Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gβγ complex underlies activation of the yeast pheromone response pathway (original) (raw)
Abstract
In the Saccharomyces cerevisiae pheromone response pathway, the Gβγ complex activates downstream responses by an unknown mechanism involving a MAP kinase cascade, the PAK-like kinase Ste20, and a Rho family GTPase, Cdc42. Here we show that Gβγ must remain membrane-associated after release from Gα to activate the downstream pathway. We also show that pheromone stimulates translocation of the kinase cascade scaffold protein Ste5 to the cell surface. This recruitment requires Gβγ function and the Gβγ-binding domain of Ste5, but not the kinases downstream of Gβγ, suggesting that it is mediated by Gβγ itself. Furthermore, this event has functional significance, as artificial targeting of Ste5 to the plasma membrane, but not intracellular membranes, activates the pathway in the absence of pheromone or Gβγ. Remarkably, although independent of Gβγ, activation by membrane-targeted Ste5 requires Ste20, Cdc42, and Cdc24, indicating that their participation in this pathway does not require them to be activated by Gβγ. Thus, membrane recruitment of Ste5 defines a molecular activity for Gβγ. Moreover, our results suggest that this event promotes kinase cascade activation by delivering the Ste5-associated kinases to the cell surface kinase Ste20, whose function may depend on Cdc42 and Cdc24.
Keywords: Heterotrimeric G protein, MAP kinase, signal transduction, PAK/Ste20 kinase, Rho/Rac/Cdc42 GTPase
The mating reaction of the yeast Saccharomyces cerevisiae provides a model signal transduction system in which a G-protein-coupled receptor activates a mitogen-activated protein (MAP) kinase cascade (for review, see Sprague and Thorner 1992; Leberer et al. 1997a). Here, two haploid cells of opposite mating types (a and α) fuse into a single diploid cell in response to secreted pheromones (a-factor and α-factor), which stimulate cell cycle arrest, transcriptional induction of mating-related genes, and morphological changes. These responses are activated through a pathway that begins with a cell surface receptor and its associated heterotrimeric G protein, Gαβγ, which is composed of Gpa1 (Gα), Ste4 (Gβ), and Ste18 (Gγ). Downstream of the G protein lies a cascade of protein kinases—Ste11, Ste7, and Fus3—that is related to mammalian MAP kinase cascades, and an associated “kinase scaffold” protein Ste5 (for review, see Herskowitz 1995; Madhani and Fink 1998). Finally, targets of this kinase cascade include Far1, which activates cell cycle arrest, and Ste12, a DNA-binding protein responsible for transcriptional induction.
The mechanism by which the kinase cascade is activated by the heterotrimeric G protein remains poorly understood. It is clear that the free Gβγ complex activates downstream responses, after binding of pheromone to the receptor stimulates its dissociation from Gα, as deletion of the gene (GPA1) encoding Gα mimics pheromone treatment (for review, see Sprague and Thorner 1992). But the identity of the immediate target of Gβγ and the molecular mechanism used by Gβγ to activate the kinase cascade are not clear. Additional participants in this process include Cdc42, a Rho family GTPase; Cdc24, a guanine nucleotide exchange factor for Cdc42; and Ste20, a member of the PAK family of Rac/Cdc42-dependent kinases (for review, see Leberer et al. 1997a; Sells and Chernoff 1997). These proteins are also required for functions other than mating, including cytoskeletal organization (for review, see Chant and Stowers 1995). The simplest model positions these proteins as intermediates in an activation pathway from Gβγ to the kinase cascade (Gβγ → Cdc24 → Cdc42 → Ste20 → Ste11 → etc.), as the Gβ subunit Ste4 binds to the activator of Cdc42, Cdc24 (Zhao et al. 1995; Nern and Arkowitz 1998), Cdc42 binds Ste20 (Simon et al. 1995; Zhao et al. 1995), and Ste20 phosphorylates Ste11 (Wu et al. 1995). This has been questioned, however, by reports that Ste20 does not need to bind Cdc42 to function in this pathway (Peter et al. 1996; Leberer et al. 1997b). In addition, this scheme does not suggest a role for a demonstrated binding interaction between Gβγ and Ste5 (Whiteway et al. 1995). Furthermore, there is no evidence that Cdc24, Cdc42, or Ste20 have their activities increased in magnitude by Gβγ, and instead Gβγ may harness these proteins to activate downstream events by means not involving their activation per se.
A clue to how yeast Gβγ signals may lie in its sensitivity to mutations in the carboxy-terminal CaaX motif of the Gγ subunit, which is a site of post-translational modifications that include addition of a hydrophobic prenyl moiety, likely a farnesyl group (Whiteway and Thomas 1994). Such modifications can either impart an affinity for membranes or contribute to affinity for other proteins (Zhang and Casey 1996). In yeast, mutations in the CaaX motif make Gβγ unable to signal, even when Gα is absent (Grishin et al. 1994; Whiteway and Thomas 1994), implying either an effect on binding to a target protein or a requirement that Gβγ perform its signaling function at the membrane. A likely target of Gβγ is Ste5, as it binds Gβγ and is required for pheromone signaling (Sprague and Thorner 1992; Whiteway et al. 1995), and because mutations in Ste5 that block binding to Gβγ disrupt signaling (Inouye et al. 1997; Feng et al. 1998), and many signaling-defective Gβ and Gγ mutants are defective at binding Ste5 (P.M. Pryciak, in prep.). Interestingly, although Gγ mutants lacking the CaaX motif are defective at signaling, they are proficient at binding Ste5 (P.M. Pryciak, in prep.), suggesting either that they disrupt binding to some other target of Gβγ or that signaling requires the Gβγ–Ste5 interaction to occur at the membrane.
In this report, we first address the requirement for the carboxy-terminal modifications in the Gγ subunit. We find they can be functionally replaced by other membrane-targeting sequences, suggesting that their role in signaling is to keep Gβγ at the membrane after it dissociates from Gα. We then address why signaling by Gβγ might require it to remain at the membrane, and provide evidence that Gβγ recruits the kinase cascade scaffold protein Ste5 to the membrane, and that this causes activation of the pathway by promoting increased proximity of Ste5 to a cell surface-associated kinase Ste20. Finally, we provide evidence that participation of Cdc24, Cdc42, and Ste20 in the pheromone response pathway does not require them to be activated by Gβγ.
Results
Role of Gγ carboxy-terminal modifications
To address whether carboxy-terminal modifications of the yeast Gγ subunit Ste18 are required for membrane association or for binding a target protein, we asked whether they could be replaced with other membrane-targeting sequences. Four potential membrane targeting domains (MTDs) were tested (Table 1): an amino-terminal myristoylation sequence (Nmyr), the first four amino-terminal transmembrane domains (NTM) from yeast Ste6, a carboxy-terminal prenylation and palmitoylation motif (Cpr), and a single carboxy-terminal transmembrane domain (CTM). Plasmids were constructed that encode fusions of these MTDs to a Ste18 product (Ste18ΔC) lacking its final five carboxy-terminal residues (normally CCTLM). These were placed under control of a galactose-inducible promoter to facilitate assays of Gβγ function in cells deleted for Gα.
Table 1.
MTD sequences
| MTD name | Source genea | Sequenceb | Localizationc | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino-terminal MTDs | |||||||||||||||||||||||||||||||||||||
| Nmyr | RSV v-src | M | G | S | S | K | S | K | P | K | D | P | S | N | R | R | H | S | L | E | P | P | D | S | T | H | H | G | G | F | P | A | S | N | T | -- | N.D. |
| NTM | S.c. STE6 | (Ste6 amino acid residues 1–236)-- | N.D. | ||||||||||||||||||||||||||||||||||
| Carboxy-terminal MTDs | |||||||||||||||||||||||||||||||||||||
| CTM | S.c. SNC2 | -- | W | W | K | D | L | K | M | R | M | C | L | F | L | V | V | I | I | L | L | V | V | I | I | V | P | I | V | V | H | F | S | PM | |||
| Snc2B | S.c. SNC2 | -- | W | W | K | D | L | K | M | R | M | g | L | F | L | V | V | I | I | L | L | V | V | I | I | V | P | I | V | V | H | F | S | PM | |||
| Snc2D | S.c. SNC2 | -- | L | F | L | V | V | I | I | L | L | V | V | I | I | V | P | I | V | V | H | F | S | PM | |||||||||||||
| Sso1B | S.c. SSO1 | -- | W | L | I | V | F | A | I | I | V | V | V | V | V | V | V | V | V | P | A | V | V | K | T | R | PM | ||||||||||
| Sed5 | S.c. SED5 | -- | N | R | W | L | A | A | K | V | F | F | I | I | F | V | F | F | V | I | W | V | L | V | N | ER/Golgi | |||||||||||
| Sec22 | S.c. SEC22 | -- | S | Q | Y | A | P | I | V | I | V | A | F | F | F | V | F | L | F | W | W | I | F | L | K | ER/Golgi/(PM) | |||||||||||
| Cpr | S.c. RAS2 | -- | A | P | G | G | N | T | S | E | A | S | K | S | G | S | G | G | C | C | I | I | S | PM | |||||||||||||
| Cpr-SS | S.c. ras2–SSIIS | -- | A | P | G | G | N | T | S | E | A | S | K | S | G | S | G | G | s | s | I | I | S | cytoplasm |
When transcriptional induction by Gβγ complexes containing the Ste18ΔC–MTD fusions was assayed in cells that lacked the Gα subunit (gpa1Δ ste18Δ), two of the four MTD fusions, Cpr and CTM, rescued the signaling defect (Fig. 1A, left). When assayed in cells that still express the Gα subunit (GPA1 ste18Δ), the same two carboxy-terminal MTD fusions rescued pheromone-responsive transcriptional induction (Fig. 1A, right), mating ability (Fig. 1B), and growth arrest (Fig. 1C). The amino-terminal MTDs (NTM and Nmyr) were ineffective, either because they did not confer membrane association, or because they interfered with Gγ function, which we did not pursue. The CTM sequence was not as effective as the native Gγ carboxyl terminus or the Cpr sequence, as mating efficiency was lower and halos were turbid in the growth arrest assay (Fig. 1B,C). This may result from conformational constraints on the Gγ carboxyl terminus, as related fusions in which linker sequence was absent and the hydrophobic sequence was placed closer to Ste18 (e.g., the Snc2D MTD; Table 1) was even less effective than the CTM fusion (not shown). Palmitoylation of a cysteine residue within the CTM sequence (Couve et al. 1995) was not required for rescue of Ste18ΔC, as a sequence mutated at this position (Snc2B; Table 1) was equally effective (data not shown). The fact that the carboxy-terminal MTDs could functionally replace the carboxyl terminus of the Gγ subunit argues that the normal requirement for this region is to localize Gβγ to the membrane.
Figure 1.
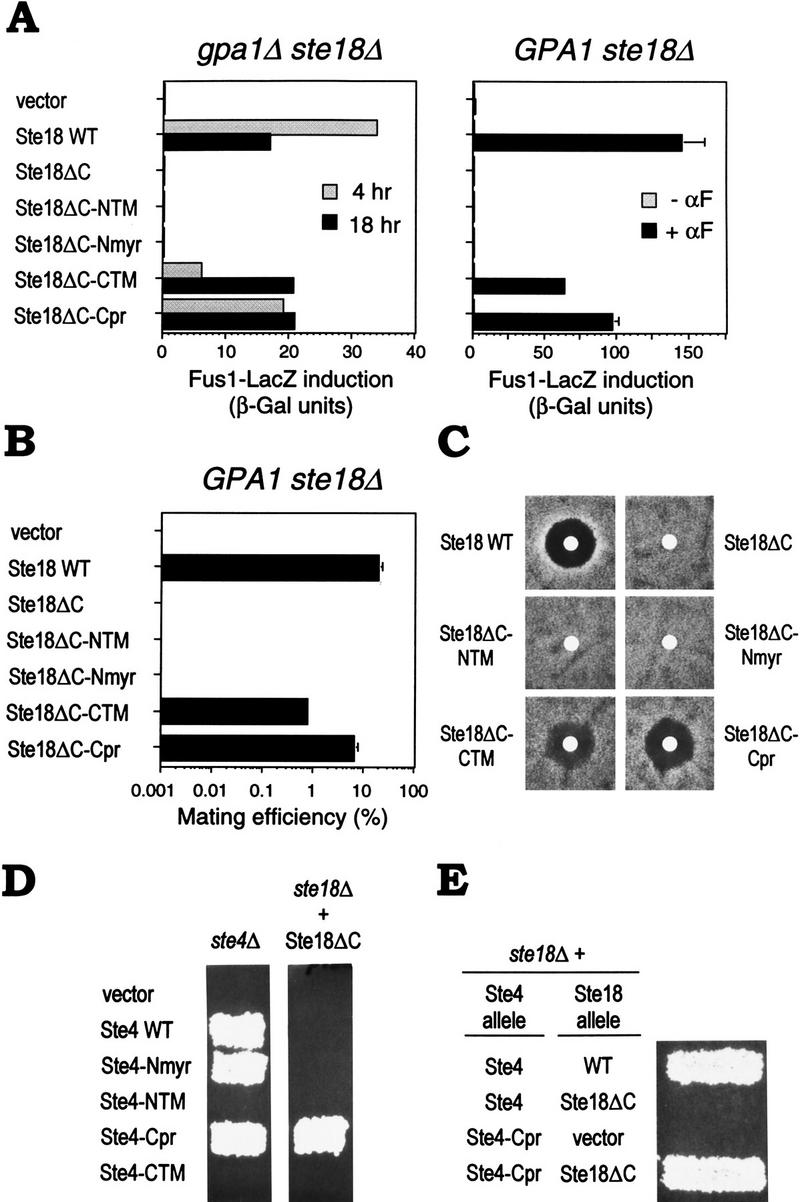
Rescue of Ste18ΔC signaling defect by fusion with heterologous membrane targeting domains. (A) Transcriptional induction. Fus1–LacZ activation is shown, after galactose induction of Ste18 derivatives for 4 or 18 hr (left) or for 4 hr ± α-factor (αF; right). Bars, average of four measurements of two transformants (left) or mean ± s.d. for three transformants (right). Strains: PPY 885, PPY 865. Plasmids, from top to bottom: pPP449, pGS18–WT, pGS18ΔC, pGS18ΔC–NTM, pGS18ΔC–Nmyr, pGS18ΔC–CTM, pGS18ΔC–Cpr. (B) Mating of strains in A, right. Partner: PPY 262. Bars, mean ± s.d. for three transformants. (C) Growth arrest. Lawns of transformants (as in A, right) were exposed for 4 days at 30°C on −TRP + RAFF + GAL to filter disks containing 25 μl of 500 μm α-factor. (D) Rescue by fusion of prenylation/palmitoylation sequence to Ste4. Patch mating tests of Ste4–MTD fusions for complementation of ste4Δ (PPY 794; left), or ability to rescue mating in a ste18Δ strain (PPY 832) that also expressed a Ste18ΔC allele from pBH21–Q98ter (right). Plasmids, from top to bottom: pPP449, pGS4, pGS4–Nmyr, pGS4–NTM, pGS4–Cpr, pGS4–CTM. (E) Signaling by Ste4–Cpr still requires the Ste18 amino terminus. Patch mating of a ste18Δ strain (PPY832) expressing the indicated plasmid-borne Ste4 and Ste18 alleles: pGS4 (Ste4), pGS4–Cpr (Ste4–Cpr), pBH21–WT (WT), pBH21–Q98ter (Ste18ΔC), pRS425 (vector).
We also tested whether the membrane targeting information for Gβγ could be provided by Gβ, rather than Gγ, by fusing the same four MTDs to Ste4 (Gβ). Unfortunately, two of the fusions (NTM and CTM) disrupted Ste4 function (Fig. 1D, left). Of the remaining two MTDs, the same one (Cpr) that rescued Gβγ function when fused directly to Ste18ΔC also rescued the Ste18ΔC defect when fused to Ste4 (Fig. 1D, right). Interestingly, the Ste4–Cpr fusion could not signal in the absence of Ste18, but only when Ste18ΔC was expressed (Fig. 1E), indicating that Gγ is required for aspects of Gβγ function other than membrane localization; this is consistent with point mutations in the Gγ amino terminus that disrupt Gβγ signaling (Grishin et al. 1994). These observations, coupled with those above, demonstrate that activation of the downstream signaling pathway by Gβγ requires it to remain at the membrane after dissociation from Gα.
Targeting of Ste5 to the membrane activates the pheromone response pathway
To explain why signaling by Gβγ might require that it remain at the membrane, we considered the possibility that it recruits its target to the membrane. Therefore, we tested whether membrane recruitment of Ste5 might play a role in activation by artificially targeting it to the membrane. For this, the carboxy-terminal MTDs used in the previous section (CTM and Cpr) were fused to full-length Ste5 as well as to a derivative of Ste5 (Ste5ΔN) lacking an amino-terminal Gβγ-binding domain (Fig. 2A). Because some fusions might cause growth arrest, they were expressed from a galactose-inducible promoter.
Figure 2.
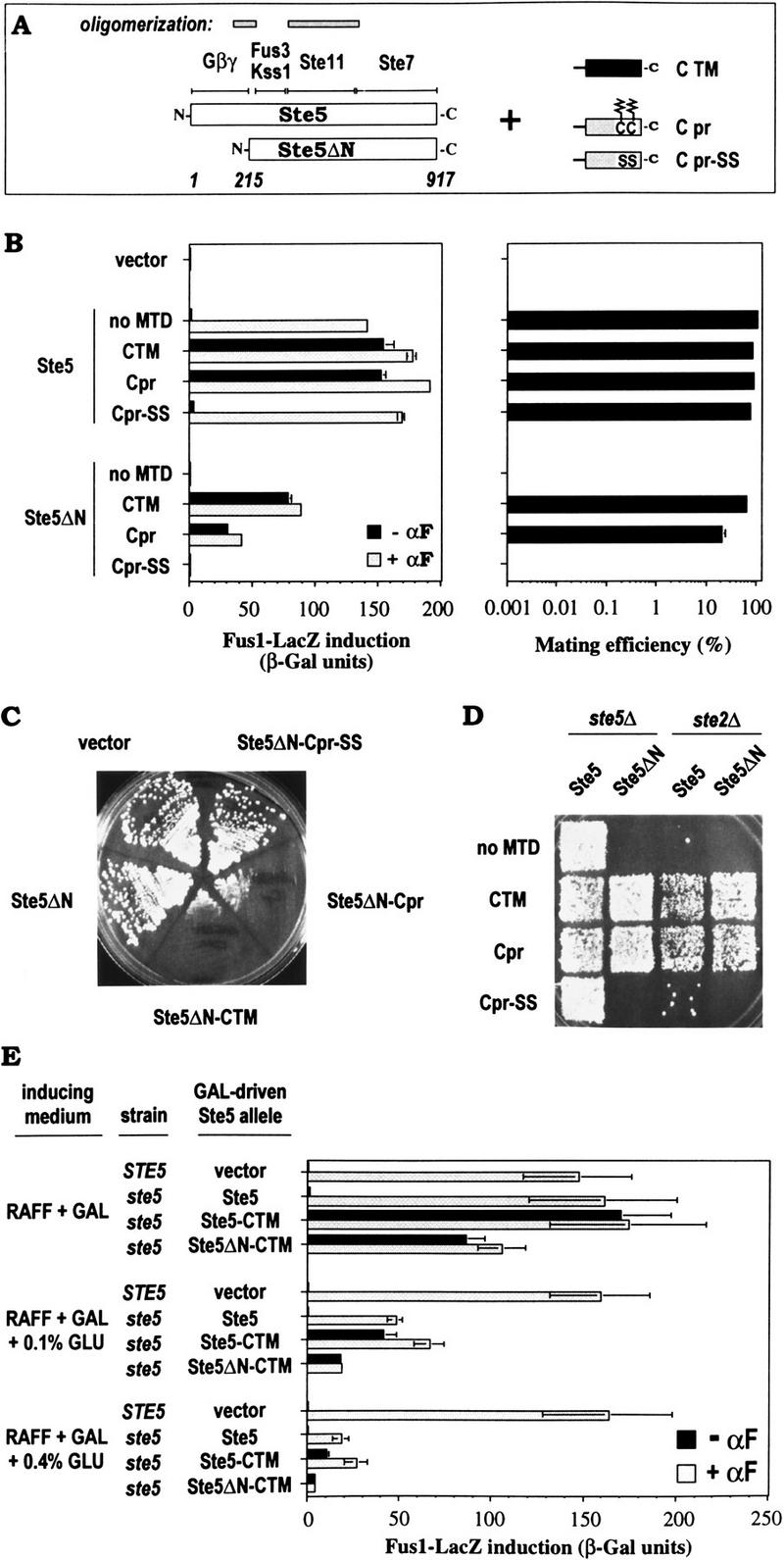
Activation of the pheromone response pathway by membrane targeting of Ste5. (A) Schematic description of Ste5 and Ste5ΔN fusions. (Top) Regions of Ste5 that bind Gβγ (Whiteway et al. 1995) or the kinases Fus3, Kss1, Ste11, and Ste7 (Choi et al. 1994), or facilitate oligomerization (Yablonski et al. 1996; Inouye et al. 1997). (Right) Carboxy-terminal MTDs. Zigzag lines: palmitoylated and farnesylated Cys Cys (CC) residues in the Cpr sequence. (B) Effects of Ste5 and Ste5ΔN MTD fusions on the pheromone response pathway. Fus1–LacZ induction ±α-factor (αF; left), and mating (right; partner: PPY 198). Bars, mean ± range for two transformants. PPY 858 (ste5Δ) harbored plasmids, from top to bottom: pPP449, pGS5, pGS5–CTM, pGS5–Cpr, pGS5–Cpr-SS, pGS5ΔN, pGS5ΔN–CTM, pGS5ΔN–Cpr, pGS5ΔN–Cpr–SS. (C) Growth arrest activated by carboxy-terminal MTD fusions to Ste5ΔN. Transformants (as in B) were streaked on −TRP + RAFF + GAL plates and incubated for 4 days at 30°C. Analogous fusions to full-length Ste5 gave similar results (not shown). (D) Membrane-targeted Ste5 derivatives rescue mating in cells lacking pheromone receptors. Strains: PPY 858, PPY 409. Plasmids, as in B. (E) Membrane-targeted Ste5 derivatives activate the pathway even when expression levels are reduced by glucose. Strains: PPY 640, PPY 858. Plasmids: pRS413, pH–GS5, pH–GS5–CTM, pH–GS5ΔN–CTM. Bars, mean ± s.d. for four transformants.
The MTD fusions to Ste5 and Ste5ΔN activated the pathway in the absence of pheromone, giving levels of Fus1–LacZ induction similar to pheromone-induced cells expressing wild-type Ste5 (Fig. 2B, left). Fusions with Ste5ΔN showed somewhat less induction than those with Ste5, and addition of pheromone did not compensate for this difference. We also created fusions with a sequence (Cpr–SS) in which the Cpr MTD contained Cys → Ser mutations at the two cysteines modified with hydrophobic palmitoyl and farnesyl groups (Mitchell and Deschenes 1995). These point mutations eliminated the effect of the Cpr fusion (Fig. 2B), arguing that it is the membrane association properties conferred by these modifications that lead to activation by Ste5. In mating assays, unfused Ste5ΔN was completely defective, but fusion with the MTDs restored mating to levels similar to wild-type Ste5 (Fig. 2B, right). These fusions also caused growth arrest (Fig. 2C) and shmoo formation (not shown), and allowed mating of cells lacking the pheromone receptor Ste2 (Fig. 2D). All phenotypes were eliminated by point mutations in the Cpr MTD (Cpr–SS; Fig. 2B–D), or by deletion of STE11 or STE7 (not shown). In summary, fusion of MTDs to Ste5 or Ste5ΔN mimics the addition of pheromone, activating responses normally induced by Gβγ, consistent with the requirement for membrane localization of Gβγ documented in the previous section.
To address whether expression levels contributed to these phenotypes, we used glucose to reduce expression of Ste5 derivatives under control of the GAL1 promoter (Fig. 2E). Pheromone-induced Fus1–LacZ levels mediated by the _GAL_-driven Ste5 construct were most similar to genomic-expressed Ste5 when induced by galactose alone, and were reduced to 30% or 11% of those levels by 0.1% or 0.4% glucose, respectively, indicating that expression levels of Ste5 became limiting. Under these conditions, Ste5–CTM and Ste5ΔN–CTM still activated Fus1–LacZ in the absence of pheromone, to at least 50% (Ste5–CTM) or 25% (Ste5ΔN–CTM) of the level mediated by similarly expressed Ste5 in response to pheromone. These data suggest that the activation phenotype does not require overproduction, although the degree of activation may depend on expression levels. Notably, the lowered expression conditions enhanced detection of pheromone responsiveness of Ste5–CTM, hinting at a pheromone-inducible event besides membrane localization that can contribute to Ste5-mediated signaling; this event depends on the amino terminus of Ste5, as Ste5ΔN–CTM was unresponsive to pheromone at all expression levels. We observed similar results by replacing the GAL1 promoter in these constructs with the native STE5 promoter, although the sequence changes remaining at the carboxyl terminus (for inserting MTD cassettes) caused poor expression of Ste5 (P.M. Pryciak and F.A. Huntress, unpubl.).
Pheromone-activated recruitment of GFP–Ste5 to the cell surface
Because artificial targeting of Ste5 to the membrane stimulated the pheromone response pathway, we tested whether Ste5 normally appears at the membrane, by tagging Ste5 with green fluorescent protein (GFP; Fig. 3). In untreated cells, GFP–Ste5 was present diffusely throughout the cytoplasm, and was enriched in the nuclei (confirmed by costaining with DAPI; not shown) of many cells. Pheromone caused some of the GFP–Ste5 to accumulate at the cell surface, at the tips of pheromone-induced projections (Fig. 3A,B), and often led to decreased nuclear localization. Cell surface localization was visible after brief pheromone treatment in cells with only slight projections, as well as in more typical pear-shaped shmoos, but became more difficult to detect at later times (Fig. 3B), for unclear reasons. Even at the earlier times, cell surface GFP–Ste5 was not visible in all cells, and therefore this localization may be transient or dynamic. Nevertheless, these results support the notion that artificially targeting Ste5 to the membrane mimics normal pathway activation.
Figure 3.

Localization of GFP–Ste5 fusions. (A) Cell surface recruitment of GFP–Ste5 in response to pheromone. PPY 858 harboring pGFP–GS5 was examined in the absence or presence of α-factor (αF; 2-hr treatment). Representative fields are shown of both DIC and fluorescence (GFP) images. (B) GFP–Ste5 localization at different times after addition of pheromone. Cells were as in A. (C) GFP–Ste5ΔN cannot translocate to the cell surface. PPY 858 harboring pGFP–GS5ΔN and either vector (pRS413; left) or a Ste5 plasmid (pH–GS5; right) is shown after treatment with α-factor. (D) Plasma membrane localization of membrane-targeted Ste5. A GFP–Ste5–CTM fusion (pGFP–GS5–CTM) was visualized without pheromone treatment in both a ste5Δ strain (PPY 858, left), where signaling was activated, and a ste11Δ strain (PPY 890, right), where signaling was blocked. GFP–Ste5–Cpr (pGFP–GS5–Cpr) gave similar results (not shown).
In contrast to full-length Ste5, a GFP fusion to Ste5ΔN, which lacks the Gβγ-binding domain and is signaling defective, was not recruited to the cell surface in either ste5Δ or STE5 backgrounds (Fig. 3C); thus, cell surface localization of Ste5 correlates with its signaling competence. We also examined the Ste5–CTM fusion, which localized to a peripheral rim around the cell indicative of plasma membrane (Fig. 3D), even in a ste11Δ strain where constitutive signaling was blocked. Interestingly, targeting Ste5 to the membrane made nuclear Ste5 undetectable, and yet the pathway became activated, as evident from the projections formed (Fig. 3D). Although we cannot exclude the possibility that a small fraction did transit to the nucleus, this may indicate that nuclear localization is not required for signaling by Ste5. In contrast, localization of Ste5 to the plasma membrane is strongly correlated with pathway activity.
Recruitment of GFP–Ste5 to the cell surface by pheromone requires Gβγ activity but not kinase cascade activity
To address how pheromone stimulates translocation of GFP–Ste5 to the cell surface, we used mutant strains lacking components of the pathway (Fig. 4A). In ste4Δ mutants, no change in GFP–Ste5 localization was detected with pheromone addition, indicating that G-protein function was required. In contrast, despite the block to downstream signaling, pheromone could still induce cell surface localization of GFP–Ste5 in the absence of the kinases Ste20, Ste11, and Ste7, although more strikingly in ste20Δ than ste11Δ and ste7Δ mutants. Because these mutants did not form projections, cell surface GFP–Ste5 was distributed more broadly than in signaling-competent cells (like the ste5Δ mutant, which was complemented by the GFP–Ste5 fusion), although it was still asymmetric in a manner possibly oriented toward an incipient bud site—where projections emanate in a uniform field of pheromone (Madden and Snyder 1992). Because translocation of GFP–Ste5 was observed in ste20Δ, ste11Δ, and ste7Δ mutants but not in ste4Δ, it requires G-protein function but not the ability of Gβγ to activate the kinases or to stimulate cell cycle arrest or transcription.
Figure 4.
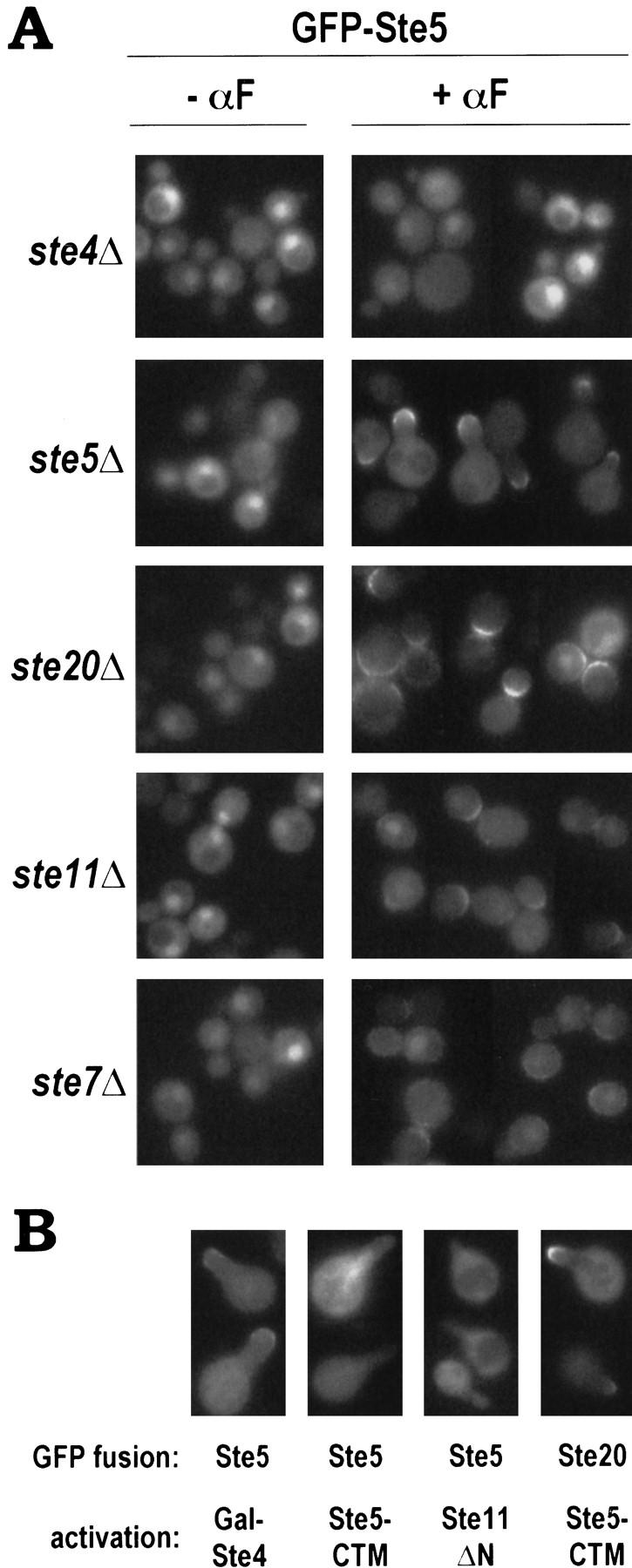
Cell surface recruitment of GFP–Ste5 is a function of the free Gβγ complex that does not require kinase cascade activity. (A) Requirement for heterotrimeric G protein but not kinases. Mutant strains expressed GFP–Ste5 (pH–GFP–GS5). For examination in the presence of α-factor (αF), a galactose-inducible Ste4 construct (pL19) was also included (except in the ste4Δ strain, as it would complement the ste4Δ mutation); although not required (not shown), this enhanced pheromone-induced cell surface localization of GFP–Ste5 in ste20Δ, ste11Δ, and ste7Δ strains. Strains, from top to bottom: PPY 889, PPY 858, PPY 860, PPY 890, PPY 891. (B) Activated Gβγ, but not activated Ste5 or Ste11, can cause translocation of GFP–Ste5 to the cell surface. GFP–Ste5 (pH–GFP–GS5) or GFP–Ste20 (pRL116) fusion proteins were visualized in projection-containing cells induced without pheromone by using galactose-inducible constructs, from left to right: pL19 (Gal–Ste4), pGS5–CTM (Ste5–CTM), pRD–STE11–H3 (Ste11ΔN), pH–GS5–CTM (Ste5–CTM). Cells were analyzed 2–8 hr after galactose induction. Representative examples are shown; GFP–Ste5 appeared at projection tips consistently when induced by Gal–Ste4, but not when induced by Ste5–CTM or Ste11ΔN.
In a complementary approach, we also examined GFP–Ste5 localization after activation at different steps in the pheromone response pathway (Fig. 4B). Like pheromone, activation at the Ste4 step (using Gal–Ste4) caused recruitment of GFP–Ste5 into projection tips in the absence of pheromone. In contrast, activation at the Ste5 and Ste11 steps (using Ste5–CTM and Gal–Ste11ΔN, respectively) did not cause GFP–Ste5 translocation (Fig. 4B). In comparison, another protein, GFP–Ste20, did localize to projection tips induced by Ste5–CTM, and therefore its localization does not require pheromone or activated Gβγ; this is consistent with the fact that GFP–Ste20, unlike GFP–Ste5, is at the cell surface before pheromone exposure (Peter et al. 1996; Leberer et al. 1997b). Our results indicate that kinase cascade activity and projection formation are neither necessary nor sufficient for translocation of GFP–Ste5 to the cell surface, whereas Gβγ activity is both necessary and sufficient. This suggests that GFP–Ste5 is brought to the membrane by Gβγ itself, and defines a molecular activity of the free Gβγ complex that can be monitored independent of kinase cascade activation.
Pathway activation requires delivery of Ste5 to the plasma membrane
To gain insight into why bringing Ste5 to the membrane activates the pheromone response pathway, we tested whether any cellular membrane, or only the plasma membrane, would produce this effect. Because transmembrane domains can target proteins to distinct membrane compartments (Rayner and Pelham 1997), we fused GFP–Ste5ΔN to carboxy-terminal transmembrane domains from four yeast proteins—Snc2, Sso1, Sed5, and Sec22 (Table 1)—that function at different stages of the secretory pathway (Ferro-Novick and Jahn 1994). We then followed both the intracellular localization of the fusions and their signaling phenotypes (Fig. 5).
Figure 5.
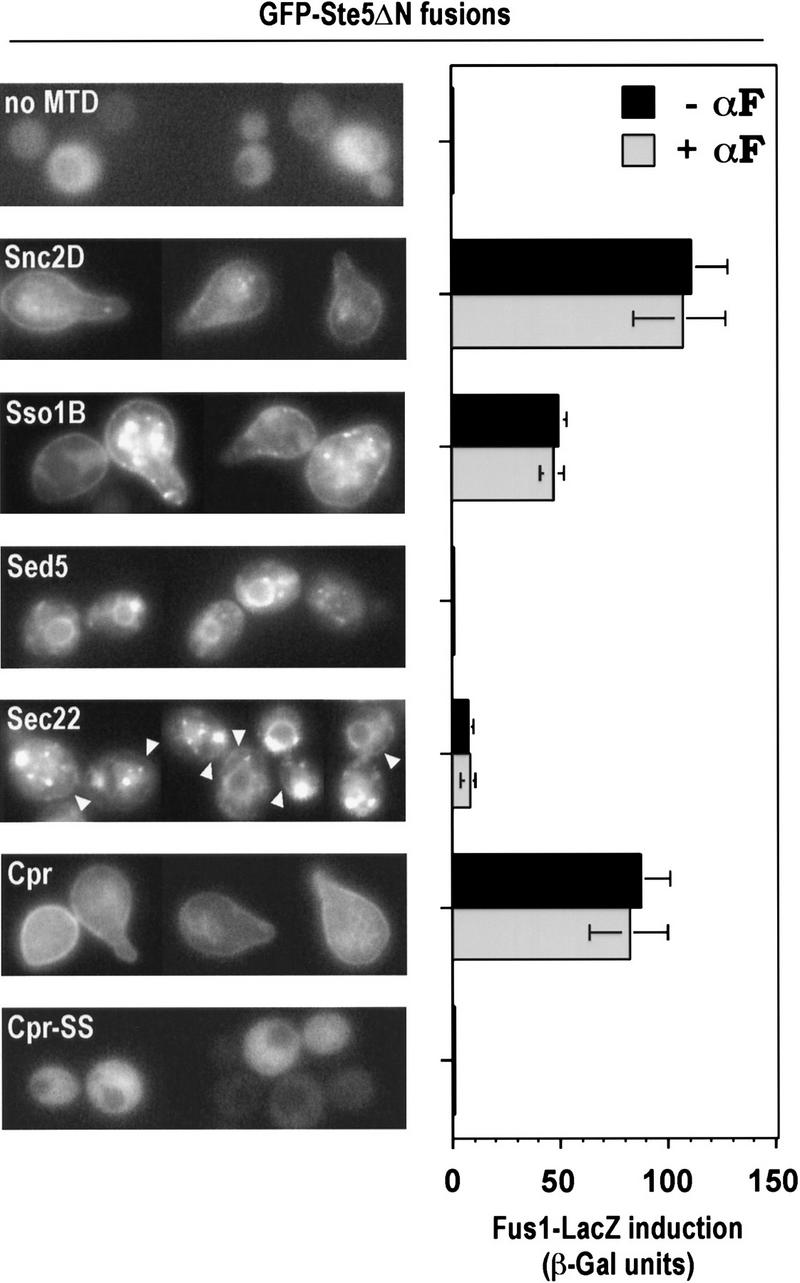
Pathway activation by Ste5ΔN fusions requires delivery to the plasma membrane. Localization (left) and Fus1–LacZ induction (right) for carboxy-terminal MTD fusions to GFP–Ste5ΔN. Snc2D, Sso1B, and Cpr MTD fusions show peripheral rim localization indicative of plasma membrane. Intracellular circular localization observed with Sed5 and Sec22 MTD fusions are reminiscent of endoplasmic reticulum, and punctate spots seen with Sso1B, Sed5, and Sec22 are reminiscent of Golgi localization. With the Sec22 MTD fusion, faint plasma membrane localization was also detectable (arrowheads). All images show cells untreated with pheromone. Strain: PPY 858. Plasmids, from top to bottom: pGFP–GS5ΔN, pGFP–GS5ΔN–Snc2D, pGFP–GS5ΔN–Sso1B, pGFP–GS5ΔN–Sed5, pGFP–GS5ΔN–Sec22, pGFP–GS5ΔN–Cpr, pGFP–GS5ΔN–Cpr–SS. Bars, mean ± s.d. for four transformants.
The Snc2 and Sso1 MTDs directed the fusions primarily to the plasma membrane, with more of the Sso1 fusion retained in a punctate location suggestive of Golgi. The Sed5 and Sec22 MTD fusions gave localizations reminiscent of endoplasmic reticulum and Golgi (see Rayner and Pelham 1997), although a small fraction of the Sec22 fusion was detectable at the plasma membrane (arrowheads). Fus1–LacZ assays showed that pathway activation by these fusions correlated with their delivery to the plasma membrane; signaling was activated strongly by the Snc2 and Sso1 MTD fusions, not at all by the Sed5 MTD fusion, and weakly by the Sec22 MTD fusion. A similar correlation was seen comparing fusions to the Cpr and Cpr–SS sequences. Note that for none of these Ste5ΔN fusions could pheromone increase pathway activity, regardless of the level activated by the fusion itself, and thus removal of the Ste5 amino terminus from the MTD fusion uncouples kinase cascade activity from pheromone input. These experiments show that pathway activation specifically requires delivery of Ste5 to the plasma membrane, and other cellular membranes will not suffice. Furthermore, they argue that the MTD sequences cause activation by their effects on subcellular location, rather than by some other, unintentional consequence, such as oligomerization (see Inouye et al. 1997; Feng et al. 1998).
Signaling activity of membrane-targeted Ste5 requires Ste20, Cdc42, and Cdc24
One explanation for why targeting Ste5 to the plasma membrane activates the pathway is that it may bring the Ste5-associated kinases into proximity of a membrane-associated activator. Therefore, we examined the dependence of this effect on Gβγ and Ste20, both of which localize at or near the plasma membrane (Hirschman et al. 1996; Peter et al. 1996; Leberer et al. 1997b) and are predicted to function upstream of Ste5 (Leberer et al. 1992; Hasson et al. 1994). Remarkably, Fus1–LacZ induction by Ste5–CTM and Ste5ΔN–CTM was independent of the Gβ subunit Ste4 (and the Gγ subunit Ste18; not shown), but was completely dependent on Ste20 (Fig. 6A). In comparison, activation of the pathway further downstream using an activated Ste11 allele (Ste11ΔN) or overproduced Ste12 (2 μm Gal–Ste12) was insensitive to the presence of both Ste4 and Ste20. Activation of growth arrest by Ste5–CTM and Ste5ΔN–CTM was also independent of Ste4 yet dependent on Ste20 (Fig. 6B), as were activation of mating and projection formation (not shown). Because these phenotypes required Ste20 but not pheromone or Gβγ, Ste20 can participate in this pathway without its kinase activity being activated by Gβγ. We saw little evidence that this role of Ste20 is substantially potentiated by Gβγ or pheromone, as pheromone stimulated minor (Ste5 fusions) or no (Ste5ΔN fusions) increase in signaling when Ste5 was targeted to the membrane (Fig. 6A; see also Figs. 2B,E, and 5).
Figure 6.


Critical role of Ste20, Cdc42, and Cdc24 in pathway activation by membrane-targeted Ste5 derivatives. (A) Analysis of requirement for Ste4 and Ste20. Fus1–LacZ induction in mutant strains expressing galactose-inducible Ste5, Ste11, or Ste12 derivatives. Strains: PPY 858, PPY 886, PPY 860. Plasmids, from top to bottom: pH–GS5, pH–GS5–CTM, pH–GS5ΔN–CTM, pRD–STE11–H3, pNC252. Bars, mean ± s.d. for four transformants. (B) Growth arrest. Streaked transformants (as in A) grown for 5 days at 30°C on a −HIS + RAFF + GAL plate are shown. (C) Dependence on Cdc24 and Cdc42. Fus1–LacZ induction in wild-type vs. cdc mutant strains was compared after pathway activation by pheromone-dependent or -independent methods. (Left) ste5Δ CDC24 (PPY 655) or ste5Δ cdc24-1 (data combined for PPY 697 and PPY 698) strains harbored, from top to bottom: pL–GS5, pL–GS5–CTM, pL–GS5ΔN–CTM, pRD–STE11–H3, pNC252. Bars, mean ± s.d. for four transformants. (Right): A bar1 cdc42-1 strain (PPY 911) harbored either pRS314–CDC42–WT (CDC42) or pRS314 (cdc42-1) plus, from top to bottom: pRS315, pL–GS5–CTM, pL–GS5ΔN–CTM, pNC252. Results using the Ste11ΔN allele in this strain are absent because they gave highly variable results, for unknown reasons, and therefore were inconclusive. Bars, mean ± s.d. for four transformants.
Because Cdc24 and Cdc42 are also required for activation of the kinase cascade by Gβγ (Simon et al. 1995; Zhao et al. 1995), and Cdc42 is membrane-associated (Ziman et al. 1993), we asked whether they were required for activation by membrane-targeted Ste5 derivatives. Their roles were tested using temperature-sensitive mutant alleles, and signaling was analyzed after shift to the restrictive temperature. As seen for response to pheromone, Cdc24 and Cdc42 were required for pathway activation by Ste5–CTM or Ste5ΔN–CTM that occurred in the absence of pheromone (Fig. 6C). In contrast, their inactivation did not interfere with Fus1–LacZ induction by Ste11ΔN or 2 μm Gal–Ste12, ensuring that the mutant cells were competent to induce transcription, and suggesting that Cdc24 and Cdc42 are required for events upstream of Ste11. Therefore, similar to our observations with Ste20, participation of Cdc24 and Cdc42 in the pheromone response pathway does not require that they have their activities increased in magnitude by pheromone or Gβγ.
To further address the requirement for Ste20 at the cell surface and the role of Cdc42, we examined the mutant Ste20Δ334–396, which lacks its Cdc42-binding site and is depleted from the cell surface (Peter et al. 1996; Leberer et al. 1997b). This mutant showed a partial defect in supporting activation by membrane-targeted Ste5 derivatives, as Fus1–LacZ levels were 13–14% (Ste5ΔN–CTM) or 31–38% (Ste5–CTM) of that promoted by Ste20WT (Table 2); this could be alleviated somewhat by addition of pheromone, but only with Ste5–CTM and not Ste5ΔN–CTM. When we assayed the Ste20Δ334–396 mutant for response to pheromone in a strain (ste20Δ) that expressed native Ste5, we observed a stronger defect (55% of wild-type Fus1–LacZ induction) than reported previously, which ranged from 80% (Peter et al. 1996) to 100% of wild type (Leberer et al. 1997b). It is unclear why we uncovered a stronger defect, but our observations were highly reproducible (see Table 2 footnotes). Our findings suggest a detectable, but subtle, role for the Cdc42-binding domain of Ste20. Because this domain is required for cell surface localization of Ste20, these results are consistent with the idea that the functional effect of targeting Ste5 to the membrane is to increase its proximity to Ste20.
Table 2.
Mutant Ste20 derivatives show defects in pathway activation by membrane-targeted Ste5 derivatives
| STE20 allelea | Strain and induction methodb | |||||||
|---|---|---|---|---|---|---|---|---|
| ste20Δ | ste5Δ ste20Δ + Ste5–CTM | ste5Δ ste20Δ + Ste5ΔN–CTM | ste4Δ ste5Δ ste20Δ (no pheromone) | |||||
| (−αF) | (+αF) | (−αF) | (+αF) | (−αF) | (+αF) | (Ste5–CTM) | (Ste5ΔN–CTM) | |
| Vector | 0.05 | 0.20 | 0.15 | 0.32 | 0.09 | 0.14 | 0.29 | 0.11 |
| wild type | 0.14 | 125.6 | 154.1 | 167.2 | 87.5 | 85.4 | 141.0 | 102.7 |
| Δ334–396c | 0.09 | 69.1 | 58.1 | 115.3 | 11.8 | 14.5 | 43.7 | 14.5 |
| S879A/S880A/P883A | 0.04 | 6.98 | 5.21 | 19.1 | 0.65 | 0.64 | 4.15 | 0.64 |
Finally, we also analyzed another Ste20 mutant, Ste20S879A/S880A/P883A, which contains mutations in a recently identified Gβγ-binding domain (Leeuw et al. 1998). This mutant is defective at response to pheromone, suggesting a critical role for a Gβγ–Ste20 interaction (Leeuw et al. 1998). In contrast, our observations using membrane-targeted Ste5 derivatives show that Ste20 can participate in this pathway in the absence of Gβγ. Therefore, we tested the Ste20S879A/S880A/P883A mutant for its ability to support pathway activation by Ste5–CTM and Ste5ΔN–CTM, and found that it shows a strong defect in this ability (Table 2). Results were similar in cells (ste4Δ ste5Δ ste20Δ) that lack the Gβ subunit Ste4, emphasizing that the defect is distinct from effects on binding Gβγ. Therefore, the Ste20S879A/S880A/P883A mutant is impaired for a signaling function that is independent of pheromone and interaction with Gβγ, and its pheromone-response defect cannot be attributed to disruption of Gβγ–Ste20 binding.
Discussion
Our observations establish five points pertaining to the mechanism by which the yeast Gβγ complex activates the signaling pathway downstream of it. First, Gβγ must remain at the membrane after dissociation from Gα to activate the pathway, as deletion of the CaaX motif in Gγ results in a signaling defect that can be rescued by fusing heterologous membrane targeting sequences to either Gβ or Gγ. Second, signaling by Gβγ can be mimicked by artificial placement of Ste5 at the plasma membrane, suggesting the functional role of Gβγ membrane localization may be to recruit Ste5. Third, a portion of the cellular population of Ste5 is recruited to the cell surface on exposure to pheromone, and this recruitment is most likely mediated by Gβγ itself. Fourth, the effect of Ste5 membrane localization is critically dependent on the cell surface-associated kinase Ste20, suggesting that membrane localization of Ste5 causes activation of its associated kinases by increasing their proximity to Ste20. Fifth, the participation of Ste20, Cdc24, and Cdc42 in the pheromone response pathway does not require their previous activation by pheromone or Gβγ, raising the possibility that Gβγ does not activate them per se but instead promotes their action on new substrates (Fig. 7A). A model that incorporates these points is presented in Figure 7B. In addition, our findings provide two ways to separate the contributions of different proteins to pathway activity: Cell surface recruitment of GFP–Ste5 provides an assay for Gβγ function independent of Ste20 and other kinases, and activation by membrane-targeted Ste5 provides an assay for Ste20 function independent of Gβγ activity.
Figure 7.


Model for pheromone response pathway activation by Gβγ. (A) General models. Cdc24, Cdc42, and Ste20 are required for Gβγ to activate the downstream kinase cascade (Ste11, Ste7, and Fus3, shown associated with the scaffold protein Ste5). A priori, Gβγ could either activate these proteins (left) or promote the action of already active proteins on the kinase cascade (right). Our observations favor the scheme on the right. Whether Cdc24/Cdc42 act by way of Ste20 is still controversial (see text). (B) Detailed model for molecular activity of Gβγ. Asterisks indicate active proteins, and Cdc24, Cdc42, and Ste20 are suggested to be active before exposure to pheromone. The model proposes that on exposure of cells to pheromone, liberated Gβγ recruits Ste5 and its associated kinases to the membrane, and thus into proximity of active Cdc24, Cdc42, and Ste20, resulting in activation of the kinase cascade by Ste20. The model also incorporates the recently described interaction of Gβγ with Ste20 (Leeuw et al. 1998), which is shown as contributing to the formation of a Ste5–Gβγ–Ste20 complex. Possible subsequent Ste5 fates are indicated below. For simplicity, we show all three kinases—Ste11, Ste7, and Fus3—accompanying Ste5 to the cell surface, but it is possible that only a subset do so.
A role for Gβγ membrane localization: recruitment of Ste5
Our findings offer a simple explanation for why Gβγ must remain at the membrane to activate the kinase cascade: Because recruitment of Ste5 to the membrane is a critical step in activation by Gβγ. Importantly, our ability to detect this recruitment in the absence of Ste20, Ste11, or Ste7 represents an assay for an immediate function of the free Gβγ complex, which does not require activity through the kinase cascade or measurable mating, cell-cycle arrest, or transcriptional induction. Although recent studies found that Ste4 (Gβ) and Ste5 coimmunoprecipitated independent of pheromone (Leeuw et al. 1998), other studies found that coimmunoprecipitation of these proteins could be stimulated by pheromone (Feng et al. 1998). Our observations support a model in which liberation of Gβγ from Gα allows Ste5 to bind Gβγ, and that this brings Ste5 to the cell surface.
There is precedent for membrane recruitment in other signal transduction pathways, including activation of Ras exchange factors and the Raf kinase (for review, see Carraway and Carraway 1995). In addition, Gβγ complexes in other systems can cause membrane recruitment of target proteins, including the β-adrenergic receptor kinase in mammalian cells (Pitcher et al. 1992) and the cytosolic regulator of adenylyl cyclase (CRAC) in Dictyostelium (Lilly and Devreotes 1995). Interestingly, in this latter example, recruitment of CRAC by Gβγ is transient. In our experiments, the fate of cell surface Ste5 is unknown, but it became difficult to detect after extended pheromone treatment, therefore it may also be transient—perhaps released on reassembly of Gαβγ or activation of the kinases—or affected by desensitization mechanisms. The small fraction of total Ste5 that accumulates there at any one time may hint at rapid exchange between different locations. The fates of the kinases presumably associated with Ste5 are also unknown (see Fig. 7B).
Kinase cascade activation
We envision membrane recruitment of Ste5 as an initial step in kinase cascade activation. The likely immediate result is phosphorylation of Ste11 by Ste20, followed by phosphorylation of Ste7, then Fus3 (for review, see Herskowitz 1995; Leberer et al. 1997a; see Fig. 7B); note, however, that we do not yet know whether the kinases Ste11, Ste7, and Fus3 accompany Ste5 to the membrane. Because some responsiveness to pheromone was retained by MTD fusions to Ste5 but not Ste5ΔN, binding of Ste5 to Gβγ may trigger additional effects on Ste5, such as a conformational change or increased association with Ste20 (see model, Fig. 7B). Because activation by Ste5ΔN–CTM was weaker than by Ste5–CTM even in the absence of Ste4, there may be roles for the Ste5 amino terminus that are not mediated by Gβγ. Other steps in activation may involve oligomerization of Ste5 (Yablonski et al. 1996; Inouye et al. 1997). Indeed, fusion of Ste5 with a domain thought to self-dimerize activated the pathway (Inouye et al. 1997), although as available evidence indicates that oligomerization of Ste5 is independent of pheromone (Yablonski et al. 1996; Feng et al. 1998), it may normally facilitate signaling without being itself induced by pheromone.
It is tempting to speculate that Ste5 carries signal from the cell surface to the nucleus, where transcription is induced, as it can be observed at both locations. However, nuclear Ste5 was observed in nonsignaling cells (Fig. 4), and Ste5 targeted to the plasma membrane was depleted from the nucleus and yet remained competent to signal, suggesting that nuclear localization of Ste5 may not be required for signaling. Instead of promoting signaling, nuclear localization of Ste5 might serve to discourage promiscuous signaling, or it may be a passive consequence of its association with kinases that bind to nuclear proteins (e.g., Tedford et al. 1997), and thus, Ste5 may accumulate there in the absence of a competing binding site—such as Gβγ at the plasma membrane. Alternative candidates for carrying signal from the cell surface to the nucleus include the Ste5-associated kinases.
Regulation of Ste20, Cdc24, Cdc42 by pheromone
We found that membrane targeting of either Ste5 or Ste5ΔN stimulates the pathway in a manner that does not require either pheromone or Gβγ activity, but still requires Ste20, Cdc42, and Cdc24. Formally, this could indicate that Ste20, Cdc42, and Cdc24 act downstream of Ste5 (but upstream of Ste11; see Fig. 6), and thus are activated by the membrane-targeted Ste5 derivative itself. We prefer the alternative explanation that they participate in a manner not requiring their activation per se (Fig. 7A, right). Moreover, we saw no clear indication that their activities are increased by pheromone, as signaling by Ste5ΔN–CTM absolutely required their function and yet could not be increased by pheromone, even when decreased expression levels were used to give submaximal signaling. Therefore, their behavior suggests that they are active before pheromone addition, but do not stimulate the pathway until they gain access to the Ste5-associated kinases, which we suggest is normally promoted by Gβγ (Fig. 7B). Note that there is no evidence that pheromone stimulates kinase activity of Ste20 (Wu et al. 1995), GTP loading of Cdc42, or exchange factor activity of Cdc24. Also, although Gβ can bind Cdc24, this appears to be dispensible for kinase cascade activation (Nern and Arkowitz 1998). Cdc42 is a membrane protein (Ziman et al. 1993) and Ste20 is enriched at the cell surface (Peter et al. 1996; Leberer et al. 1997b), supporting the notion that they await Ste5 at the plasma membrane. Although previous studies found that Ste5 copurified with Ste20 in extracts of cells that had not been exposed to pheromone (Leeuw et al. 1995), microscopic analysis suggests that localization similar to Ste20 (Peter et al. 1996; Leberer et al. 1997b) is apparent for Ste5 only after pheromone treatment (Figs. 3 and 4).
Because Cdc24, Cdc42, and Ste20 participate in functions other than mating, including bud formation and cytokinesis (Adams et al. 1990; Cvrckova et al. 1995), perhaps it is not surprising that they do not need to be activated by Gβγ. Instead of activating them, Gβγ may cause them to act on new substrates. This may clarify several related issues: (1) Activation of Gβγ has no obvious consequence in cells lacking a functional kinase cascade, which might otherwise be deleterious as hyperactivity of Cdc24, Cdc42, and Ste20 causes growth and morphological defects (Ziman et al. 1991; Leberer et al. 1997b); (2) mutations thought to activate Cdc42 (Simon et al. 1995; Akada et al. 1996) or Ste20 (Leberer et al. 1997b) only weakly activate the pheromone response pathway, which may be explained if proximity of Cdc42 and Ste20 to the Ste5-associated kinases is more limiting than their activity levels; (3) pheromone induces only mating genes and not filamentation genes that are induced when Ste20, Ste11, and Ste7 are activated by other methods (Madhani and Fink 1998), perhaps because Gβγ does not activate Ste20 but merely directs it to act on those kinase molecules that are associated with Ste5.
Recently it was found that Gβγ can bind Ste20, and that defects in this binding correlate with impaired pheromone response (Leeuw et al. 1998). In our experiments with membrane-targeted Ste5, however, Ste20 can function in the absence of Gβγ, and the Gβγ binding-deficient mutant Ste20S879A/S880A/P883A cannot support pathway activation even by Gβγ-independent means. Nevertheless, a role for a Gβγ–Ste20 interaction remains supported by their pheromone-inducible coimmunoprecipitation as well as by signaling-defective Ste4 mutants that disrupt binding to Ste20 (Leeuw et al. 1998). Therefore, to unite these various observations, we suggest that Gβγ normally performs two functions: It recruits Ste5 to the membrane and brings Ste20 close to the recruited Ste5 (see Fig. 7B), thereby nucleating a complex that promotes phosphorylation of Ste11 by Ste20. A requirement for Gβγ–Ste20 binding may be bypassed in our experiments either because the lifetime of Ste5 at the cell surface is sufficiently increased that Ste20 can find Ste5 without help from Gβγ, or because expression levels of our membrane-targeted Ste5 derivatives are sufficiently high to present all cell surface Ste20 molecules with a nearby Ste5 molecule. Consistent with this latter suggestion, when membrane-targeted Ste5 was expressed at limiting levels (Fig. 2E), pathway activation became submaximal and responsive to further input by pheromone. In either scenario, the role of the Gβγ–Ste20 interaction would not be to stimulate the kinase activity of Ste20, but to nucleate a complex including both Ste20 and Ste5. This view is suggested by our observation that Ste20 can support pathway activation by membrane-targeted Ste5 in the absence of Gβγ.
The precise role for Cdc24 and Cdc42 in pheromone response remains unclear. Some evidence suggests that they are required to generate active Ste20, as the requirement for Cdc24 can be alleviated by activated Ste20 (Zhao et al. 1995) and Ste20 kinase activity can be stimulated by GTP-loaded Cdc42 (Simon et al. 1995). Other evidence argues that the role of Ste20 is independent of Cdc42, as Ste20 mutants lacking a Cdc42-binding site still transmit pheromone response (Peter et al. 1996; Leberer et al. 1997a). Our results support the role of Cdc24 and Cdc42 being related to Ste20 function, as they are required for pathway activation by means that require Ste20 (i.e., pheromone or membrane-targeted Ste5) and not by those that do not (i.e., Ste11ΔN or Ste12 overproduction). In addition, we uncovered noticeable defects in the Ste20 mutant lacking its Cdc42-binding site. It is possible that Cdc24 and Cdc42 are required less for the kinase activity of Ste20 than for its cell surface localization, and that this role is obscured in some experiments because of the ability of Ste20 to associate with other cell surface molecules such as Gβγ, Ste5, actin, and Bem1 (Leeuw et al. 1995, 1998).
Relevance to chemotropism and polarity control
Finally, our observations are also relevant to chemotropic cell orientation during mating (e.g., Schrick et al. 1997; Nern and Arkowitz 1998), as Cdc24 and Cdc42 are involved in cytoskeletal and cell polarity control, and as membrane recruitment of proteins by Gβγ could potentially mark sites on the cell surface where extracellular signal is received. Our results suggest that Cdc24 and Cdc42 may not be activated per se by Gβγ, as previously suspected. Because there is evidence that Gβγ may control chemotropism in a manner requiring neither Ste5 nor Ste20 (Schrick et al. 1997; P.M. Pryciak, unpubl.), there may be other recruitment targets of Gβγ, which may include or function with Cdc24 and Cdc42. Whether Gβγ alters Cdc24 and Cdc42 function, and whether this involves recruitment of other proteins into their vicinity, remains a topic for future studies.
Materials and methods
Yeast strains and media
Synthetic complete medium (SC), lacking nutrients as appropriate to maintain selection for plasmids, was used (Sherman 1991). Carbon sources included GLU (2% glucose), RAFF (2% raffinose), and RAFF + GAL (2% raffinose, 2% galactose). Yeast strains (Table 3) were constructed using standard genetic techniques.
Table 3.
Yeast strains used in this study
| Strain | Genotype |
|---|---|
| PPY 409a | MATa ade2 his3 leu2 trp1 ura3 can1 ste2Δ::URA3 |
| PPY 496a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste20-1::TRP1 |
| PPY 640a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 |
| PPY 794a | MATa ade2 his3 leu2 trp1 ura3 can1 ste4Δ::ura3FOA |
| PPY 832a | MATa ade2 his3 leu2 trp1 ura3 can1 ste18::URA3 |
| PPY 858a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste5::ADE2 |
| PPY 860a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste5::ADE2 ste20-1::TRP1 |
| PPY 865a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste18::URA3 |
| PPY 866a | MATα ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste4Δ::ura3FOA ste5::ADE2 ste20-1::TRP1 |
| PPY 886a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste5::ADE2 ste4Δ::ura3FOA |
| PPY 889a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste4::ADE2 |
| PPY 890a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste11::ADE2 |
| PPY 891a | MATa ade2 his3 leu2 trp1 ura3 can1 FUS1::FUS1–lacZ::LEU2 ste7::ADE2 |
| PPY 262b | MATα cry1 leu2 lys2 trp1 ura3 SUP4-3 FUS1::FUS1–lacZ::URA3 |
| PPY 655b | MATa cry1 ade2 ade3 his4 leu2 lys2 trp1 ura3 SUP4-3 FUS1::FUS1–lacZ::LYS2 ste5Δ1::LYS2 |
| PPY 697b | MATa cry1 ade2 ade3 his4 leu2 lys2 trp1 ura3 SUP4-3 FUS1::FUS1–lacZ::LYS2 ste5Δ1::LYS2 cdc24-1 |
| PPY 698b | MATa cry1 ade2 ade3 his4 leu2 lys2 trp1 ura3 SUP4-3 FUS1::FUS1–lacZ::LYS2 ste5Δ1::LYS2 cdc24-1 |
| PPY 885b | MATa cry1 ade2 his4 leu2 lys2 trp1 ura3 SUP4-3 FUS1::FUS1–lacZ::LYS2 ste18::LEU2 gpa1Δ::URA3 |
| PPY 198c | MATα his7 lys9 trp1 ura3 can1 cyh2 |
| PPY 911d | MATa bar1 ade1 his2 leu2 trp1 ura3 cdc42-1 FUS1::FUS1–lacZ::HIS2 |
| PT2αe | MATα hom3 ilv1 can1 |
Plasmids
Plasmids described previously include the following: pRD–STE11–H3 (CEN URA3 GAL1p–GST–STE11ΔN) (Neiman and Herskowitz 1994); pL19 (CEN URA3 GALp–STE4) (Whiteway et al. 1990); pBTL150 (CEN URA3 STE20S879A/S880A/P883A) (Leeuw et al. 1998); pNC252 (2 μm URA3 GALp–STE12) (Pryciak and Hartwell 1996); pBH21–WT (2 μm LEU2 ADHp–STE18) and pBH21–Q98ter (2 μm LEU2 ADHp–ste18–Q98ter) (Grishin et al. 1994); pRL116 (CEN URA3 GFP–STE20) and pBTL56 (CEN URA3 GFP–STE20Δ334–396) (Leberer et al. 1997b); pRS314, pRS315, pRS316, and pRS413 (Sikorski and Hieter 1989); pRD53 is a CEN URA3 vector with a _GAL1/10 Eco_RI–_Bam_HI fragment between _Spe_I and _Bam_HI of pRS316 (R. Deshaies, pers. comm.). pRS(−p)SSIIS (CEN LEU2 ras2–SSIIS) was a gift from R. Deschenes. pRS314–CDC42–WT (CEN TRP1 CDC42) was a gift from D. Lew.
Plasmids created for this study are described below. PCR amplifications used polymerase Pfu (Stratagene). pPP449 (CEN TRP1 GAL1p) and pPP450 (CEN LEU2 GAL1p) contain the _Sac_I–_Xho_I fragment from pRD53 in pRS314 and pRS315, respectively. STE4, STE18–WT, ste18ΔC (STE18 lacking its last 5 codons), STE5, and ste5ΔN (STE5 lacking its first 214 codons) were amplified by PCR; the primers incorporated two unique restriction sites at each end (_Bam_HI, _Nco_I upstream; _Mlu_I, _Pst_I downstream or _Mlu_I, _Eco_RI for STE4) so that MTD or GFP fragments could be introduced. Downstream primers eliminated native stop codons (except for STE18–WT) and introduced a new stop codon between the two restriction sites, therefore, inserting carboxy-terminal MTDs at the _Mlu_I site extended the open reading frame into the MTD. Digested PCR products were ligated into pPP449 as a _Bam_HI–_Eco_RI fragment to create pGS4 (CEN TRP1 GAL1p–STE4), or into pPP450 or pPP449 as _Bam_HI–_Pst_I fragments to create the CEN TRP1 plasmids pGS18–WT (GAL1p–STE18–WT), pGS18ΔC (GAL1p–ste18ΔC), pGS5 (GAL1p–STE5), and pGS5ΔN (GAL1p–ste5ΔN), or the CEN LEU2 plasmids pL–GS5 (GAL1p–STE5) and pL–GS5ΔN (GAL1p–ste5ΔN).
MTD sequences (Nmyr, NTM, CTM, Cpr, Cpr-SS, Snc2B, Snc2D, Sso1B, Sed5, Sec22) were ampified by PCR. Amino-terminal MTDs were inserted as _Bam_HI–_Nco_I fragments, and carboxy-terminal MTDs as _Mlu_I–_Apa_I fragments, using primer-introduced sites. In this way, pGS4, pGS18ΔC, pGS5, pGS5ΔN, pL–GS5, and pL–GS5ΔN served as recipients to create the following: pGS4–NTM, –Nmyr, –CTM, and –Cpr; pGS18ΔC–NTM, –Nmyr, –CTM, –Snc2B, –Snc2D, and –Cpr; pGS5–CTM, –Cpr, and –Cpr-SS; pGS5ΔN–CTM, –Cpr, and –Cpr-SS; pL–GS5–CTM; and pL–GS5ΔN–CTM. The _Sac_I–_Apa_I fragments from pGS5, pGS5–CTM, pGS5ΔN, and pGS5ΔN–CTM were transferred into pRS413 to create the HIS3-marked derivatives pH–GS5, pH–GS5–CTM, pH–GS5ΔN, and pH–GS5ΔN–CTM, respectively. GFP with signal-enhancing mutations S65A, V68L, S72A was amplified from pGFP-mut#2 (Cormack et al. 1996) and inserted as a _Bam_HI fragment into pGS5, pGS5–CTM, pGS5ΔN, pGS5ΔN–CTM, and pH–GS5 to create pGFP–GS5, pGFP–GS5–CTM, pGFP–GS5ΔN, pGFP–GS5ΔN–CTM, and pH–GFP–GS5, respectively. pGFP–GS5ΔN served as recipient of carboxy-terminal MTD _Mlu_I–_Apa_I fragments to create pGFP–GS5ΔN–Snc2D, –Sso1B, –Sed5, –Sec22, –Cpr, and –Cpr-SS.
β-Galactosidase assays
Fus1–LacZ assays were performed on 1-ml culture and units calculated as described (Pryciak and Hartwell 1996). Fresh RAFF cultures, inoculated from RAFF stocks, were grown overnight at 30°C to an OD660 of 0.3–0.5, then GAL (2%) was added ±10 μm α-factor (determined by dose-response assays to be saturating for W303 and 381G strains under these conditions; not shown). Unless indicated otherwise, β-galactosidase activity was then measured after 4 hr of incubation at 30°C. For assays in Figure 2E, cells were grown in RAFF, then treated with GAL either alone or with 0.1% or 0.4% GLU ± 10 μm α-factor. For cdc24-1 strains, transformants were grown at 28°C in RAFF, preincubated for 90 min at 37.5°C, then induced for 5 hr at 37.5°C with GAL ± 10 μm α-factor. For bar1 cdc42-1 strains, cotransformants were grown at 28°C in RAFF containing 0.1% GLU (growth was poor in RAFF), preincubated for 2 hr at 38.5°C, then induced for 4 hr with GAL ± 0.1 μm α-factor.
Mating assays
For quantitative mating (Schrick et al. 1997), overnight RAFF cultures were used, 1 × 107 a transformants were mixed with 2 × 107 α partner cells, collected onto sterile filters, and the filters placed on SC + RAFF + GAL plates for 18 hr at 30°C. Dilutions of harvested cells were plated on media selective for either diploids or total plasmid-containing cells. Mating efficiency was the percentage of total plasmid-containing (a + a/α) cells that were diploids. For patch mating, a transformants were patched onto lawns of PT2α on SC + RAFF + GAL plates, incubated at 30°C overnight, and replicated to plates selective for diploids.
Microscopy
Fresh colonies (2- to 3-day-old) from selective glucose plates were suspended in selective RAFF + GAL liquid and incubated at 30°C; when included, α-factor (10 μm) was added 2 hr later. Live cells were generally examined after 4 hr in RAFF + GAL by spotting onto poly-lysine coated slides, using a Nikon E600 epifluorescence microscope with a 100× Plan Fluor oil immersion objective. Images were collected using a cooled, black and white, CCD camera (DAGE-MTI).
Acknowledgments
We thank R. Deschenes, T. Leeuw, E. Leberer, and D. Lew for plasmids and strains, as well as C. Boone, D. McCollum, and A. Neiman for comments on the manuscript. This work was supported by grants to P.M.P. from the Worcester Foundation, The Millipore Foundation, and the National Institutes of Health (GM57769). P.M.P. dedicates this study to the memory of Maria Pryciak.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
A recent study (L.J.W.M. Oehlen and F.R. Cross. 1998. The role of Cdc42 in signal transduction and mating of the budding yeast Saccharomyces cerevisiae. J. Biol. Chem. 273: 8556–8559) suggests that decreased pheromone-induced transcription in cdc24 and cdc42 mutant cells can be attributed largely to inhibitory Cln1/2-Cdc28 kinase activity that accumulates during cell cycle arrest.
Footnotes
E-MAIL peter.pryciak@ummed.edu; FAX (508) 856-8774.
References
- Adams AE, Johnson DI, Longnecker RM, Sloat BF, Pringle JR. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J Cell Biol. 1990;111:131–142. doi: 10.1083/jcb.111.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akada R, Kallal L, Johnson DI, Kurjan J. Genetic relationships between the G protein βγcomplex, Ste5p, Ste20p and Cdc42p: Investigation of effector roles in the yeast pheromone response pathway. Genetics. 1996;143:103–117. doi: 10.1093/genetics/143.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway KL, Carraway CA. Signaling, mitogenesis and the cytoskeleton: Where the action is. Bioessays. 1995;17:171–175. doi: 10.1002/bies.950170212. [DOI] [PubMed] [Google Scholar]
- Chant J, Stowers L. GTPase cascades choreographing cellular behavior: Movement, morphogenesis, and more. Cell. 1995;81:1–4. doi: 10.1016/0092-8674(95)90363-1. [DOI] [PubMed] [Google Scholar]
- Choi KY, Satterberg B, Lyons DM, Elion EA. Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell. 1994;78:499–512. doi: 10.1016/0092-8674(94)90427-8. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Couve A, Protopopov V, Gerst JE. Yeast synaptobrevin homologs are modified posttranslationally by the addition of palmitate. Proc Natl Acad Sci. 1995;92:5987–5991. doi: 10.1073/pnas.92.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvrckova F, DeVirgilio C, Manser E, Pringle JR, Nasmyth K. Ste20-like protein kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes & Dev. 1995;9:1817–1830. doi: 10.1101/gad.9.15.1817. [DOI] [PubMed] [Google Scholar]
- Feng Y, Song LY, Kincaid E, Mahanty SK, Elion EA. Functional binding between Gβ and the LIM domain of Ste5 is required to activate the MEKK Ste11. Curr Biol. 1998;8:267–278. doi: 10.1016/s0960-9822(98)70108-3. [DOI] [PubMed] [Google Scholar]
- Ferro-Novick S, Jahn R. Vesicle fusion from yeast to man. Nature. 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- Grishin AV, Weiner JL, Blumer KJ. Biochemical and genetic analysis of dominant-negative mutations affecting a yeast G-protein γ subunit. Mol Cell Biol. 1994;14:4571–4578. doi: 10.1128/mcb.14.7.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson MS, Blinder D, Thorner J, Jenness DD. Mutational activation of the STE5 gene product bypasses the requirement for G protein beta and gamma subunits in the yeast pheromone response pathway. Mol Cell Biol. 1994;14:1054–1065. doi: 10.1128/mcb.14.2.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: For mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Hirschman J, DeZutter G, Simonds W, Jenness DD. The Gβγ complex of the yeast pheromone response pathway: Subcellular fractionation and protein–protein interactions. J Biol Chem. 1996;272:240–248. doi: 10.1074/jbc.272.1.240. [DOI] [PubMed] [Google Scholar]
- Inouye C, Dhillon N, Thorner J. Ste5 RING-H2 domain: Role in Ste4-promoted oligomerization for yeast pheromone signaling. Science. 1997;278:103–106. doi: 10.1126/science.278.5335.103. [DOI] [PubMed] [Google Scholar]
- Leberer E, Dignard D, Harcus D, Thomas DY, Whiteway M. The protein kinase homologue Ste20p is required to link the yeast pheromone response G-protein βγ subunits to downstream signalling components. EMBO J. 1992;11:4815–4824. doi: 10.1002/j.1460-2075.1992.tb05587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E, Thomas DY, Whiteway M. Pheromone signalling and polarized morphogenesis in yeast. Curr Opin Genet Dev. 1997a;7:59–66. doi: 10.1016/s0959-437x(97)80110-4. [DOI] [PubMed] [Google Scholar]
- Leberer E, Wu C, Leeuw T, Fourest-Lieuvin A, Segall JE, Thomas DY. Functional characterization of the Cdc42p binding domain of yeast Ste20p protein kinase. EMBO J. 1997b;16:83–97. doi: 10.1093/emboj/16.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuw T, Fourest-Lieuvin A, Wu C, Chenevert J, Clark K, Whiteway M, Thomas DY, Leberer E. Pheromone response in yeast: Association of Bem1p with proteins of the MAP kinase cascade and actin. Science. 1995;270:1210–1213. doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- Leeuw T, Wu C, Schrag JD, Whiteway M, Thomas DY, Leberer E. Interaction of a G-protein β-subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature. 1998;391:191–195. doi: 10.1038/34448. [DOI] [PubMed] [Google Scholar]
- Lilly PJ, Devreotes PN. Chemoattractant and GTPγS-mediated stimulation of adenylyl cyclase in Dictyostelium requires translocation of CRAC to membranes. J Cell Biol. 1995;129:1659–1665. doi: 10.1083/jcb.129.6.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K, Snyder M. Specification of sites for polarized growth in Saccharomyces cerevisiae and the influence of external factors on site selection. Mol Biol Cell. 1992;3:1025–1035. doi: 10.1091/mbc.3.9.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Fink GR. The riddle of MAP kinase signaling specificity. Trends Genet. 1998;14:151–155. doi: 10.1016/s0168-9525(98)01425-5. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Deschenes RJ. Characterization of protein prenylation in Saccharomyces cerevisiae. Methods Enzymol. 1995;250:68–78. doi: 10.1016/0076-6879(95)50063-4. [DOI] [PubMed] [Google Scholar]
- Neiman AM, Herskowitz I. Reconstitution of a yeast protein kinase cascade in vitro: Activation of the yeast MEK homologue STE7 by STE11. Proc Natl Acad Sci. 1994;91:3398–3402. doi: 10.1073/pnas.91.8.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA. A GTP-exchange factor required for cell orientation. Nature. 1998;391:195–198. doi: 10.1038/34458. [DOI] [PubMed] [Google Scholar]
- Peter M, Neiman AM, Park H-O, van Lohuizen M, Herskowitz I. Functional analysis of the interaction between the small GTP binding protein Cdc42 and the Ste20 protein kinase in yeast. EMBO J. 1996;15:7046–7059. [PMC free article] [PubMed] [Google Scholar]
- Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Role of βγ subunits of G proteins in targeting the β-adrenergic receptor kinase to membrane-bound receptors. Science. 1992;257:1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- Pryciak PM, Hartwell LH. AKR1 encodes a candidate effector of the Gβγ complex in the Saccharomyces cerevisiae pheromone response pathway and contributes to control of both cell shape and signal transduction. Mol Cell Biol. 1996;16:2614–2626. doi: 10.1128/mcb.16.6.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner JC, Pelham HRB. Transmembrane domain-dependent sorting of proteins to the ER and plasma membrane in yeast. EMBO J. 1997;16:1832–1841. doi: 10.1093/emboj/16.8.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrick K, Garvik B, Hartwell LH. Mating in Saccharomyces cerevisiae: The role of the pheromone signal transduction pathway in the chemotropic response to pheromone. Genetics. 1997;147:19–32. doi: 10.1093/genetics/147.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Chernoff J. Emerging from the Pak: The p21-activated protein kinase family. Trends Cell Biol. 1997;7:162–167. doi: 10.1016/S0962-8924(97)01003-9. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M-N, DeVirgilio C, Souza B, Pringle JR, Abo A, Reed SI. Role for the Rho-family GTPase Cdc42 in yeast mating-pheromone signal pathway. Nature. 1995;376:702–705. doi: 10.1038/376702a0. [DOI] [PubMed] [Google Scholar]
- Sprague GF, Thorner JW. Pheromone response and signal transduction during the mating process of Saccharomyces cerevisiae. In: Jones EW, Pringle JR, Broach JR, editors. The molecular and cellular biology of the yeast Saccharomyces. Vol. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 657–744. [Google Scholar]
- Tedford K, Kim S, Sa D, Stevens K, Tyers M. Regulation of the mating pheromone and invasive growth responses in yeast by two MAP kinase substrates. Curr Biol. 1997;7:228–238. doi: 10.1016/s0960-9822(06)00118-7. [DOI] [PubMed] [Google Scholar]
- Whiteway MS, Thomas DY. Site-directed mutations altering the CAAX box of Ste18, the yeast pheromone-responsive pathway Gγ subunit. Genetics. 1994;137:967–976. doi: 10.1093/genetics/137.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteway M, Hougan L, Thomas DY. Overexpression of the STE4 gene leads to mating response in haploid Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:217–222. doi: 10.1128/mcb.10.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteway MS, Wu C, Leeuw T, Clark K, Fourest-Lieuvin A, Thomas DY, Leberer E. Association of the yeast pheromone response G protein βγ subunits with the MAP kinase scaffold Ste5p. Science. 1995;269:1572–1575. doi: 10.1126/science.7667635. [DOI] [PubMed] [Google Scholar]
- Wu C, Whiteway M, Thomas DY, Leberer E. Molecular characterization of Ste20p, a potential mitogen-activated protein or extracellular signal-regulated kinase kinase (MEK) kinase kinase from Saccharomyces cerevisiae. J Biol Chem. 1995;270:15984–15992. doi: 10.1074/jbc.270.27.15984. [DOI] [PubMed] [Google Scholar]
- Yablonski D, Marbach I, Levitzki A. Dimerization of Ste5, a mitogen-activated protein kinase cascade scaffold protein, is required for signal transduction. Proc Natl Acad Sci. 1996;93:13864–13869. doi: 10.1073/pnas.93.24.13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FL, Casey PJ. Protein prenylation: Molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- Zhao Z-S, Leung T, Manser E, Lim L. Pheromone signalling in Saccharomyces cerevisiae requires the small GTP-binding protein Cdc42p and its activator CDC24. Mol Cell Biol. 1995;15:5246–5257. doi: 10.1128/mcb.15.10.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziman M, O’Brien JM, Ouellette LA, Church WR, Johnson DI. Mutational analysis of CDC42Sc, a Saccharomyces cerevisiae gene that encodes a putative GTP-binding protein involved in the control of cell polarity. Mol Cell Biol. 1991;11:3537–3544. doi: 10.1128/mcb.11.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziman M, Preuss D, Mulholland J, O’Brien JM, Botstein D, Johnson DI. Subcellular localization of Cdc42p, a Saccharomyces cerevisiae GTP-binding protein involved in the control of cell polarity. Mol Biol Cell. 1993;4:1307–1316. doi: 10.1091/mbc.4.12.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]