antibodies produced by stable human hybridoma cell lines constructed with Epstein-Barr virus transformants. J Invest Dermatol. 1993;100:310–315. doi: 10.1111/1523-1747.ep12469916. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Wiche G. Plectin and IFAP-300K are homologous proteins binding to microtubule-associated proteins 1 and 2 and to the 240-kilodalton subunit of spectrin. J Biol Chem. 1987;262:1320–1325. [PubMed] [Google Scholar]
- Hieda Y, Nishizawa Y, Vematsu J, Owaribe K. Identification of a new hemidesmosomal protein HD1: A major high molecular mass component of isolated hemidesmosomes. J Cell Biol. 1992;116:1497–1506. doi: 10.1083/jcb.116.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Constantini F, Lacy E. Manipulating the mouse embryo. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Kennel SJ, Foote LJ, Falcioni R, Sonnenberg A, Stringer CD, Crouse C, Hemler ME. Analysis of the tumor-associated antigen TSP-180. Identity with α6-β4 in the integrin superfamily. J Biol Chem. 1989;264:15515–15521. [PubMed] [Google Scholar]
- Koss-Harnes D, Jahnsen FL, Wiche G, Søyland E, Brandtzaeg P, Gedde-Dahl Jr T. Plectin abnormality in Epidermolysis Bullosa Simplex Ogna: Non-responsiveness of basal keratinocytes to some anti-rat plectin antibodies. Exp Dermatol. 1996;6:41–48. doi: 10.1111/j.1600-0625.1997.tb00144.x. [DOI] [PubMed] [Google Scholar]
- Liu C-g, Maercker C, Castañón MJ, Hauptmann R, Wiche G. Human plectin: Organization of the gene, sequence analysis, and chromosome localization (8q24) Proc Natl Acad Sci. 1996;93:4278–4283. doi: 10.1073/pnas.93.9.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliocco AM, Mitchell LB, Brownwell AK, Lester WM. Dilated cardiomyopathy in multicore myopathy. Am J Cardiol. 1989;63:150–151. doi: 10.1016/0002-9149(89)91108-9. [DOI] [PubMed] [Google Scholar]
- McLean WHI, Pulkkinen L, Smith FJD, Rugg EL, Lane EB, Bullrich F, Burgeson RE, Amano S, Hudson DL, Owaribe K, McGrath JA, McMillan JR, Eady RAJ, Leigh IM, Christiano AM, Uitto J. Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes & Dev. 1996;10:1724–1735. doi: 10.1101/gad.10.14.1724. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramov-Newerly W, Roder JC. Derivation of completely cell culture derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci. 1992;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic B, Mac Nulty E, Mir B, Wiche G. Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin-vimentin network junctions. J Cell Biol. 1996;134:1455–1467. doi: 10.1083/jcb.134.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penegyres PK, Kakulas BA. The natural history of minicore-multicore myopathy. Muscle Nerve. 1991;14:411–415. doi: 10.1002/mus.880140505. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Smith FJD, Shimizu H, Murata S, Yaoita H, Hachisuka H, Nishikawa T, McLean WHI, Uitto J. Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol Gen. 1996;5:1539–1546. doi: 10.1093/hmg/5.10.1539. [DOI] [PubMed] [Google Scholar]
- Rugg EL, McLean IWH, Lane EB, Pitera R, McMillan RJ, Heppenstal PJC, Navsaria HA, Leigh IM, Eady AJ. A functional “knockout” of human keratin 14. Genes & Dev. 1994;8:2563–2573. doi: 10.1101/gad.8.21.2563. [DOI] [PubMed] [Google Scholar]
- Ruhrberg C, Nasser Hajibagheri MA, Simon M, Dooley TP, Watt FM. Envoplakin, a novel precursor of the cornified envelope that has homology to desmoplakin. J Cell Biol. 1996;134:715–729. doi: 10.1083/jcb.134.3.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Günther U, Franke WW, Birchmeier W. Targeted mutation of plakoglobin in mice reveals essential function of desmosomes in the embryonic heart. J Cell Biol. 1996;135:215–225. doi: 10.1083/jcb.135.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Aparicio P, de Velasco M, Niessen CM, Borradori CM, Kuikman L, Hulsman EHM, Fässler R, Owaribe K, Sonnenberg A. The subcellular distribution of the high molecular mass protein HD-1 is determined by the cytoplasmic domain of integrin β4 subunit. J Cell Sci. 1997;10:169–178. doi: 10.1242/jcs.110.2.169. [DOI] [PubMed] [Google Scholar]
- Sawamura D, Li K, Chou M-L, Uitto J. Human bullous pemphigoid antigen (BPAG1) J Biol Chem. 1991;266:17784–17790. [PubMed] [Google Scholar]
- Seifert GJ, Lawson D, Wiche G. Immunolocalization of the intermediate filament-associated protein plectin at focal contacts and actin stress fibers. Eur J Cell Biol. 1992;59:138–147. [PubMed] [Google Scholar]
- Smith FJD, Eady RAJ, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WHI, Lane EB. Plectin deficiency results in muscular dystrophy with Epidermolysis bullosa. Nature Genet. 1996;13:450–457. doi: 10.1038/ng0896-450. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J, Pulkkinen L, Smith FJD, McLean WHI. Plectin and human genetic disorders of the skin and muscle. Exp Dermatol. 1996;5:237–246. doi: 10.1111/j.1600-0625.1996.tb00124.x. [DOI] [PubMed] [Google Scholar]
- van der Neut R, Krimpenfort P, Calafat J, Niessen CM, Sonnenberg A. Epithelial detachment due to the absence of hemidesmosomes in integrin β4 null mice. Nature Genet. 1996;13:366–369. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- Vassar R, Coulombe PA, Degenstein L, Albers K, Fuchs E. Mutant keratin expression in transgenic mice causes marked abnormalities resembling a human genetic skin disease. Cell. 1991;64:365–380. doi: 10.1016/0092-8674(91)90645-f. [DOI] [PubMed] [Google Scholar]
- Wiche G. Plectin: General overview and appraisal of its potential role as a subunit protein of the cytomatrix. CRC Crit Rev Biochem Mol Biol. 1989;24:41–67. doi: 10.3109/10409238909082551. [DOI] [PubMed] [Google Scholar]
- Wiche G, Krepler R, Artlieb U, Pytela R, Denk H. Occurrence and immunolocalization of plectin in tissues. J Cell Biol. 1983;97:887–901. doi: 10.1083/jcb.97.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G, Krepler R, Artlieb U, Pytela R, Aberer W. Identification of plectin in different human cell types and immunolocalization at epithelial basal cell surface membranes. Exp Cell Res. 1984;155:43–49. doi: 10.1016/0014-4827(84)90766-3. [DOI] [PubMed] [Google Scholar]
- Wiche G, Becker B, Luber K, Weitzer G, Castañón MJ, Hauptmann R, Stratowa C, Stewart M. Cloning and sequencing of rat plectin indicates a 466-kD polypeptide chain with a three-domain structure based on a central alpha-helical coiled coil. J Cell Biol. 1991;114:83–99. doi: 10.1083/jcb.114.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Dowling J, Yu QC, Kouklis P, Cleveland DW, Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 1996;86:655–665. doi: 10.1016/s0092-8674(00)80138-5. [DOI] [PubMed] [Google Scholar]