Autocrine growth and anchorage independence: two complementing Jun-controlled genetic programs of cellular transformation (original) (raw)
Abstract
Cellular transformation can be achieved by constitutive activation of growth-regulatory signaling pathways, which, in turn, activate nuclear transcription factors thought to execute a transformation-specific program of gene expression. Members of the dimeric transcription factor family AP-1 are at the receiving end of such growth-regulating pathways and the viral form of the AP-1 subunit Jun establishes one important aspect of transformation in chick embryo fibroblasts (CEFs): enhanced growth in agar and in low serum. Enhanced Jun activity is likely to target several different genetic programs as Jun forms heterodimers with one of several members of the Fos and ATF2 subfamilies, resulting in transcription factors with different sequence specificities. To identify the programs relevant for transformation, we have reduced the complexity of AP-1 factors by constructing Jun bZip mutants that can efficiently dimerize and transactivate with only a restricted set of partner subunits. Upon introduction into CEFs, a Jun mutant selective for the Fos family induced anchorage-independent growth but no growth factor-independence. In contrast, a c-Jun mutant with preference for ATF2-like proteins caused growth factor-independence, but no growth in agar. Coexpression of both mutants reestablished the combined transformation program as induced by wild-type Jun. These data show that Jun-dependent cell transformation can be resolved into at least two distinct and independent processes, anchorage and growth factor independence, obviously triggered by two classes of Jun heterodimers likely regulating different sets of target genes.
Keywords: ATF2, Fos, bZip mutants, transcription, transformation, chick embryo fibroblasts, anchorage independence, growth factor independence
The c-Jun proto-oncoprotein is a major component of the AP-1 family of dimeric transcription factors. c-Jun can, in principle, homodimerize, but is commonly associated with other transcription factor partners. Heterodimer partners include members of the Jun, Fos, and ATF/CREB subfamilies. Dimerization occurs by coiled-coil interaction through the leucine zipper domain, and this interaction is a prerequisite for DNA-binding (for review, see Angel and Karin 1991; Hurst 1994). Dimers containing c-Jun regulate transcription in response to a multitude of extracellular signals that cause c-Jun phosphorylation. Also, the dimer partners are subject to activation in response to extracellular stimuli, and, therefore, the extent to which c-Jun dimers are activated by upstream protein kinases can depend on the dimer partner (Deng and Karin 1994; van Dam et al. 1995). c-Jun dimers are thought to play a decisive role in embryonal development (Hilberg et al. 1993; Johnson et al. 1993), in cell proliferation and tumorigenesis (for review, see Vogt 1994), in the cellular response to genotoxic stress (Dévary et al. 1992; Schreiber et al. 1995), and in apoptosis (Ham et al. 1995).
Deregulated expression of c-Jun or of its mutated viral counterpart v-Jun can trigger transformation in primary embryo fibroblasts of avian and mammalian origin. Transformation requires presence of the carboxy-terminal bZip domain as well as of the amino-terminal transcriptional regulatory domain(s) of Jun (for review, see Vogt 1994). The identification of Jun target genes involved in transformation has been complicated by the fact that Jun-containing dimers differ in their DNA-binding specificities (Benbrook and Jones 1990; Ivashkiv et al. 1990; Hai and Curran 1991). For instance, c-Jun:Fos dimers, which become abundant in response to extracellular signals, bind with high affinity to the 7-bp consensus AP-1 binding site 5′-TGAGTCA as present in the human collagenase promoter, but relatively weakly to the 8-bp c-Jun:ATF2 motif 5′-TTACCTCA as present in the c-jun promoter. In contrast, c-Jun:ATF2 heterodimers have very low affinity for the Jun:Fos site and bind efficiently only to the 8-bp element (Benbrook and Jones 1990; Ivashkiv et al. 1990; Hai and Curran 1991; van Dam et al. 1993).
Here, we address the contribution to Jun-dependent transformation of the dimer partner and thus of the type of target gene activated. We have generated c-Jun mutants that preferentially dimerize and bind to DNA either as Jun:Fos or as Jun:ATF2. A mutant that only efficiently bound to DNA in association with Fos (or a Fos-like protein), caused chicken embryo fibroblasts (CEFs) to grow in soft agar, but could not induce serum independence. An ATF2-seeking mutant allowed CEF cells to grow in low serum, but did not induce growth in agar. These data demonstrate the existence of two independent and separable Jun-induced genetic programs controlling either anchorage independence or autocrine growth.
Results
c-Jun zipper mutants with dimerization preference for Fos or ATF-2
To restrict dimerization by Jun to one or few partners, we constructed mutants in its dimerization domain. Following proposed interaction rules (Schuermann et al. 1991; O’Shea et al. 1992; Vinson et al. 1993), we mutated the amino acids neighboring the hydrophobic leucines of the zipper, the so-called e and g positions (see helical wheel representation in Fig. 1), thought to be crucial for the specificity of dimerization. We expect two types of specificity of bZip dimerization: (1) the first type would be determined solely by the coiled-coil interaction of the two leucine zippers; (2) the second type represents dimerization in the presence of the cognate DNA element, which depends on the conformation of both the zipper and the adjacent basic region responsible for contacting the DNA. Binding to the cognate DNA element induces major conformational changes depending on the heterodimer partner (Kerppola et al. 1993). It is this second type, the dimerization specificity at the DNA element, which is relevant for the selection of target genes and, as will be the goal of this study, for the transformation program. We report on two of our human c-Jun mutants that exhibited interesting properties: m0 and m1. These mutants were obtained by replacing and introducing glutamate and lysine residues at positions e1, g1, e2, e4, and g4 (Fig. 1).
Figure 1.
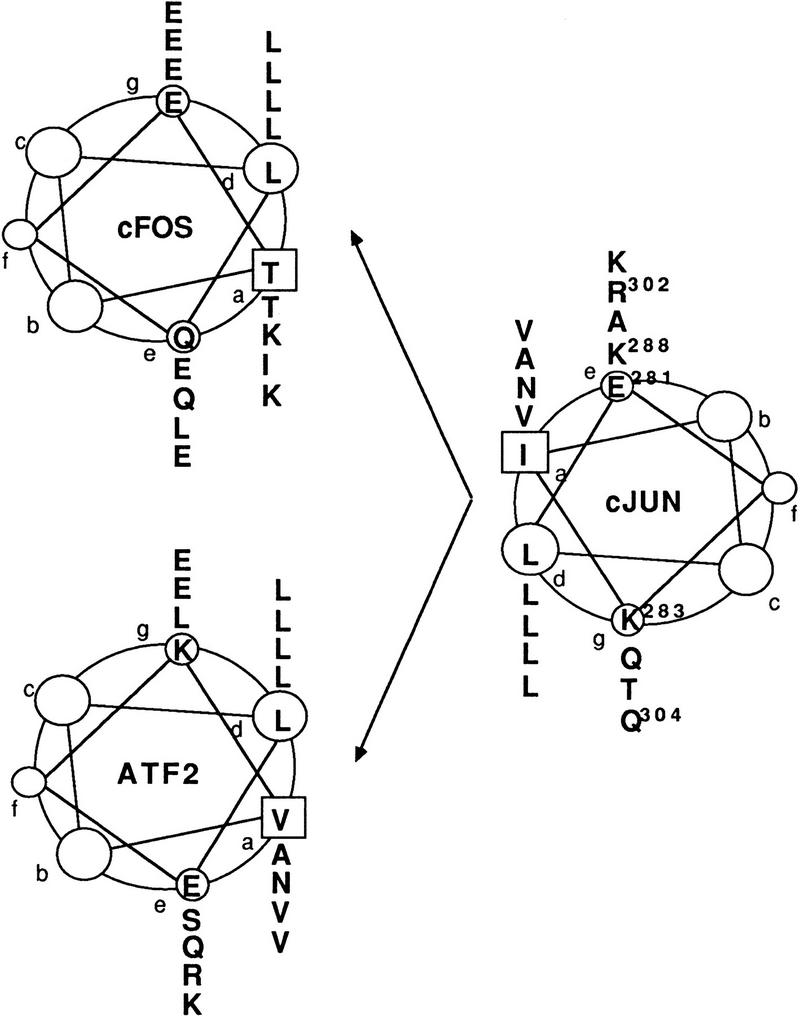

c-Jun mutants with altered Jun:Fos and Jun:ATF2 dimerization specificities. (Top) Helical wheel representations of the c-Jun:c-Fos and c-Jun:ATF2 leucine zipper dimers. The amino acids at the e and g positions determining dimer specificity are located adjacent to the hydrophobic interface that is formed by the leucines at the d positions and the a residues. (Bottom) c-Jun mutants m0 and m1 were constructed by replacement of amino acid residues at the e and/or g positions as depicted. In m0, Glu-281 of c-Jun is replaced by a lysine. In m1, Lys-283, Lys-288, Arg-302, and Gln-304 are replaced by four glutamates.
Dimerization specificity in solution (in the absence of DNA binding) was measured in a mammalian one-hybrid type of analysis. The bZip domains of wild-type or mutant c-Jun were fused to the DNA-binding domain (DBD) of the yeast transcription factor Gal4, and the hybrid proteins were examined in F9 teratocarcinoma cells for their transactivating potential at a Gal4-dependent reporter. In this assay, the hybrid proteins cannot transactivate unless they can recruit through their zipper domain a transactivating subunit. As unstimulated F9 cells contain neither c-Fos nor c-Jun, but contain ATF2, which is a poor transactivator in these cells (data not shown; van Dam et al. 1995), recruitment of a transactivating subunit can only occur if c-Fos or ATF2–VP16 are introduced into the same cell. ATF2–VP16 is a hybrid protein in which the potent transactivation domain of the viral transactivator VP16 is fused to the carboxyl terminus of ATF2 (Liu and Green 1990). As shown in Figure 2, neither the Gal4–DBD nor the c-Jun–bZip domain on their own can transactivate the Gal4-dependent reporter when transiently cotransfected. Gal4-DBD–c-Jun-bZip wild type, as expected, transactivated upon introduction of either c-Fos or ATF2–VP16 (activation with c-Fos was about 2.0-fold better than with ATF2 under the conditions used). To assess the specificities of the bZip mutants, we compared their relative partner preference. The leucine zipper of c-Jun–m0 preferred c-Fos as its dimer partner (ratio c-Fos:ATF2 = 3.0), whereas, in contrast, c-Jun–m1 had very low affinity for c-Fos and interacted preferentially with ATF2–VP16 (ratio c-Fos:ATF2 = 0.4). We conclude that the bZip domain mutants of c-Jun differ from wild type by their dimerization preference in solution. Jun mutant m0 prefers c-Fos; m1 prefers ATF2.
Figure 2.

The interaction of mutant c-Jun leucine zipper domains with c-Fos and ATF2–VP16 as measured by mammalian one-hybrid analysis. Undifferentiated F9 cells were transiently transfected with 2 μg of 5×GAL4–E4–luciferase reporter plasmid together with 0.5 μg of the indicated wild-type or mutant RSV–Gal4–DBD–c-Jun–bZip expression vectors or Gal4–DBD or c-Jun–bZip control constructs in the presence of the indicated amounts of RSV–c-Fos, RSV–ATF2–VP16 expression vectors and/or an RSV control plasmid (pUC–RSV:−) to equalize the total amount of RSV–LTR sequences. Fold activation represents luciferase activity induced by the Gal4 fusion proteins in the presence of the c-Fos or ATF2–VP16 expression vectors. The Gal4 fusion proteins themselves did not significantly affect the activity of the reporter. The data represent the mean of four independent experiments in which the constructs were tested in parallel. Standard deviations are <25%. Relative induction ratios for m0: (m0 + Fos/m0 + ATF2–VP16)/(wt + Fos/wt + ATF2–VP16) = 1.5; for m1: (m1 + Fos/m1 + ATF2–VP16)/(wt + Fos/wt + ATF2–VP16) = 0.2.
Because the relevant dimerization specificity for target gene selection is that on DNA, major efforts were undertaken to document c-Jun mutant behavior at appropriate DNA elements. We first tested dimerization specificity in gel retardation experiments using in vitro-translated proteins. The DNA-binding assays were performed with oligonucleotides representing either a classical c-Jun:c-Fos site [TGAGTCA from the collagenase promoter (Angel et al. 1987), designated coll-TRE], or a high-affinity c-Jun:ATF2-binding site [TTACCTCA, jun2 site of the c-_jun_ promoter (van Dam et al. 1993)]. The decisive gel-retardation features of the mutants are shown in Figure 3: (1) in the absence of a partner subunit, no DNA binding is detectable at the coll-TRE under the experimental conditions used, not even with wild-type c-Jun (Fig. 3, left panel), reflecting the low stability or affinity of Jun homodimers for the coll-TRE; at the jun2 site, no complex of only wild-type c-Jun or of one of the mutants is formed (Fig. 3, right panel); (2) in the presence of c-Fos, wild-type c-Jun and mutant c-Jun–m0, but not c-Jun–m1, bound strongly to the coll-TRE (left panel of Fig. 3); (3) in the presence of ATF2 (note that a truncated bZipATF2 was used here for better gel resolution), wild-type c-Jun and mutant c-Jun–m1 formed gel retardation complexes at the jun2 element; again as the most interesting result, c-Jun–m0 did not interact with ATF2 on DNA (Fig. 3; right panel).
Figure 3.

Dimer-specific DNA-binding properties of c-Jun leucine zipper mutants m0 and m1 in vitro. Gel retardation assays showing the DNA-binding affinities for the collagenase AP-1 binding site (coll–TRE) and the jun2 site of in vitro-translated wild-type and mutant c-Jun proteins in the absence or presence of in vitro-translated c-Fos or bZip–ATF2 (amino acids 335–505). c-Jun proteins were quantified by measurement of the amount of incorporated [35S]methionine, and similar amounts were mixed on ice with control reticulocyte lysate (−) or lysates containing c-Fos or bZip–ATF2 after which 32P-labeled DNA probe was added. The DNA–protein complexes were resolved on 4% polyacrylamide gels and visualized by autoradiography. Note that the increased mobility of the c-Jun–m1:ATF2–DNA complex as compared with that of c-Jun wild-type is explained by a higher negative charge of the c-Jun–m1 protein than of wild-type c-Jun, owing to the replacement of two lysines, an arginine, and a glutamine by four glutamic acid residues.
We conclude that in the DNA-bound state, the dimerization preferences of the c-Jun mutants are accentuated in comparison to dimerization in solution as measured in the one-hybrid experiment of Figure 2. The accentuation was particularly evident for c-Jun–m0. c-Jun–m0 prefers c-Fos; c-Jun–m1 binds ATF2. The experiments do not indicate exclusive preference for either c-Fos or ATF2. Because the amino acid sequences of the bZip regions are highly conserved within the Fos family, and between ATF2 and ATFa, mutant preferences likely extend to other members of the two families. All c-Jun proteins tested here bind to DNA strongly only as heterodimers.
Preference of c-Jun–m0 and c-Jun–m1 for Jun:Fos and Jun:ATF2-dependent promoters in vivo
The behavior of a c-Jun subunit in vivo will depend not only on the nature of its dimerization domain but also on the availability and concentration of partner proteins. In vitro gel retardation conditions have, therefore, their severe limitations for the affinity analysis of AP-1 heterodimers in a given cell. With an excess of oligonucleotide, only careful determinations of the on and off rates would be informative. This is almost impossible with mixtures of heterodimers that can, under certain in vitro conditions, bind to any similar element. For instance, Fos:Jun heterodimers, which do not utilize the c-fos promoter CRE site in vivo, can, however, bind to the CRE in vitro if DNA is offered in excess (Schönthal et al. 1989). Therefore, we put most efforts into examining transactivation of model promoters in cells. To this end, we compared transactivation of Jun:Fos- and Jun:ATF2-controlled promoters upon overexpression of wild-type and mutant c-Jun proteins in undifferentiated F9 teratocarcinoma cells. As mentioned above, these cells have lower complexity of endogenous AP-1 subunits than other cell lines. The reporter genes used were 5×coll–TRE–tata (Jonat et al. 1990) and 5×jun2–tata (van Dam et al. 1993) which have been shown to specifically respond to overexpressed Jun:Fos and Jun:ATF2, respectively (van Dam et al. 1993, 1995; data not shown). Because F9 cells lack endogenous c-Fos and c-Jun proteins, but contain ATF2 (van Dam et al. 1995), transactivation by the Jun proteins was determined in the absence or in the presence of a cotransfected expression vector encoding c-Fos. An example of these analyses is shown in Figure 4. Under conditions where basal transactivation of 5×coll–TRE–tata by wild-type Jun homodimers was marginal, the c-Fos expression construct enhanced the transactivation by wild-type c-Jun 2.4-fold (set as 1 in Fig. 4). Even stronger than that by wild-type c-Jun, Fos enhanced the transactivation by c-Jun–m0 (Fig. 4). c-Jun–m1 exerted even a slightly inhibitory effect on this promoter, both in the presence or absence of c-Fos. With the 5×jun2 reporter, c-Jun–m0 was inactive. In contrast, c-Jun–m1 activated even more strongly than wild-type c-Jun (Fig. 4). Overexpression of ATF2 did not further enhance the activation of the 5×jun2 promoter by either wild type or mutant Jun (data not shown), indicating that ATF2 (or ATF2-like) factor levels in F9 cells are saturating. Western blot analysis confirmed that all c-Jun proteins accumulated to the same extent in these cells (data not shown; also see Fig. 7, below). Essentially similar data on promoter-specific transactivation were obtained in HeLa cells, which under nonstimulated conditions contain low c-Fos levels but relatively high amounts of ATF2 and ATFa (Offringa et al. 1990; data not shown): with the 5×jun2-dependent reporter c-Jun–m1 was the best activator, followed by wild-type c-Jun, while c-Jun–m0 did not transactivate at all. c-Fos enhanced coll–TRE-dependent promoter activity in the presence of wild-type c-Jun or of c-Jun–m0, but not c-Jun–m1 (data not shown). In both cell types, F9 and HeLa, we also tested the natural collagenase promoter, which depends critically on AP-1 acting on a single site. Results were similar to those obtained with 5×coll–TRE–tata (data not shown).
Figure 4.

Jun:Fos and Jun:ATF2-specific transactivation by c-Jun leucine zipper mutants c-Jun m0 and m1 in vivo. Comparison of transactivation by wild-type c-Jun and by the Jun mutants m0 and m1 of Jun:Fos- and Jun:ATF2-dependent minimal promoters. F9 teratocarcinoma cells were transiently transfected with 2 μg of either the 5×coll–TRE–tata–luciferase, 5×jun2–tata–luciferase, or tata–luciferase reporter plasmids together with 0.5 μg of wild-type or mutant RSV–c-Jun expression vector in the presence or absence of 25 ng pRSV–c-Fos, or an RSV control plasmid (pUC–RSV). Fold activation represents activity obtained in the presence of the c-Jun expression vectors relative to the activity in the presence of the pUC–RSV control vector, and normalized to the activation on the tata-luciferase control. The experiments were performed at least three times and each time had very similar results. One of them is shown. s.d. < 25%.
Figure 7.

Expression of wild-type and of mutant human c-Jun proteins in infected CEF cells measured by Western blot analysis. Total cell extract (10 μg) was subjected to SDS-PAGE, blotted onto nitrocellulose and incubated with a polyclonal antibody raised against bacterially expressed mouse c-Jun. The two arrows indicate the positions of the endogenous avian c-Jun protein (c-cJun, 314 amino acids; calculated molecular mass of 34.4 kD) and the virally expressed human c-Jun proteins (h-c-Jun, 331 amino acids; calculated molecular mass of 35.7 kD), respectively.
Promoter specificity of the c-Jun mutants in chick embryo fibroblasts
CEFs are particularly suitable for the study of a transformation program because overexpression of exogenous c-Jun without the need for a second oncogene induces a growth-promoting phenotype, including growth in agar and in low serum (Bos et al. 1990; Castellazzi et al. 1990). Because CEFs will be the decisive cell type in the present study of transformation, it is important to know how the model promoters behave in CEFs supplied with Jun and to confirm the selective choice of partners by Jun mutant proteins. Cotransfection of the 5×coll–TRE–tata reporter and human wild-type c-Jun into CEFs led to an expression level set at 1 (Fig. 5). Mutant c-Jun–m0, the Fos-seeking mutant, strongly enhanced transcription from the 5×coll–TRE promoter, whereas c-Jun–m1 slightly reduced transcription (Fig. 5). Cotransfection with a c-Fos-expressing contruct enhanced transactivation by wild-type c-Jun 3.6-fold (set as 1). The relative activation by the mutants was not changed much. In the presence of c-Fos, c-Jun–m1 did not reduce 5×coll–TRE–tata promoter activity, perhaps because of the release of residual endogenous c-Jun from complexes with factors other than c-Fos, resulting in its availability to c-Fos. With the 5×jun2–tata reporter, the reverse behavior of the mutants became apparent. c-Jun–m0 did not activate, rather reduced transcription as compared with wild-type c-Jun, irrespective of additional ATF2 expression (Fig. 5). Transactivation by wild-type c-Jun was not increased further by expression of additional ATF2 (factor of enhancement close to 1), indicating that ATF2 in CEFs is relatively abundant. c-Jun–m1, however, enhanced promoter activity, particularly when cotransfected with an ATF2 construct, which suggests that ATF2 becomes limiting relative to Jun if an ATF2-seeking mutant is overexpressed. We conclude that the c-Jun mutants exhibit in CEFs, just as in F9 or HeLa cells, strong preference for c-Fos or ATF2 (or family members), respectively. The model promoters suggest that endogenous promoters would also be selected accordingly.
Figure 5.
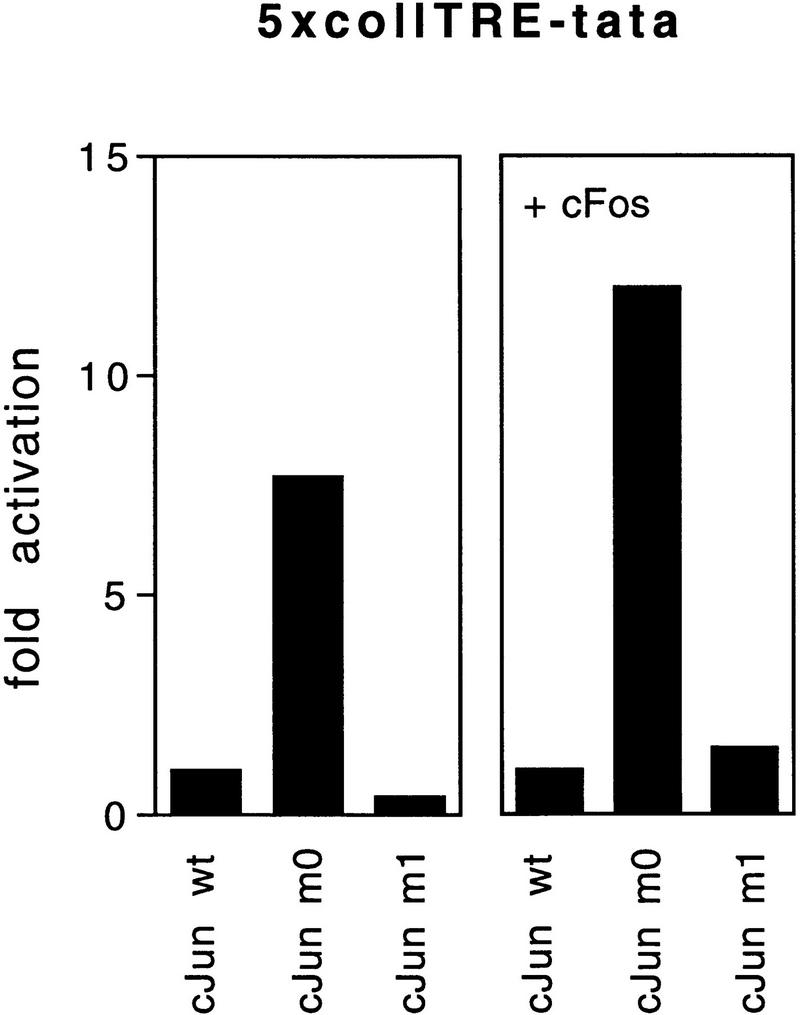

Promoter-selective activation by c-Jun mutants in CEF cells. CEFs (5 × 105) were transiently cotransfected with 2 μg of either the 5×coll–TRE–tata–luciferase, 5×jun2–tata–luciferase, or tata–luciferase reporter plasmids together with 1 μg of either wild-type or mutant RSV–c-Jun expression vector in the presence or absence of 50 ng of pRSV–c-Fos, 25 ng of pCMV–ATF2 or empty control plasmid (pUC18 carrier up to 13 μg per plate). Activity relative to that achieved in CEF transfected by wild-type Jun is plotted. Five independent experiments, using three independently generated series of primary cultures, were performed. One of these is shown.
Although of limited predictive value, but to confirm that endogenous subunits can act as partners of exogenously expressed c-Jun in CEFs, we analyzed extracts of CEFs that had been infected by a retroviral vector encoding wild-type or mutant c-Jun, or from noninfected control CEFs for protein binding to the Jun:Fos- and Jun:ATF2-specific DNA elements, coll–TRE, and jun2. Complexes formed in vitro were UV cross-linked, immunoprecipitated with antibody to AP-1 subunits and resolved by SDS-PAGE (Fig. 6). Chicken c-Jun and slightly slower migrating human c-Jun were cross-linked to both probes, while the endogenous chicken ATF2 could only be obtained as cross-linked to jun2 (Fig. 6A). Protein cross-linked to the coll–TRE sequence was examined for the presence of Fra2, which is the major member of the Fos family expressed in nonstimulated CEFs (Nishina et al. 1990; Suzuki et al. 1994; Sonobe et al. 1995). Cross-linked Fra2 was immunoprecipitated as a heterogeneous mixture of protein–DNA complexes of 46- to 55-kD apparent molecular mass (trailing behind c-Jun) from extracts of both nontransformed and c-Jun-transformed CEFs (Fig. 6B, left panel, lanes 3,4). This finding is in agreement with the previously described heterogeneous electrophoretic mobility and abundance of Fra2 in CEF (Nishina et al. 1990; Suzuki et al. 1994; Sonobe et al. 1995). Under nonstringent conditions that do not disrupt the heterodimer association, wild-type c-Jun and c-Jun–m0, but not c-Jun–m1 (nor Jun–eb1, which can only homodimerize), could be efficiently coprecipitated with Fra2 (Fig. 6B, right panel). Thus, these data are in agreement with the dimerization preference of DNA-bound c-Jun mutants synthesized in vitro (Fig. 3) and with the mutant behavior at model promoters (Fig. 5). Antibodies to ATF2 coprecipitated wild-type c-Jun and c-Jun–m1 only from complexes cross-linked to the jun2 element and treated under nonstringent conditions (not shown).
Figure 6.


c-Jun:ATF2 and c-Jun:Fra2 heterodimeric complexes in CEF cells. Immunoprecipitation of DNA-bound c-Jun, Fra2, and ATF2 from untransformed (CEF) and human c-Jun-transformed CEFs [wild-type c-Jun (cJ-CEF); c-Jun–m0 (m0-CEF); c-Jun–m1 (m1-CEF); c-Jun–eb1 chimera (Castellazzi et al. 1993, eb1-CEF)] extracts after covalent cross-linking to either the jun2 or coll–TRE oligonucleotides. CEF cell extracts were incubated with BrdU- and 32P-labeled DNA probes, cross-linked by UV-irradiation, and diluted in a mild (B) or stringent (A,B) immunoprecipitation buffer (see Materials and Methods). Immunocomplexes with antibodies as indicated were resolved on 12% SDS-PAGE and visualized by autoradiography. (A) The addition of antibody or nonimmune control antibody is shown above the gel plot. Note that endogenous chicken c-Jun migrates faster than human c-Jun. In cJ-CEF, the endogenous expression is repressed (see also Fig. 7). (B) To show the difference in c-Jun levels coprecipitating with Fra2 under nonstringent condition (right), relatively short exposures of the nonstringent precipitations are presented. Note that anti-Fra2 coprecipitates heterodimers of Fra2 with endogenous chicken c-Jun if the cross-links were done with CEF or m1-CEF extracts, heterodimers with human c-Jun only with cJ-CEF and m0-CEF extracts. No heterodimers with Fra2 are formed in the control extracts from Jun–eb1-CEF transformants as eb1–bZip mediates only homodimerization (Castellazzi et al. 1993). (−) Nonimmune serum.
We conclude that in CEF cells the c-Jun mutants also select their heterodimer partners with preference: c-Jun–m0 binds to a member of the c-Fos family, in this case Fra2, and c-Jun–m1 binds to ATF2. The zipper mutants are thus suitable tools to dissect the c-Jun-dependent transformation program.
Jun–m0 and Jun–m1 address distinct and different parts of the transformation program
To analyze the transforming ability of the Jun zipper mutants, CEF cultures stably expressing the mutants were generated by retroviral infection [derivatives of both c-Jun and v-Jun, the mutated form derived from avian sarcoma virus 17 (Maki et al. 1987), were tested in parallel]. Western blot analysis confirmed that the resulting chronically infected cell cultures expressed similar levels of the exogenous human c-Jun proteins (Fig. 7) or of v-Jun proteins (data not shown). As in the case of cells transformed by v-Jun and wild-type c-Jun [Bos et al. 1990; Castellazzi et al. 1990; Kilbey et al. 1996; although Jun autoregulates the c-jun promoter positively, excess amounts repress (K.-K. Park and H. Ponta, unpubl.)], the endogenous chicken c-Jun protein was down-regulated in CEF cells expressing either one of the human c-Jun proteins (chicken c-Jun migrates slightly faster than human c-Jun, see Fig. 6A). We also verified that in these retrovirally infected cultures the endogenous transactivation activity on the 5×jun2–TRE promoter is higher in Jun–m1-transformed CEFs than in Jun–m0 transformants. The opposite result was obtained on the 5×coll–TRE promoter (Fig. 8).
Figure 8.

Promoter-selective activation by v-Jun mutants in stably transformed CEF cells. CEFs (3 × 105) chronically infected with either R–v-Jun–m0 (hatched bar) or R–v-Jun–m1 (solid bar) were transiently transfected with 2 μg of either 5×coll–TRE–TK–luciferase, 5×jun2–TK–luciferase, or TK–luciferase reporter plasmid together with 10 μg of pUC18 as a carrier. Fold activation is the ratio of the activation on 5×jun2–TK/TK or on 5×coll–TRE–TK/TK.
The most interesting result of the transformation analysis was that the mutants conferred only partial transformation phenotypes. Under normal growth conditions (6% serum, liquid medium), all Jun-overexpressing cell cultures proliferated to the same extent (shown for v-Jun derivatives in Fig. 9B, left panel; identical results were obtained with c-Jun derivatives, not shown). However, cells expressing Jun–m0 and Jun–m1 differed in their abilities to grow in solid and low serum media. Jun–m0-expressing CEF cells grew well in soft agar, to the same extent as those expressing wild-type Jun [shown for both c-Jun and v-Jun and their mutant derivatives (Table 1; Fig. 9A)], but could not grow at low serum concentrations (Fig. 9B, right panel). In contrast, Jun–m1-expressing cells could not grow in agar (Table 1; Fig. 9A), but did proliferate at low serum (Fig. 9B, right panel). Thus, Jun mutants m0 and m1, with specificity for the Fos subfamily (Fra2) and the ATF2 subfamily, respectively, each establish one distinct aspect of the transformed phenotype induced by Jun.
Figure 9.



Different transformed phenotypes induced by dimer-specific Jun mutants in CEF cells. (A) Representative microscopic fields from wild-type and mutant c-Jun-expressing CEF cells grown for 2 weeks in agar (2 × 103 cells per 60 mm petri dish were seeded). Bar, 0.1 mm. (B) Serum-dependence of R–Jun-, R–ATF2-, and R–Fra2-infected CEF cultures overexpressing ATF2, Fra2, wild-type Jun or the mutants m0 and m1 in a v-Jun background. (R) Control cells infected with empty retrovirus. (6% serum; left) Infected CEF cells were plated at 1.5 × 105 cells per plate in duplicates and supplied with medium containing 6% FCS (Castellazzi et al. 1990). (0.6% serum; right) CEF cultures were plated at 6 × 105 cells per plate in medium containing 0.6% serum for 4 days to ascertain correct depletion of serum; the cultures were then replated at day 0 in the same medium at 1.5 × 105 cells per plate in duplicates. In each experiment, viable cells were counted by use of trypan blue at the days indicated. R control cells behaved identically to uninfected CEF (data not shown). (C) Induction of serum-independent growth in Jun–m0 cells superinfected by Jun–m1. The procedures were as in B except that CEFs were preinfected with either RD–m0 or RD–m1 as indicated. Success of the double infection was ascertained by Western blotting (not shown). Note that the ability of an m0 infectant is not improved much by reinfection with m0 but is enhanced by m1. Jun–m1-transformed cells grow in low serum (as shown in B). This cannot be further improved by additional m1 and only slightly by m0.
Table 1.
Anchorage-independent growth of CEF-expressing c-Jun and v-Jun bZip mutants
| Culture | Number of cells seededa | Plating efficiency (%) | ||
|---|---|---|---|---|
| 1 × 104 | 2 × 103 | 1 × 103 | ||
| Uninfected | − | − | — | |
| R vector | − | − | — | |
| R–c-Jun wild type | + | 444/442 | 222/168 | 21 |
| R–c-Jun-m0 | + | 198/258 | 142/108 | 13 |
| R–c-Jun-m1 | 92/86* | 26/23# | − | <1 |
| R–v-Jun | + | 550/496 | 142/164 | 19.5 |
| R–v-Jun–m0 | + | 210/178 | 102/88 | 9.6 |
| R–v-Jun–m1 | 24/32* | 10/14* | 4/8* | <1 |
| R–Fra2 | +** | 52/32** | 14/16** | 1.8 |
| R–ATF2 | − | − | − | |
| RD–v-Jun–m1 + R vector | − | 16/12* | N.D. | |
| RD–v-Jun–m1 + R–v-Jun–m0 | N.D. | 64/44 | N.D. | |
| RD–v-Jun–m0 + R vector | N.D. | 194/220 | N.D. | |
| RD–v-Jun–m0 + R–v-Jun–ml | N.D. | 398/346 | N.D. |
In agreement with the fact that in Jun-transformed CEFs, ATF2 is in relative abundance and the heterodimer partner is limiting, enhanced expression of ATF2 by retroviral infection did not induce either transformation property (Table 1; Fig. 9B), whereas retroviral expression of Fra2 caused microcolony development in agar at a low frequency [but no serum independence (Table 1; Fig. 9B)]. This suggests that enhanced levels of Fra2, complexed with endogenous Jun or with another partner, can trigger, although with low efficiency, the program specificity also seen with the Fos/Fra-seeking mutant Jun–m0.
One would predict that coinfection with both mutants would re-establish the combination of both transformation properties—growth factor independence and agar growth—just like that induced by wild-type Jun. Indeed, this was the case: Superinfection with distinguishable retroviruses of cells infected by v-Jun–m0 with either the control vector or a retroviral vector encoding v-Jun–m0 did not or only slightly increase growth in low serum. Superinfection with v-Jun–m1 enhanced growth considerably (Fig. 9C, left). Enhanced growth was accompanied by a change in cell cycle distribution: a decrease in G1 and increase of cells in S phase (not shown). Superinfection of cells containing v-Jun–m1 with empty vector of additional v-Jun–m1 had no effect. Superinfection with a retrovirus encoding v-Jun–m0 increased low-serum growth slightly over vector control (Fig. 9C, right). Corresponding complementation was obtained for the formation of agar colonies (Table 1).
Full transformation of cells comprises a complex set of properties (Jove and Hanafusa 1987; Benjamin and Vogt 1990). Transfection with cell-membrane-associated oncoproteins such as Src and Ras induces a large spectrum of phenotypic alterations including cytoskeletal rearrangement, enhanced hexose transporter synthesis and hexose uptake, and induction of gelatinase and urokinase plasminogen activator (uPA) activity. The nuclear oncoprotein Jun induces only part of the spectrum. Glucose transporter-3 mRNA synthesis (not shown) and deoxyglucose uptake (Fig. 10) and cytoskeletal organization as determined by immunofluorescence with anti-actin and anti-vinculin (not shown) were not affected by either wild-type or mutant Jun. For our comparison of Jun mutants only those transformation properties are relevant that are differentially affected by Jun or the zipper mutants. For instance, wild-type Jun weakly induced gelatinase and strongly induced uPA activity as measured by zymograms (Fig. 11). Jun–m0 also induced both activities, while Jun–m1 induced uPA activity only slightly and even inhibited gelatinase activity. Reintroduction of v-Jun–m0 into v-Jun–m1 transformed CEFs, or the opposite, led to complementation in that it restored a v-Jun phenotype for uPA activity, and to a lesser degree, for gelatinase activity (Fig. 11). Gelatinase and uPA thus represent endogenous direct or indirect targets that are positively regulated by Jun:Fra2.
Figure 10.

Deoxyglucose uptake by Src and Jun transformants. Conditions are as in Figs. 9 and 11.
Figure 11.

Jun-induced uPA and 72-kD gelatinase activities. Zymographs showing uPA and 72-kD gelatinase activity were obtained with conditioned medium of CEF cultures infected with either the empty R vector, or with R vector encoding v-Jun, v-Jun–m0, v-Jun–m1, or v-Src (NY72-4 variant; Mayer et al. 1986). (m0 + m1) and (m1 + m0) are cultures coexpressing v-Jun–m0 and v-Jun–m1 generated by sequential infection (see Table 1 and Fig. 9C). The gels were evaluated by densitometry. Maximum induction was obtained with v-Src and set to 100.
Discussion
In this report, we provide strong evidence for the existence of two complementing genetic programs, autocrine growth and anchorage independence, which are controlled independently by different Jun-containing dimeric transcription factors. As a prerequisite for such analyses we developed a methodology for dissecting the complexity of the AP-1 family of transcription factors in cells carrying their normal endogenous complement of AP-1 subunits. The principle of the method is based on overexpression of a bZip mutant that selects and sequesters a preferred partner subunit, thus shifting the equilibrium in the AP-1 complement toward this specific heterodimer and allowing us to ask whether this heterodimer participates in a reaction to be studied. The AP-1 family is of central importance to many cellular processes, from proliferation to differentiation and apoptosis. In most cases, it has not yet been possible to distinguish which heterodimer is responsible. We propose to use this methodology in dissecting the AP-1 factors responsible for instance for the interaction of AP-1 with nuclear receptors, for apoptosis, and for tumor promotion. bZip mutants can be used in cells from different species as there is a high degree of conservation of c-Fos and Fra2 bZip sequences between chick and man (Mölders et al. 1987; Nishina et al. 1990) and between ATF2 bZip sequences of Xenopus, chick, and humans (Villarreal and Richter 1995; J. Baguet and M. Castellazzi, unpubl.). Here, we demonstrate the validity of our approach in examining the role of Jun heterodimers in the transformation of CEFs. Importantly, the experiments yielded interesting information on the AP-1-mediated program of genes involved in transformation of CEFs. To what extent can the CEF system be generalized? Possibly, the abundance of partner subunits in a given cell and the condition of the cell that is a reflection of the extracellular stimuli at a given time, will determine the phenotypic outcome of a transfection and transformation attempt by Jun. The heterogeneity of the endogenous complement of AP-1 subunits puts some limit on the generalization of the CEF data, but the transcription analyses in F9 and HeLa cells give us confidence that the mutants will be useful in several more complex systems as described above.
bZip specificity
The bZip mutants of Jun used in this study, m0 and m1, prefer a subset of partners each. The zipper specificity is already quite significant if protein–protein interaction is analyzed in the absence of DNA. Specificity is, however, accentuated with the appropriate DNA element present, in particular in the case of Jun–m0, suggesting that interaction of the adjacent basic region with DNA affects the stability of dimers. Nevertheless, there is still considerable choice that depends on the expression of AP-1 subunits in the cell type examined. In CEFs, the major partners appear to be Fra2 and ATF2 for mutants m0 and m1, respectively.
Two complementing programs of transformation genes
The decisive result of the transformation by Jun mutants m0 and m1 is the clear-cut partial transformation phenotype established by each one individually. Following the corresponding Jun:Fos and Jun:ATF2 model promoter results, the mutants select different target genes. Both types of target genes are needed for the transformation phenotype as induced by wild-type Jun, in that double infection with both mutants results in complementation to the full program. Induction of both growth in agar and low serum can also be achieved if Jun homodimers are stabilized artificially (Jun/eb1 and Jun/gcn4, Castellazzi et al. 1993). Stabilized Jun homodimers accept both heptameric and octameric DNA elements (H. van Dam et al., unpubl.) and could, like wild-type Jun, activate both parts of the transformation program.
It is conceivable that the overexpression of Jun or Jun mutants creates a loss and/or gain of function. Exogenous overexpression of Jun represses the endogenous level of Jun. Thus, for instance, overexpression of Jun–m1 will sequester ATF2, but will not bind Fra2, thus activating Jun:ATF2 dependent target genes (gain of function) and reducing Jun:Fra2-dependent expression (loss of function). On their own, ATF2 and Fra2 will then not contribute to gain or loss of function as they did not, or very inefficiently, transform when overexpressed individually. The importance of a gain of function with respect to target gene activation is demonstrated by previously published data showing that an amino-terminal deletion mutant of Jun, lacking the transactivation domain, does not induce transformation but acts as a dominant-negative (Castellazzi et al. 1991, 1993).
Jun:Fos or Jun:Fra2(-like) dimers regulate a program of target genes critical for anchorage-independent growth, while Jun:ATF2(-like) dimers regulate target genes relevant for growth in reduced concentrations of serum (Fig. 12). In comparison to Jun, the upstream acting oncoproteins exert a more pleiotropic effect on cell growth. Nevertheless their influence on Jun heterodimers can be assigned to the two different portions of the Jun-induced genetic program in that Jun:Fos is predominantly at the receiving end of the Ras–Raf–Erk pathway and Jun:ATF2 is mostly regulated through SEK–JNK and p38 but not Erk (van Dam et al. 1995; Liu et al. 1996; Canman and Kastan 1996; Wilhelm et al. 1997). Our interpretation is compatible with what is known about the regulation of putative target genes that could be responsible for these two facets of cellular transformation (Jove and Hanafusa 1987). Jun:Fos (Fra2) target genes are possibly concerned with the extracellular matrix, thus enabling cells to grow in soft agar, whereas Jun:ATF2 may target factors relevant for autocrine stimulation. Promoter analyses and studies on c-fos −/− and c/v-_fos_-overproducing cells and mice have revealed that genes involved in anchorage independence are regulated by Jun:Fos, for example, genes coding for adhesion molecules, cytoskeleton components, and matrix-degrading enzymes (Angel et al. 1987; Matrisian 1990; Reichman et al. 1992; Hofmann et al. 1993; Hu et al. 1994; Jooss and Müller 1995; Saez et al. 1995; Schreiber et al. 1995). The Fos-seeking mutant Jun–m0, but not Jun–m1, indeed caused elevated activity of gelatinase and uPA. On the other side, certain growth factor and growth factor receptor encoding genes are regulated by ATF-binding sites and thus possibly Jun:ATF2 (Gray et al. 1993; Newell et al. 1994; Morishita et al. 1996).
Figure 12.
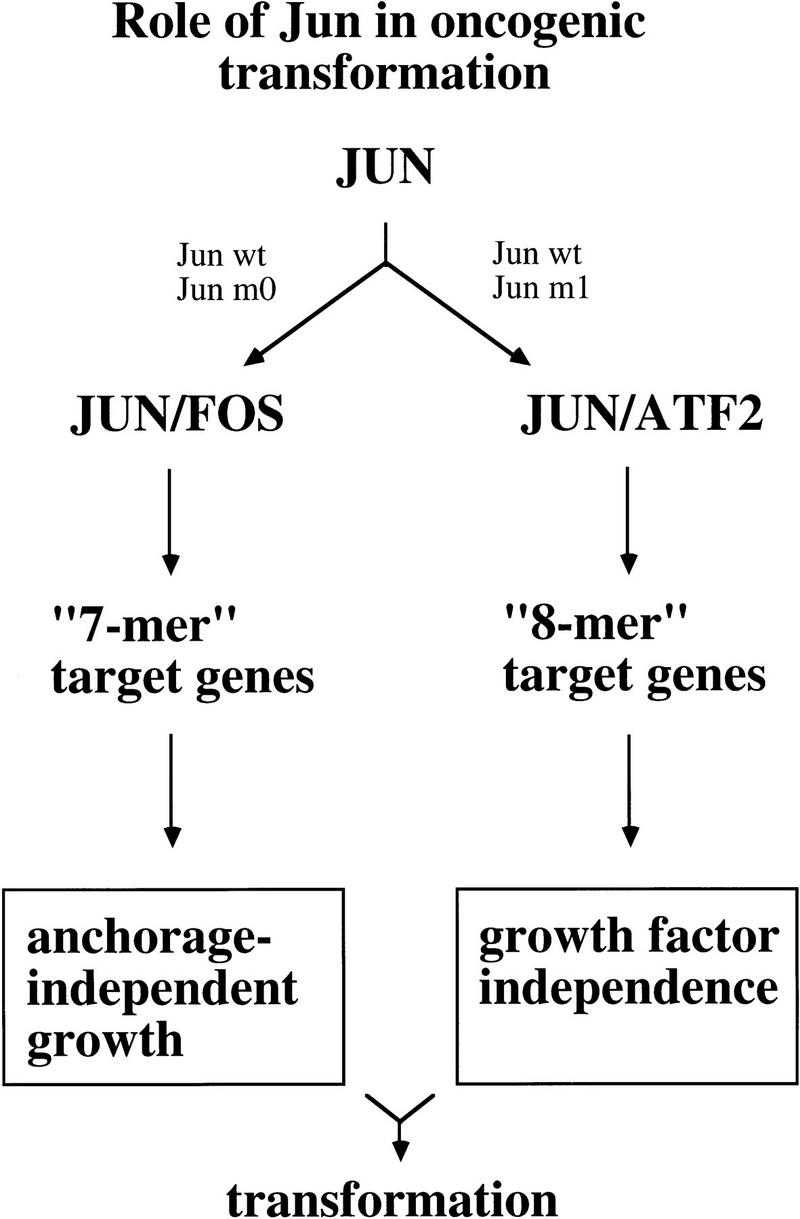
Model of Jun-dependent transformation. Note that Fos stands for Fos-like. In CEFs the major Jun partner is Fra2.
Materials and methods
DNA constructs
The human c-Jun zipper mutants m0 and m1 were generated by PCR mutagenesis and mutations were confirmed by sequencing. Outer primers were 5′-CTAGAAGCTTCTCTGTTCTATGAGTGCAAAGATGGG-3′ and 5′-CCTCTAGATCTAAATGTTTGCAACTGCTGCGTTAGC-3′. Inner primers were 5′-GAGAGAATCGCGCGCCTGAAGGAAAA-3′ (m0) and 5′-AACATGCTCGAGGAAGAGGTGGC-3′ (m1), and their complementary equivalents. Templates were pRSV–Δ6-194c-Jun (m0) or pRSV–Δ6-194/m35 (m1; H. van Dam, unpubl.) and pRSV–c-Jun (Angel et al. 1988). The PCR-generated fragments were subsequently introduced into pRSV–c-Jun as _Bst_XI–_Hpa_I fragments.
In pRSV–Gal4–DBD–c-Jun–bZip, amino acids 224–331 of human c-Jun are fused to the DNA-binding domain of Gal4 (amino acids 1–147): _Pst_I-linearized pRSV–c-Jun was blunt-ended and ligated to an _Xho_I linker. The _Xho_I–_Sac_I fragment was subsequently introduced into _Xho_I–_Sac_I digested pRSV–Gal4–DBD. pRSV–Gal4–DBD–c–Jun–bZip–m0 and –m1 were obtained by replacement of the _Bst_XI–_Bam_HI fragment of pRSV–Gal4–DBD–c-Jun–bZip with the corresponding fragments of pRSV–c-Jun–m0 and pRSV–cJun–m1. pRSV–ATF2–VP16 was obtained first by replacement of the _Xba_I fragment of pBAT–ATF2 (see below) with the corresponding fragment of pATF2–VP16 (Liu and Green 1990) and subsequently by replacement of the ATF2 region of pRSV–ATF2 (van Dam et al. 1995) with ATF2–VP16 via exchange of _Sal_I–_Not_I (partial) fragments. The expression vectors for c-Fos, Gal4–DBD, and c-Jun-bZip (pRSV–d6–194cJun) have been described previously (Angel et al. 1989; Sadowski et al. 1988; Offringa et al. 1990). The 5×GAL4–E4–tata–luciferase reporter plasmid was kindly provided by C. Livingstone and N.C. Jones (ICRF, London, UK). pCMV–ATF2, containing the human ATF2 cDNA inserted into pCMV–neo–Bam, was kindly provided by M. Duyndam (University of Leiden, The Netherlands). The reporter plasmids 5×coll–TRE–tata–luciferase, 5×jun2–tata–luciferase, and tata–luciferase were constructed by introduction of the _Xho_I–_Sty_I fragment of pGl2 into _Xho_I–_Sty_I-digested tata–CAT, 5×coll–TRE–tata–CAT, and 5×jun2–tata–CAT (Jonat et al. 1990; van Dam et al. 1993). TK–luciferase, 5×coll–TRE–TK–luciferase and 5×jun2–TK–luciferase were constructed by introduction of the _Pvu_II–_Bgl_II fragments of the corresponding CAT plasmids (Angel et al. 1987; van Dam et al. 1995) into _Sma_I–_Bgl_II-digested pGl3 (Promega).
For in vitro transcription/translation, wild-type and mutant (human) c-Jun and bZip–ATF2 (amino acids 335–505) were cloned into the pBluescript KS-derivative pBAT (Annweiler et al. 1991). For translation of c-Fos (mouse), a pGEM3-derived expression vector (kindly provided by H.Th.M. Timmers, University of Utrecht, The Netherlands) was used.
For construction of the retroviral expression vectors, the various c-Jun-, Fra2-, and ATF2-coding sequences were first introduced into the polylinker of the adaptor plasmid CLA12 (Hughes et al. 1987). The respective coding sequences were subsequently introduced as _Cla_I fragments into the retroviral vector RCAS (Hughes et al. 1987) to generate R–c-Jun, R–m0 and R–m1, R–ATF2 and R–Fra2. R–c-Jun/eb1 has been described previously (Castellazzi et al. 1993; Vandel et al. 1995).
Introduction of the mutant c-Jun zipper sequences into v-Jun coding sequences was performed by PCR (S. Huguier and M. Castellazzi, unpubl.).
Cell culture, transient transfections, and recombinant retroviruses
Transient transfection of undifferentiated F9 cells and HeLa tk− cells by use of the calcium phosphate and DEAE–dextran methods, respectively, was performed as described (Angel et al. 1988). Preparation of cell extracts and subsequent analysis for luciferase activity was performed as described in Duyndam et al. (1996). F9 cells were harvested 24 hr after transfection, HeLa cells 48 hr after transfection.
Primary CEF cultures were prepared from 8-day-old C/E SPAFAS chicken embryos and grown in medium containing 6% serum as described (Castellazzi et al. 1990). Human c-Jun or v-Jun-expressing CEF cultures were obtained by chronic retroviral infection with the replication-competent retrovirus R–c-Jun or R–v-Jun. R–m0 and R–m1 viruses and control retrovirus (R) were generated similarly. For superinfection, a second replication-competent retrovirus with different envelope, RD, was used, carrying the same gene constructs. Analysis of anchorage- and serum-independent growth has been described (Castellazzi et al. 1993). For promoter activity analysis 3–5×105 cells were seeded on 60-mm dishes and transiently transfected with luciferase reporter plasmids by use of the calcium phosphate method. For each reporter plasmid, a single precipitate was made and subsequently divided over the different CEF cultures to avoid variation in precipitate quality. After 14 hr, cells were washed with PBS and incubated for another 30 hr, after which the still subconfluent cells were harvested for luciferase assays (see above).
Deoxyglucose uptake and zymography for gelatinase and uPA
Deoxyglucose uptake was determined according to Frost and Lane (1985) with 90% confluent CEF cells washed with PBS and incubated in PBS in a mixture of 0.1 mm 2-deoxy-glucose/2-deoxy-D-[1-14C] glucose (specific activity, 54 mCi/mmole, Amersham) at 0.3 μCi/ml for 10 min at room temperature.
Protease activities were detected according to the substrate–SDS-PAGE assay system described by Heussen and Dowdle (1980) with 24 hr conditioned supernatant from monolayer CEF cultures. For gelatinases, the gel contained 0.3 mg/ml gelatin (Sigma), and for uPA activity 1 mg/ml α-casein (Sigma) and 8 mg/ml lysine–Sepharose affinity-purified chicken plasminogen. Incubations were at 37°C for 4 hr with buffer conditions as described (for gelatinases, see Heussen and Dowdle 1980; for uPA, see Marshall et al. 1990). Proteolytic activities were detected as clear bands against the blue background of stained substrate. Scanning of the gels and quantification were performed by computer methods.
In vitro translation, DNA binding assays, and immunoprecipitation analysis
In vitro transcription/translation of pBAT–c-Jun, pBAT–bZip–ATF2 and pEP–c-Fos was performed by use of a Promega kit according to the manufacturer’s procedure. Plasmids were linearized, transcribed with T3 or SP6 RNA polymerase, respectively, and translated in reticulocyte lysate with [35S]methionine. The translation efficiency was determined by SDS-PAGE. Proteins were quantified by use of a Fuji Bio-Imaging Analyser. For gel retardation analysis, similar amounts of proteins were mixed in a total volume of 4 μl of reticulocyte lysate per 20-μl reaction mixture.
Preparation of cell extracts for Western analysis and DNA-binding assays were performed as described previously (van Dam et al. 1993). DNA-binding assays, UV cross-linking, and the coll–TRE and jun2 DNA probes used, have also been described (van Dam et al. 1993). In vitro translated proteins were incubated with 10 fmoles of 32P-labeled probe for 30 min at room temperature, after which DNA–protein complexes were resolved on 4% Tris–glycine gels.
For immunoprecipitation of cross-linked proteins, the DNA-binding reactions included 30 μg of CEF cell extract and 400 fmoles of BrdU-containing DNA-probe. Immunoprecipitation under stringent conditions was performed in 10 mm HEPES (pH 7.9), 150 mm NaCl, 0.1% sodium–deoxycholate (DOC), 0.1% NP-40, 0.1% SDS; under mild conditions in 50 mm Tris (pH 7.5), 5 mm EDTA, 125 mm NaCl, 0.1% Triton X-100. Antibodies used for immunoprecipitation were c-Jun–pep1 (Oncogene Science), which recognizes both chicken and human c-Jun, the ATF2-specific antibody C19, and the Fra2-specific antibody L15 (Santa Cruz).
Acknowledgments
We thank S. Mueller, A. Steffen, R. Schiffelers, H. Wikman, N. Beerens, L. Vandel, S. Noguet, and P. Blancou for contributions to experiments, Drs. M. Duyndam, M. Timmers, M. Green, and N. Jones for DNA constructs. This work was supported by an EMBO long-term fellowship and a fellowship of the Royal Netherlands Academy of Arts and Sciences (H.v.D.), by grants from Deutsche Forschungsgemeinschaft (P.A. and P.H., He 551/8-2), from Association pour la Recherche sur le Cancer, the Fédération Nationale des Centres de Lutte Contre le Cancer, and the Mutelle Générale de l’Education Nationale (M.C.), and by the European Economic Community Biomedicine and Health Program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL genetik@igen.fzk.de; FAX 49 7247 823354.
References
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- Angel P, Smeal T, Meek J, Marin M. Jun and vJun contain multiple regions that participate in transcriptional activation in an interdependent manner. New Biol. 1989;1:35–43. [PubMed] [Google Scholar]
- Annweiler A, Hipskind RA, Wirth T. A strategy for efficient in vitro translation of cDNAs using the rabbit β-globin leader sequence. Nucleic Acids Res. 1991;19:3750. doi: 10.1093/nar/19.13.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrook DM, Jones NC. Heterodimer formation between CREB and JUN proteins. Oncogene. 1990;5:295–302. [PubMed] [Google Scholar]
- Benjamin T, Vogt PK. Cell transformation by viruses. In: Fields BN, editor. Virology. New York, NY: Raven Press; 1990. [Google Scholar]
- Bos TJ, Monteclaro FS, Mitsunobu F, Ball Jr AR, Chang CHW, Nishimura T, Vogt PK. Efficient transformation of chicken embryo fibroblasts by c-Jun requires structural modification in coding and noncoding sequences. Genes & Dev. 1990;4:1677–1687. doi: 10.1101/gad.4.10.1677. [DOI] [PubMed] [Google Scholar]
- Canman CE, Kastan MB. Signal transduction. Three paths to stress relief. Nature. 1996;384:213–214. doi: 10.1038/384213a0. [DOI] [PubMed] [Google Scholar]
- Castellazzi M, Dangy J-P, Mechta F, Hirai S-I, Yaniv M, Samarut J, Lassailly A, Brun G. Overexpression of avian or mouse c-jun in primary chick embryo fibroblasts confers a partially transformed phenotype. Oncogene. 1990;5:1541–1547. [PubMed] [Google Scholar]
- Castellazzi M, Spyrou G, La Vista N, Dangy J-P, Piu F, Yaniv M, Brun G. Overexpression of c-jun, junB, or JunD affects cell growth differently. Proc Natl Acad Sci. 1991;88:8890–8894. doi: 10.1073/pnas.88.20.8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellazzi M, Loiseau L, Piu F, Sergeant A. Chimeric c-Jun containing an heterologous homodimerization domain transforms primary chick embryo fibroblasts. Oncogene. 1993;8:1149–1160. [PubMed] [Google Scholar]
- Deng T, Karin M. c-Fos transcriptional activity stimulated by H-Ras-activated protein kinase distinct from JNK and ERK. Nature. 1994;371:171–175. doi: 10.1038/371171a0. [DOI] [PubMed] [Google Scholar]
- Dévary Y, Gottlieb RA, Smeal T, Karin M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- Duyndam MCA, van Dam H, van der Eb AJ, Zantema A. The CR1 and CR3 domains of the adenovirus type 5 E1A proteins can independently mediate activation of ATF-2. J Virol. 1996;70:5852–5859. doi: 10.1128/jvi.70.9.5852-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost SC, Lane MD. Evidence for the involvement of vicinal sulfhydryl groups in insulin-activated hexose transport by 3T3-L1 adipocytes. J Biol Chem. 1985;260:2646–2652. [PubMed] [Google Scholar]
- Gray JG, Chandra G, Clay WC, Stinnett SW, Haneline SA, Lorenz JJ, Patel IR, Wisely GB, Furdon PJ, Taylor JD, Kost TA. A CRE/ATF-like site in the upstream regulatory sequence of the human interleukin 1β gene is necessary for induction in U937 and THP-1 monocytic cell lines. Mol Cell Biol. 1993;13:6678–6689. doi: 10.1128/mcb.13.11.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Heussen C, Dowdle EB. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Anal Biochem. 1980;102:196–202. doi: 10.1016/0003-2697(80)90338-3. [DOI] [PubMed] [Google Scholar]
- Hilberg F, Aguzzi A, Howells N, Wagner EF. c-Jun is essential for normal mouse development and hepatogenesis. Nature. 1993;365:179–181. doi: 10.1038/365179a0. [DOI] [PubMed] [Google Scholar]
- Hofmann M, Rudy W, Günthert U, Zimmer SG, Zawadzki V, Zöller M, Lichtner RB, Herrlich P, Ponta H. A link between RAS and metastatic behavior of tumor cells: ras induces CD44 promoter activity and leads to low-level expression of metastasis-specific variants of CD44 in CREF cells. Cancer Res. 1993;53:1516–1521. [PubMed] [Google Scholar]
- Hu E, Mueller E, Oliviero S, Papaioannou VE, Johnson R, Spiegelman BM. Targeted disruption of the c-fos gene demonstrates c-fos-dependent and -independent pathways for gene expression stimulated by growth factors or oncogenes. EMBO J. 1994;13:3094–3103. doi: 10.1002/j.1460-2075.1994.tb06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave PS. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987;61:3004–3012. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst HC. Transcription factors: 1: bZIP proteins. Protein Profile. 1994;1:123–168. [PubMed] [Google Scholar]
- Ivashkiv LB, Liou H-C, Kara CJ, Lamph WW, Verma IM, Glimcher LH. mXBP/CRE-BP2 and c-Jun form a complex which binds to the cyclic AMP, but not to the 12-0-tetradecanoylphorbol-13-acetate, response element. Mol Cell Biol. 1990;10:1609–1621. doi: 10.1128/mcb.10.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RS, van Lingen B, Papaioannou VE, Spiegelman BM. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes & Dev. 1993;7:1309–1317. doi: 10.1101/gad.7.7b.1309. [DOI] [PubMed] [Google Scholar]
- Jonat C, Rahmsdorf HJ, Park K-K, Cato ACB, Gebel S, Ponta H, Herrlich P. Anti-tumor promotion and antiinflammation: Down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62:1189–1204. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- Jooss KU, Müller R. Deregulation of genes encoding microfilament-associated proteins during Fos-induced morphological transformation. Oncogene. 1995;10:603–608. [PubMed] [Google Scholar]
- Jove R, Hanafusa H. Cell transformation by the viral src oncogene. Annu Rev Cell Biol. 1987;3:31–56. doi: 10.1146/annurev.cb.03.110187.000335. [DOI] [PubMed] [Google Scholar]
- Kerppola TK, Luk D, Curran T. Fos is a preferential target of glucocorticoid receptor inhibition of AP-1 activity in vitro. Mol Cell Biol. 1993;13:3782–3791. doi: 10.1128/mcb.13.6.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbey A, Black EJ, Unlu M, Gillespie DA. The v-Jun oncoprotein replaces p39 c-Jun as the predominant AP-1 constituent in ASV17-transformed fibroblasts: Implications for SAPK/JNK-mediated signal transduction. Oncogene. 1996;12:2409–2418. [PubMed] [Google Scholar]
- Liu F, Green MR. A specific member of the ATF transcription factor family can mediate transcription activation by the adenovirus E1a protein. Cell. 1990;61:1217–1224. doi: 10.1016/0092-8674(90)90686-9. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, Wang JY. Three distinct signaling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- Maki Y, Bos TJ, Davis C, Starbuck M, Vogt PK. Avian sarcoma virus 17 carries the jun oncogene. Proc Natl Acad Sci. 1987;84:2848–2852. doi: 10.1073/pnas.84.9.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BC, Sageser DS, Rao NY, Emi M, Hoidal JR. Alveolar epithelial cell plasminogen activator. J Biol Chem. 1990;265:8198–8204. [PubMed] [Google Scholar]
- Matrisian LM. Metalloproteinases and their inhibitors in matrix remodeling. Trends Genet. 1990;6:121–125. doi: 10.1016/0168-9525(90)90126-q. [DOI] [PubMed] [Google Scholar]
- Mayer BJ, Jove R, Krane JF, Poirier F, Calothy G, Hanafusa H. Genetic lesions involved in temperature sensitivity of the src gene products of four Rous sarcoma virus mutants. J Virol. 1986;60:858–867. doi: 10.1128/jvi.60.3.858-867.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölders H, Jenuwein T, Adamkiewicz J, Müller R. Isolation and structural analysis of a biologically active chicken c-fos cDNA: Identification of evolutionarily conserved domains in fos protein. Oncogene. 1987;1:377–385. [PubMed] [Google Scholar]
- Morishita K, Johnson DE, Williams LT. A novel promoter for vascular-endothelial growth factor receptor (flt-1) that confers endothelial-specific gene expression. J Biol Chem. 1996;270:27948–27953. doi: 10.1074/jbc.270.46.27948. [DOI] [PubMed] [Google Scholar]
- Newell CL, Deisseroth AB, Lopez-Berestein G. Interaction of nuclear proteins with an AP-1/CRE-like promoter sequence in the human TNF-alpha gene. J Leukocyte Biol. 1994;56:27–35. doi: 10.1002/jlb.56.1.27. [DOI] [PubMed] [Google Scholar]
- Nishina H, Sato H, Suzuki T, Sato M, Iba H. Isolation and characterization of fra-2, an additional member of the fos gene family. Proc Natl Acad Sci. 1990;87:3619–3623. doi: 10.1073/pnas.87.9.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea E, Rutkowski R, Kim PS. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell. 1992;68:699–708. doi: 10.1016/0092-8674(92)90145-3. [DOI] [PubMed] [Google Scholar]
- Offringa R, Gebel S, van Dam H, Timmers M, Smits A, Zwart R, Stein B, Bos JL, van der Eb A, Herrlich P. A novel function of the transforming domain of E1a: Repression of AP-1 activity. Cell. 1990;62:527–538. doi: 10.1016/0092-8674(90)90017-9. [DOI] [PubMed] [Google Scholar]
- Reichmann E, Schwarz H, Deiner EM, Leitner I, Eilers M, Berger J, Busslinger M, Beug H. Activation of an inducible c-FosER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell. 1992;71:1103–1116. doi: 10.1016/s0092-8674(05)80060-1. [DOI] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Saez E, Rutberg SE, Mueller E, Oppenheim H, Smoluk J, Yuspa SH, Spiegelman BM. c-fos is required for malignant progression of skin tumors. Cell. 1995;82:721–732. doi: 10.1016/0092-8674(95)90469-7. [DOI] [PubMed] [Google Scholar]
- Schönthal A, Büscher M, Angel P, Rahmsdorf HJ, Ponta H, Hattori K, Chiu R, Karin M, Herrlich P. The Fos and Jun/AP-1 proteins are involved in the downregulation of Fos transcription. Oncogene. 1989;4:629–636. [PubMed] [Google Scholar]
- Schreiber M, Baumann B, Cotten M, Angel P, Wagner EF. Fos is an essential component of the mammalian UV response. EMBO J. 1995;14:5338–5349. doi: 10.1002/j.1460-2075.1995.tb00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuermann M, Hunter JB, Hennig G, Müller R. Non-leucine residues in the leucine repeats of Fos and Jun contribute to the stability and determine the specificity of dimerization. Nucleic Acids Res. 1991;19:739–746. doi: 10.1093/nar/19.4.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonobe MH, Yoshida T, Murakami M, Kameda R, Iba H. fra-2 promoter can respond to serum-stimulation through AP-1 complexes. Oncogene. 1995;10:689–696. [PubMed] [Google Scholar]
- Suzuki T, Murakami M, Onai N, Fukuda E, Hashimoto Y, Sonobe MH, Kameda T, Ichinose M, Miki K, Iba H. Analysis of AP-1 function in cellular transformation pathways. J Virol. 1994;68:3527–3535. doi: 10.1128/jvi.68.6.3527-3535.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam H, Duyndam M, Rottier R, Bosch A, de Vries-Smits L, Herrlich P, Zantema A, Angel P, van der Eb AJ. Heterodimer formation of c-Jun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12:479–487. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandel L, Pfarr CM, Huguier S, Loiseau L, Sergeant A, Castellazzi M. Increased transforming activity of JunB and JunD by introduction of an heterologous homodimerization domain. Oncogene. 1995;10:495–507. [PubMed] [Google Scholar]
- Villareal XC, Richter JD. Analysis of ATF2 gene expression during early Xenopus laevis development. Gene. 1995;153:225–229. doi: 10.1016/0378-1119(94)00770-s. [DOI] [PubMed] [Google Scholar]
- Vinson CR, Hai T, Boyd SM. Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: Prediction and rational design. Genes & Dev. 1993;7:1047–1058. doi: 10.1101/gad.7.6.1047. [DOI] [PubMed] [Google Scholar]
- Vogt PK. Oncogenic transformation by Jun. In: Angel PE, Herrlich PA, editors. The Fos and Jun families of transcription factors. Boca-Raton, FL: CRC Press; 1994. pp. 203–219. [Google Scholar]
- Wilhelm D, Bender K, Knebel A, Angel P. The level of intracellular glutathione is a key regulator for the induction of stress-activated signal transduction pathways including p38 and JNK/SAPK kinases by alkylating agents. Mol Cell Biol. 1997;17:4792–4800. doi: 10.1128/mcb.17.8.4792. [DOI] [PMC free article] [PubMed] [Google Scholar]