The type 2C Ser/Thr phosphatase PP2Cγ is a pre-mRNA splicing factor (original) (raw)
Abstract
To identify activities involved in human pre-mRNA splicing, we have developed a procedure to separate HeLa cell nuclear extract into five complementing fractions. An activity called SCF1 was purified from one of these fractions by assaying for reconstitution of splicing in the presence of the remaining four fractions. A component of SCF1 is shown to be PP2Cγ, a type 2C Ser/Thr phosphatase of previously unknown function. Previous work suggested that dephosphorylation of splicing factors may be important for catalysis after spliceosome assembly, although the identities of the specific phosphatases involved remain unclear. Here we show that human PP2Cγ is physically associated with the spliceosome in vitro throughout the splicing reaction, but is first required during the early stages of spliceosome assembly for efficient formation of the A complex. The phosphatase activity is required for the splicing function of PP2Cγ, as an active site mutant does not support spliceosome assembly. The requirement for PP2Cγ is highly specific, as the closely related phosphatase PP2Cα cannot substitute for PP2Cγ. Consistent with a role in splicing, PP2Cγ localizes to the nucleus in vivo. We conclude that at least one specific dephosphorylation event catalyzed by PP2Cγ is required for formation of the spliceosome.
Keywords: PP2Cγ, phosphatase, splicing, pre-mRNA processing, spliceosome
Pre-mRNA splicing is a complex reaction that requires five small nuclear ribonucleoprotein particles (snRNPs) and numerous non-snRNP protein factors that assemble into a spliceosome (for review, see Krämer 1996; Will and Lührmann 1997). Spliceosome assembly is an ordered process that involves sequential formation of complexes E → A → B → C (for review, see Reed and Palandjian 1997). The E, A, and B complexes are precursors to the spliceosome, and the C complex is the functional spliceosome.
A large number of components required for mammalian splicing have yet to be identified. One successful method to identify factors is complementation of either fractionated or deficient extracts. An important advantage of this approach is that the complementation assay used to isolate a factor can also be used for further mechanistic studies. For example, the human protein SF2/ASF was purified as an essential factor that complements splicing in an inactive cytoplasmic S100 extract (Krainer et al. 1990). SF2/ASF has since been shown to be a member of a family of structurally and functionally related splicing factors, the SR proteins, named after a domain rich in arginine and serine dipeptides (for review, see Fu 1995). SR proteins have multiple functions in splicing, and the S100 complementation assay was an important tool in determining these functions. Three other factors were identified by fractionation of nuclear extracts: SF1 (Krämer 1992), SF3a, and SF3b (Brosi et al. 1993). SF1, also called mBBP, is involved in branch site recognition (Berglund et al. 1997), and SF3a and SF3b participate in the binding of U2 snRNP to the branch site (Gozani et al. 1996).
In this work we describe a new procedure for separating HeLa nuclear extract into five complementing fractions that are competent for in vitro splicing when combined. By use of four of these crude fractions for biochemical complementation assays, an activity in the remaining fraction has been identified and named SCF1 (Splicing Complementing Factor 1). Purification and characterization of this activity showed that a component of SCF1 is the type 2C Ser/Thr protein phosphatase PP2Cγ (Travis and Welsh 1997).
Ser/Thr phosphatases can be divided into four major classes (PP1, PP2A, PP2B, and PP2C) on the basis of their substrate specificity, metal ion requirements, and inhibitor sensitivity (for review, see Shenolikar 1994). Type 2C phosphatases require metal ions for activity and are resistant to okadaic acid, an inhibitor of the PP1 and PP2A enzymes. Previous studies with phosphatase inhibitors showed that type 1 and type 2A activities are required for splicing in vitro (Mermoud et al. 1992; Tazi et al. 1992). However, type 1 and type 2A phosphatases are a highly diverse group, as multiple catalytic and regulatory subunits can associate with one another to dictate substrate specificity (Shenolikar 1994). Which specific forms of these phosphatases are involved in splicing is not known. The role of type 2C Ser/Thr protein phosphatases was not addressed, as specific inhibitors are not available. The protein phosphatase PP1 has also been shown to affect alternative splice-site choice (Cardinali et al. 1994) and the subnuclear localization of splicing factors (Misteli and Spector 1996).
Multiple cycles of phosphorylation and dephosphorylation may be required for splicing. A number of mammalian kinases have been implicated in splicing, including SRPK1 and SRPK2 (Gui et al. 1994; Kuroyanagi et al. 1998; Wang et al. 1998b), Clk/Sty (Colwill et al. 1996), DNA topoisomerase I (Rossi et al. 1996), a CaMK II-like kinase (Parker and Steitz 1997), and cyclin E–cdk2 (Seghezzi et al. 1998). In vitro, most of these kinases phosphorylate the carboxy-terminal RS domains of SR proteins, which are extensively phosphorylated in vivo (for review, see Fu 1995). Phosphorylation of the RS domain appears to be required for some functions of SR proteins (Mermoud et al. 1994; Roscigno and Garcia-Blanco 1995; Cao et al. 1997; Tacke et al. 1997; Xiao and Manley 1997) and for their localization (Misteli and Spector 1998). In addition, experiments with thiophosphorylated U1-70K protein and SF2/ASF suggested that a specific dephosphorylation event(s) is required for splicing (Tazi et al. 1993; Cao et al. 1997). Although these studies have established that phosphorylation and dephosphorylation are important for splicing, the specific substrates and enzymes involved, as well as the mechanistic consequences of these modifications, are poorly understood.
Results
Establishment of a reconstituted system
Selective precipitation by ammonium sulfate was chosen as a first step in fractionating nuclear extract into complementing fractions. Two fractions were generated at a given ammonium sulfate concentration, and the corresponding high and low ammonium sulfate fractions were tested for splicing activity with β-globin pre-mRNA. The low ammonium sulfate fraction did not have significant splicing activity, unless the corresponding high ammonium sulfate fraction was added (Fig. 1, cf. lanes 1 and 3, 4 and 6, 7 and 9). As SR proteins are soluble in high concentrations of ammonium sulfate (Krainer et al. 1990; Zahler et al. 1992), the activity present in the high ammonium sulfate fraction could be due to SR proteins. To test this possibility, purified SR proteins were added to the low ammonium sulfate fractions and splicing activity was assayed. SR proteins were able to complement the 20%–45% and 20%–50% saturation fractions (lanes 7 and 11) but not the 20%–40% fraction (lane 3). This result shows that the 40%–90% ammonium sulfate fraction contains one or more splicing factors, in addition to SR proteins, which are needed to complement the 20%–40% fraction. Therefore, to purify the activity present in the high ammonium sulfate fraction, the 20%–40% cut was chosen as a complementing fraction. Further characterization showed that the 20%–40% fraction contained the vast majority of the abundant spliceosomal snRNPs (data not shown). The initial 0%–20% step removed large aggregates and improved the signal-to-background ratio in the splicing reaction (data not shown).
Figure 1.
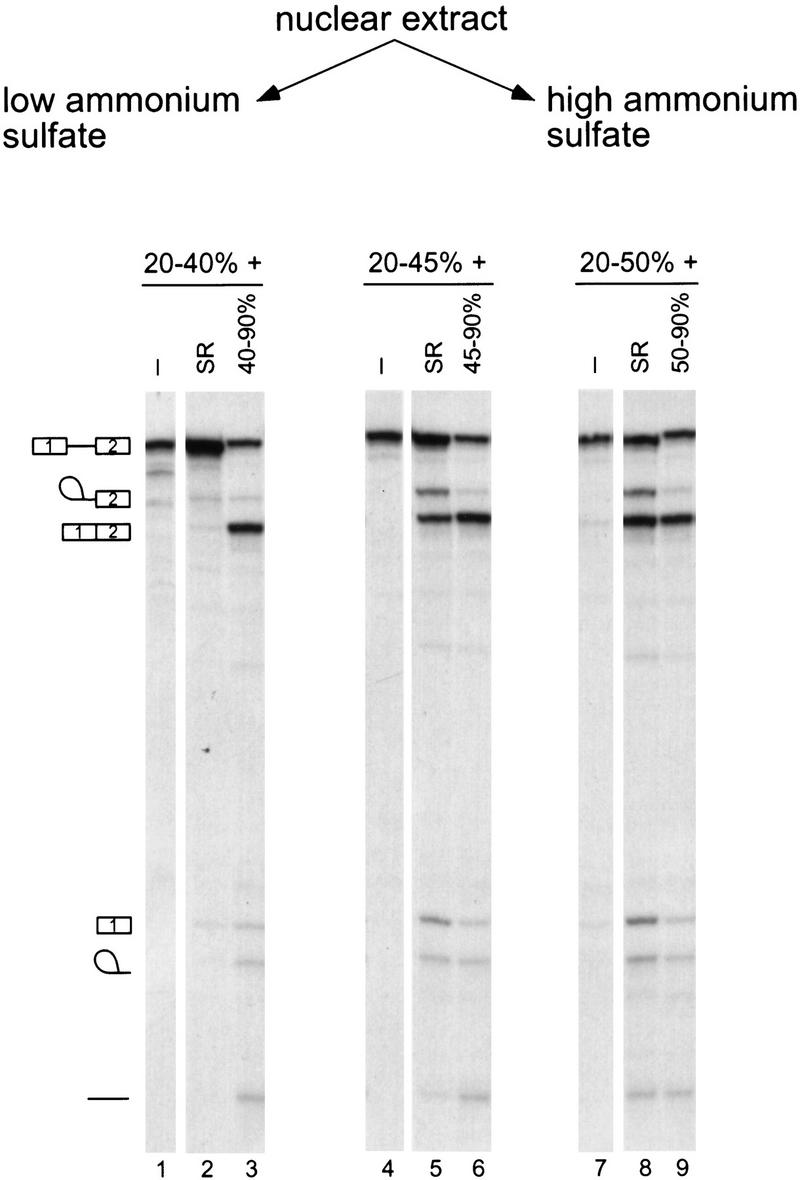
Fractionation of nuclear extract and reconstitution by ammonium sulfate precipitation. Nuclear extract was fractionated into high and low ammonium sulfate fractions, which were tested alone or in combination for pre-mRNA splicing. β-globin pre-mRNA was incubated with the indicated fractions for 2 hr. Three sets of fractionation experiments are shown, representing three different cuts, 20%–40%, 20%–45%, and 20%–50% saturation. Either purified HeLa SR proteins or the corresponding high ammonium sulfate cut was added to each of these fractions. The positions of the pre-mRNA, mRNA, lariat–exon 2, lariat intron, and debranched intron are shown.
To assay for factors in the high ammonium sulfate fraction other than SR proteins, splicing assays were carried out in the presence of the 20%–40% fraction and purified SR proteins. In addition, purified human creatine kinase was added to ensure that ATP levels remained constant in the splicing reactions throughout the fractionation procedure. To purify the high ammonium sulfate activity, we found it necessary to separate this fraction on a CsCl equilibrium density gradient (Fig. 2A). This step served multiple purposes. First, nucleic acids sediment at the bottom of the gradient, whereas proteins remain near the top. The removal of nucleic acids improved the subsequent ion-exchange chromatography step. Second, an enrichment of splicing activity was obtained. By Bradford assay, the majority of the protein was found in the top five fractions, whereas splicing activity was found in fractions 4–9. Under these conditions, some separation of proteins was obtained, as SDS-PAGE confirmed that the protein distribution is not uniform throughout the upper ten fractions (data not shown). Finally, a splicing inhibitor was detected in the top three to four fractions, as assayed by inhibition of β-globin splicing on addition to nuclear extract (data not shown). The identity of this inhibitor is presently unknown. Removal of the inhibitory activity and nucleic acid greatly aided further purification of the high ammonium sulfate activity.
Figure 2.
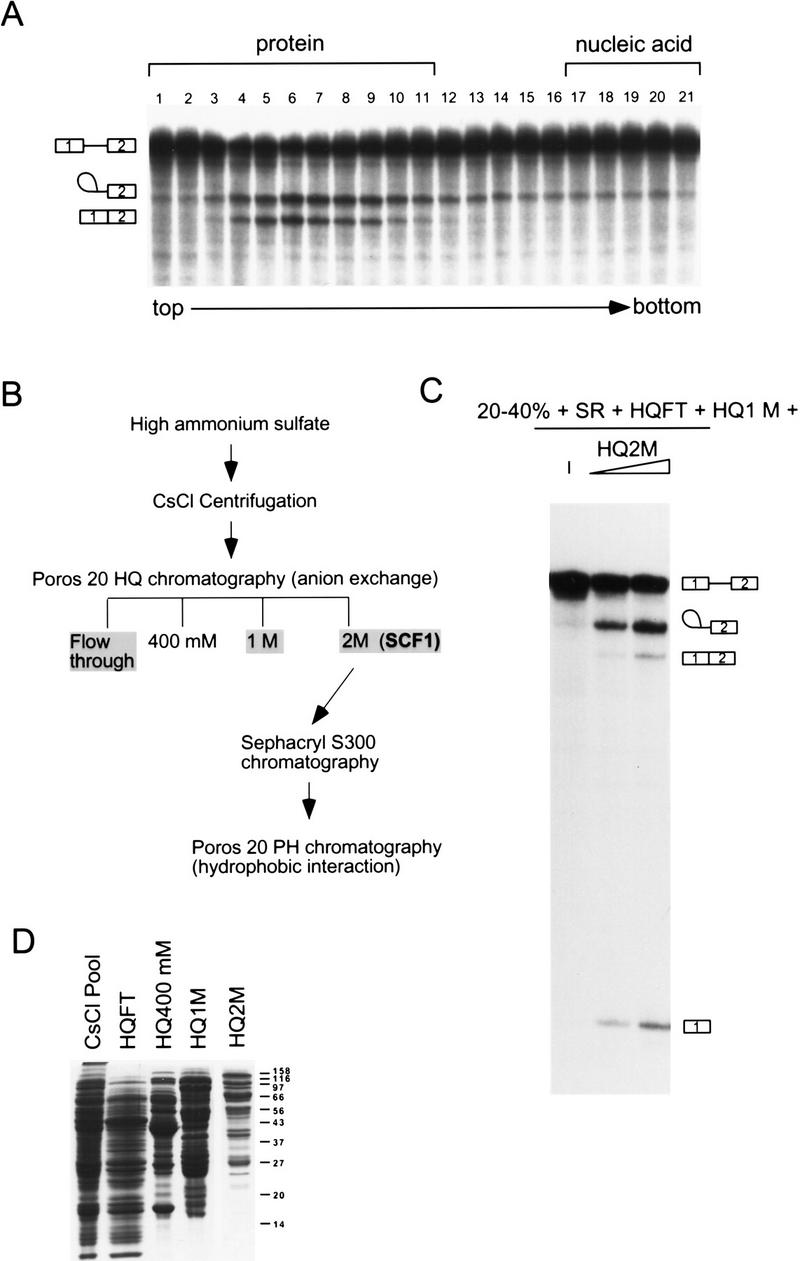
Separation of the high ammonium sulfate fraction. (A) CsCl equilibrium density centrifugation of the high ammonium sulfate fraction. Fractions from the gradient were assayed for splicing activity with β-globin pre-mRNA in reactions that also contained the low ammonium sulfate fraction (20%–40%) and SR proteins. The sedimentation of bulk protein and nucleic acid are indicated. (B) Flow chart of SCF1 purification. (C) Fractions from the Poros 20 HQ step were assayed for splicing in combination with the 20%–40% fraction and SR proteins. In this assay, none of the HQ fractions had complementing activity in isolation (data not shown). (D) Coomassie-stained SDS gel of pooled fractions from the flow-through (FT) and the salt eluates of the Poros 20 HQ step.
Pooled CsCl fractions were loaded onto a Poros 20 HQ column, and the bound proteins were eluted by stepwise salt washes (Fig. 2B). Three of the four fractions (HQFT, HQ1M, and HQ2M) were required to reconstitute efficient splicing. The HQFT plus the HQ1M fractions were unable to complement the 20%–40% fraction plus SR proteins for splicing activity. If, in addition to the HQFT and HQ1M fractions, the HQ2M fraction was also included, splicing activity was restored (Fig. 2C). In this complementation assay, the most abundant RNA species are lariat–exon 2 and free exon 1 from the first catalytic step of splicing, perhaps because a factor required for the second step of splicing was partially depleted. The HQ2M fraction had <2% of the total protein recovered and had the lowest protein complexity by SDS-PAGE compared with the other HQ fractions (Fig. 2D). Therefore, the activity in this fraction, named SCF1, was chosen for further purification.
At this point in the procedure, five complementing fractions from nuclear extracts have been generated: 20%–40% ammonium sulfate, SR proteins, HQFT, HQ1M, and HQ2M (SCF1). In summary, the HQ2M fraction defines an activity required for in vitro splicing, called SCF1, which can be assayed in a reconstituted system containing the other four complementing fractions.
Purification of SCF1
SCF1 was further purified from the HQ2M fraction, as shown in Figure 2B. By gel-filtration chromatography, SCF1 eluted at an apparent native molecular mass of 150 kD (data not shown). The pool from this step was separated into multiple peaks by hydrophobic interaction chromatography (Fig. 3A). A peak of SCF1 was detected in fraction 29, trailing into fractions 30–31 and, to a lesser extent, fractions 32–33 (Fig. 3B). Three major protein bands, of 76, 40, and 20 kD, and four minor bands, of 48, 30, 25, and 23 kD, cochromatographed with splicing activity (Fig. 3C). Fractions 29 and 30 were combined, and peptide sequence was obtained individually for all seven bands. All of the sequenced peptides belong to a single protein, the type 2C Ser/Thr phosphatase PP2Cγ (Travis and Welsh 1997). No other bands cochromatographed with SCF1 activity, although some bands trailed into the active fractions. The multiple bands of PP2Cγ resulted from proteolytic cleavages during purification, which apparently did not lead to dissociation of the resulting fragments. In a later purification of SCF1, in which protease inhibitor concentrations were increased and the slow gel-filtration step was omitted, only a 76-kD band was observed to cochromatograph with SCF1 (data not shown). However, the data in Figure 3C correspond to the actual fractions used to determine the peptide sequences.
Figure 3.



Hydrophobic interaction chromatography. (A) Column profile. SCF1 from the Sephacryl S-300 chromatography step was loaded onto a Poros 20 PH column equilibrated in high salt, and the proteins were eluted by a reverse salt gradient. The _A_280 and conductivity tracing are shown. The splicing activity detected in B is indicated. (B) Splicing activity with β-globin pre-mRNA. Fractions from the gradient and flow-through were assayed in a reaction that contained 20%–40% fraction, SR proteins, HQ1M, and HQFT fractions. As the concentration of protein in the fractions is very low, activity is weaker than in previous steps. (C) Fractions at or flanking the peak of splicing activity were analyzed on a 10% SDS–polyacrylamide gel stained with a fluorescent protein stain, Sypro Orange (Molecular Probes). (*) Bands that comigrated with splicing activity.
PP2Cγ
Type 2C phosphatases share sequence homology, which was the basis for the identification of PP2Cγ from the human EST database (Travis and Welsh 1997). As shown in Figure 4, PP2Cγ shares significant homology in the amino- and carboxy-terminal thirds with other type 2C phosphatases, including PP2Cα, whose crystal structure has been solved (Das et al. 1996). PP2Cα is a metalloenzyme, and the specific amino acids shown to coordinate two Mn2+ ions in the active site of PP2Cα are also conserved in PP2Cγ, as well in other PP2C phosphatases. A unique feature of PP2Cγ is a region of ∼200 amino acids rich in acidic residues, which interrupts the conserved PP2C phosphatase domains. A predicted ORF from Caenorhabditis elegans encoding a PP2C phosphatase also has an acidic domain at the same position (Fig. 4). It is not clear that these acidic domains have equivalent functions, because they only show 27% identity and 35% similarity, compared with 44% and 60% identity for the flanking amino- and carboxy-terminal domains, respectively. However, the conservation of the position, composition, and size of the acidic domains suggests that these two proteins are orthologs.
Figure 4.

Homology of PP2Cγ with other 2C Ser/Thr phosphatases. The amino acid sequence of human PP2Cγ (GenBank accession no. Y13936) was compared with those of human PP2Cα (S87759), S. cerevisiae PTC3 (U72346), and an ORF from C. elegans (U00051) by the PILEUP program (GCG) and manual adjustment. Identities are indicated by black shading and similarities by gray shading. PP2Cγ has an acidic domain from residues 117–319, and a similar domain is found in the C. elegans ORF. In this region, homology is not indicated, but the acidic residues are boxed. The crystal structure of PP2Cα showed that six highly conserved amino acids are involved in coordinating two active site metal ions. (•) Five of the six residues involved in metal ion coordination; (♦) the sixth residue, _Asp_496, which was mutated to Ala to make the active site mutant D496A.
Copurification of PP2C activity with SCF1
If PP2Cγ were simply a contaminant in the fractions of the Poros 20 PH column, then it would be likely to partially separate from splicing activity at some other point in the purification. A bovine ortholog of PP2Cγ, called MCPP, was detected in brain homogenates by assaying for a phosphatase that requires Mn2+ or Mg2+, is resistant to okadaic acid, and is inhibited by Ca2+ (Wang et al. 1995). We used the same assay conditions with myelin basic protein (MBP) to follow this activity in fractions generated during purification of human SCF1. Under these conditions, type 1, 2A, and 2B phosphatase activities are not measured. Type 2C activity cofractionated with SCF1 splicing activity during the initial ammonium sulfate fractionation (data not shown), the CsCl step (Fig. 5A,B), and the Poros 20 HQ separation (Fig. 5C). The precise copurification of type 2C phosphatase activity with SCF1 over multiple steps strongly suggests that PP2Cγ is not a contaminant in the final purification step.
Figure 5.
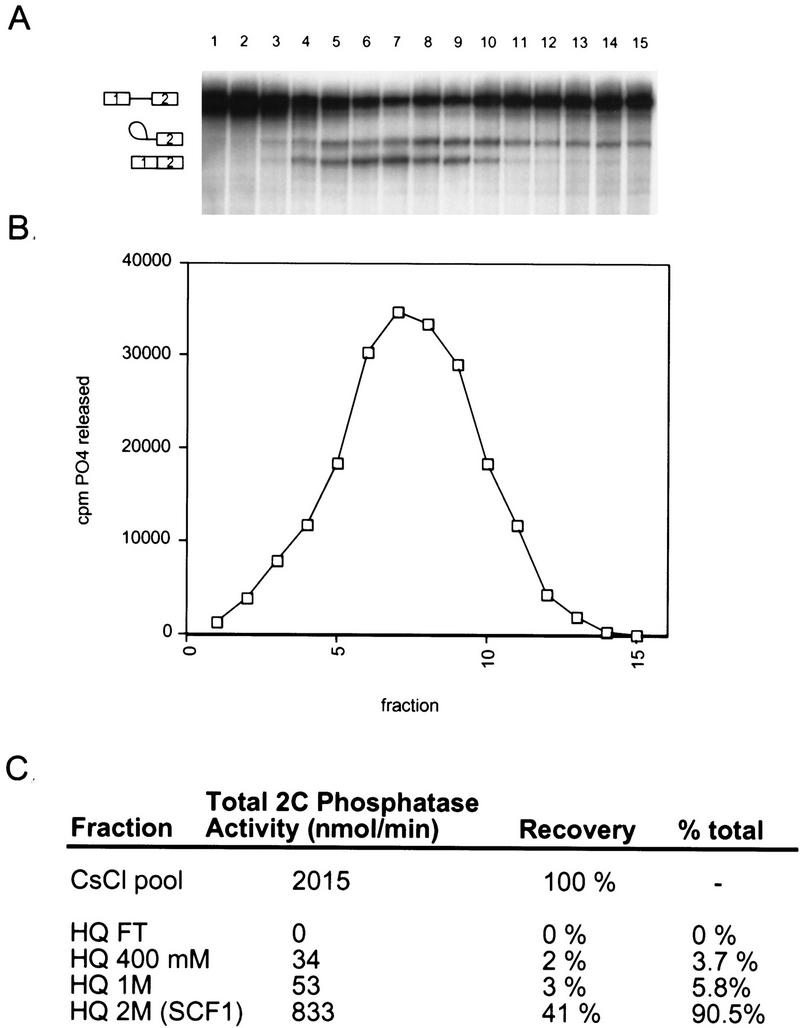
Copurification of type 2C phosphatase activity with splicing activity. (A) Fractions from the upper half of a CsCl gradient were assayed for splicing activity with β-globin pre-mRNA in reactions that also contained the low ammonium sulfate fraction (20%–40%) and SR proteins. (B) The same fractions were assayed for type 2C phosphatase activity by incubating with 32P-labeled MBP. (C) Type 2C phosphatase activity in fractions from the HQ column step.
Splicing activity of recombinant PP2Cγ
Recombinant forms of PP2Cγ were expressed in Escherichia coli or baculovirus-infected Sf9 cells as soluble proteins, and were purified to homogeneity (Fig. 6A). Both the E. coli and Sf9-derived rPP2Cγ had phosphatase activity (Fig. 6B). A mutant of rPP2Cγ was also prepared, with an active-site mutation, D496A, on the basis of the crystal structure of PP2Cα, a related phosphatase (Das et al. 1996). This substitution is predicted to disrupt the coordination of one of two active-site metal ions. The mutation reduced the specific activity of rPP2Cγ for phosphorylated MBP by 1500-fold (Fig. 6B). These proteins were tested for activity in splicing by use of two different assays.
Figure 6.
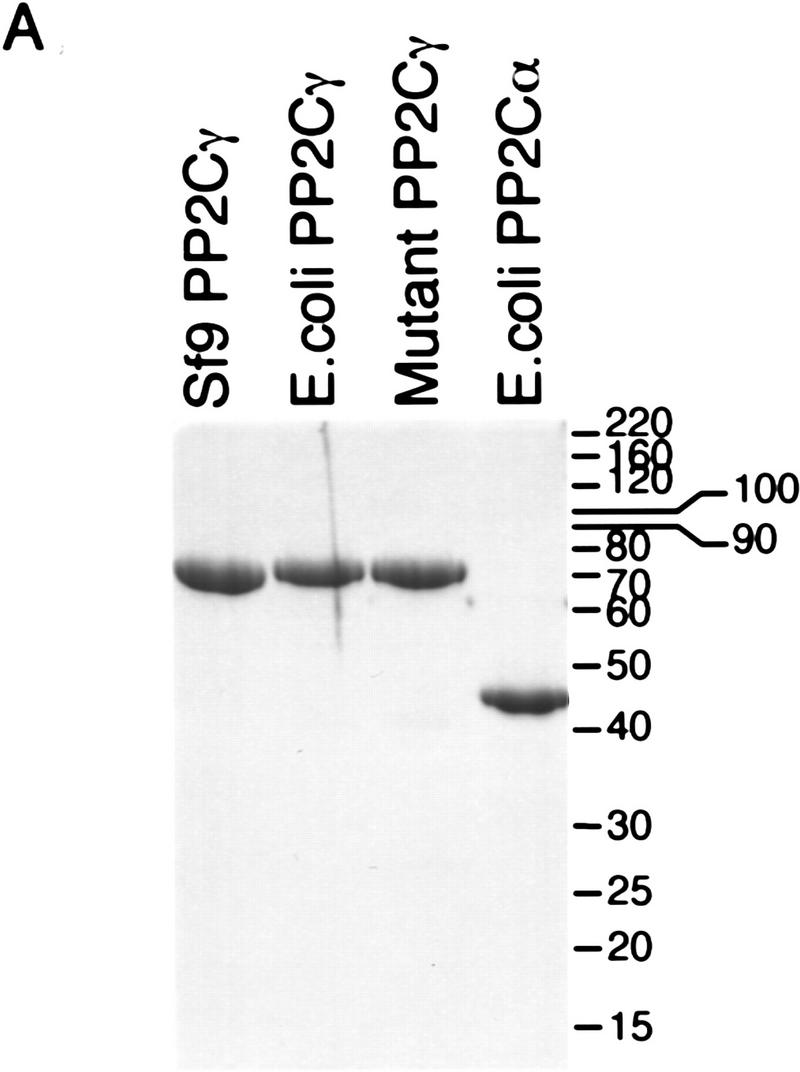

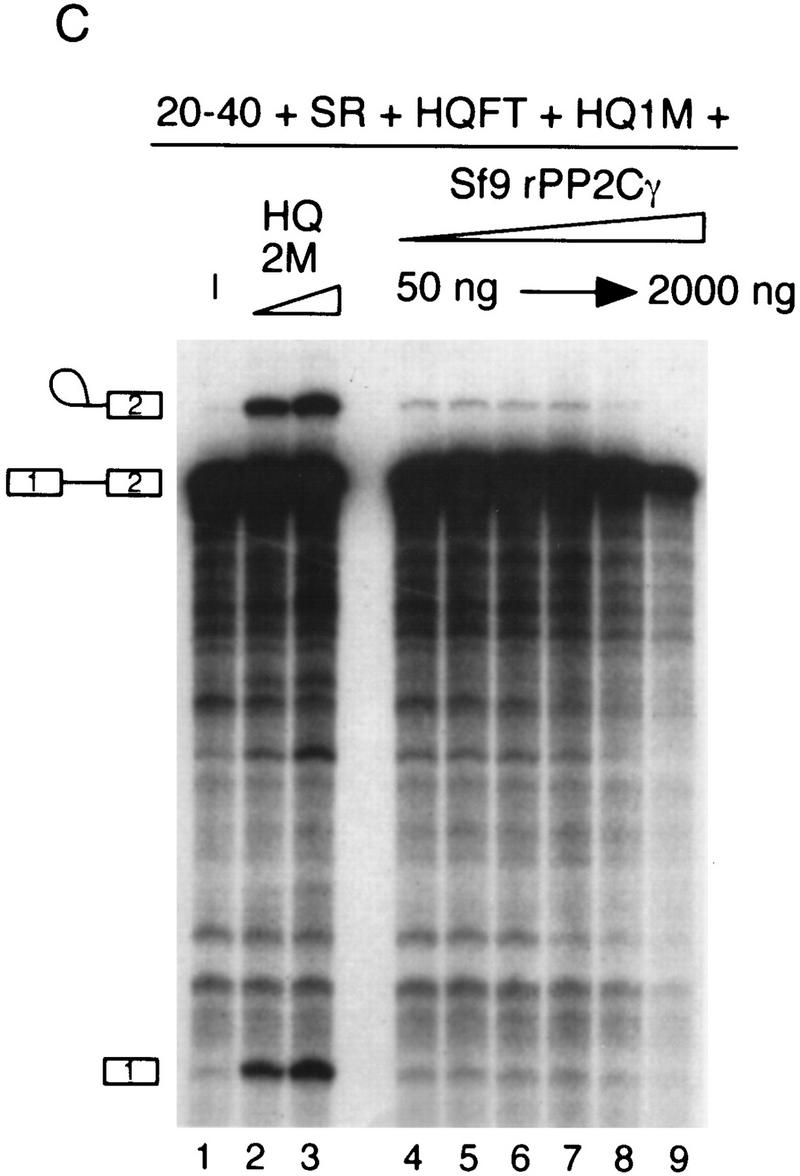

Phosphatase and splicing activity of recombinant PP2Cγ. (A) Coomassie-stained SDS gel of recombinant proteins. Mutant PP2Cγ has an alanine substitution for a highly conserved aspartate at position 496 (D496A). (B) Specific phosphatase activity of recombinant protein with 32P-labeled MBP as a substrate. The D496A mutation results in a nearly inactive form of the phosphatase. (C) rPP2Cγ was assayed for splicing with β-globin pre-mRNA in a reaction that contained the 20%–40% fraction, SR proteins, and HQFT and HQ1M fractions. Lanes 2 and 3 contain 1 and 2 μl, respectively, of the HQ2M fraction, which contains 0.1 U/μl of phosphatase activity (1 unit = 1 nmole/min). rPP2Cγ was added in lane 4 (50 ng, 0.02 unit), lane 5 (100 ng), lane 6 (200 ng), lane 7 (500 ng), lane 8 (1000 ng), and lane 9 (2000 ng). (D) Native gel analysis of spliceosomal complexes formed on β-globin pre-mRNA. The indicated combinations of fractions were incubated for 1 hr under splicing conditions and analyzed by native gel electrophoresis. Reactions contained either 0.4, 1, and 2 μl of the HQ2M fraction or 20, 50, and 100 ng of the indicated recombinant proteins. As a control, the complexes formed in a 30-min incubation with nuclear extract are also shown.
Recombinant proteins were tested in an in vitro splicing assay that demands that catalysis occur. When all of the complementing fractions except the HQ2M fraction were added to a splicing reaction, only a very low level of splicing was observed. On addition of the HQ2M fraction, which defines SCF1 activity, splicing was restored (Fig. 6C). When rPP2Cγ from Sf9 cells was added to this assay in place of the HQ2M fraction, a weak but detectable increase in splicing was observed (Fig. 6, cf. lane 1 with lanes 4–9), although it was considerably less than observed with the HQ2M fraction (Fig. 6, cf. lanes 2–3 with 4–9). However, the Poros 20 PH fractions from the final step of SCF1 purification also had weak activity (see Fig. 3B). Mutant rPP2Cγ did not have any detectable splicing activity (data not shown). Adding less rPP2Cγ than shown did not increase the amount of splicing observed, and preincubation of rPP2Cγ in nuclear extract did not improve the splicing activity (data not shown). These results suggest that either another factor in addition to PP2Cγ, or a modification of PP2Cγ, is required for the level of activity seen in the HQ2M fraction. An inhibitory effect on splicing was observed at high levels of rPP2Cγ, which may be due to nonspecific effects (e.g., nonspecific dephosphorylation of splicing factors).
To further characterize SCF1, the requirement for spliceosome assembly was investigated. Splicing reactions containing either nuclear extract or the complementing fractions were analyzed by native gel electrophoresis (Konarska 1989). In whole nuclear extract, the A, B, and C splicing complexes were resolved (Fig. 6D, lane 17). The A and B complexes are prespliceosomal complexes, whereas the C complex is the functional spliceosome (for review, see Reed and Palandjian 1997). With complementing fractions, but without the HQ2M fraction, only a small amount of A complex formation was observed, which was greatly stimulated on addition of the HQ2M fraction (lanes 1–4). Stimulation of B complex was also observed, whereas detectable C complex did not accumulate. These data show that SCF1 is required for efficient A complex formation.
To determine whether the A complex stimulation by SCF1 is due to PP2Cγ, recombinant proteins were tested in the spliceosome assembly assay. rPP2Cγ had full SCF1 activity in this assay, and recombinant proteins expressed in Sf9 cells or E. coli showed equivalent activities (Fig. 6D, lanes 5–10). Any additional factor or modification of PP2Cγ required for splicing catalysis is not required for A complex, and probably B complex, assembly. The catalytically inactive form of rPP2Cγ, D496A, was unable to substitute for SCF1 (lanes 11–13), showing that phosphatase activity is required. Another type 2C phosphatase was tested for SCF1 activity in the complex assembly assay. PP2Cα is homologous to PP2Cγ, except that it lacks an acidic domain (Fig. 4). Although related at the sequence and biochemical level, PP2Cα was unable to stimulate A complex formation, and was actually somewhat inhibitory (Fig. 6D, lanes 14–16). As this protein has in vitro phosphatase activity with MBP (Fig. 6B), phosphatase activity alone is not sufficient for SCF1 activity. In addition, PP1 and PP2A activities are not required for A complex assembly (Mermoud et al. 1992; Tazi et al. 1992). We conclude that the A complex formation activity of SCF1 can be fully accounted for by PP2Cγ, requires phosphatase activity, and is specific for PP2Cγ.
Association of PP2Cγ with the spliceosome
Monoclonal antibodies were raised to recombinant PP2Cγ. A Western blot of HeLa nuclear extract probed with three different monoclonal antibodies detected a single band of 76 kD (Fig. 7A). This size is consistent with the observed mobility of rPP2Cγ and the reported mobilities of bovine and mouse PP2Cγ (Wang et al. 1995; Guthridge et al. 1997). Although the antibodies recognized at least some of the proteolytic fragments in purified preparations of PP2Cγ (data not shown), only the full-length protein was detected in nuclear extract, confirming that the multiple bands observed in the final purification of SCF1 (Fig. 3C) arose from proteolysis during purification.
Figure 7.

Association of PP2Cγ with the spliceosome and intracellular localization. (A) Western blot of HeLa nuclear extract probed with three different anti-PP2Cγ monoclonal antibodies. (B) Immunoprecipitations of β-globin splicing reactions after a 1-hr incubation. Radiolabeled RNA was recovered from the immunoprecipitates and analyzed by denaturing PAGE. The amount of input RNA shown is equivalent to 10% of the amount used for each immunoprecipitation. Anti-Sm (mAb Y12) and anti-trimethyl guanosine (mAb K121) immunoprecipitations were included as positive controls, and an anti-maltose-binding protein (α-MalE) antibody (mAb105) served as a negative control. (C) The intracellular localization of PP2Cγ in HeLa cells was determined by use of an anti-PP2Cγ monoclonal antibody (7-53) and FITC-conjugated secondary antibody. SC35 localization is included as a positive control and as a marker for the speckle region of the nucleus.
To determine whether PP2Cγ is physically associated with the spliceosome, immunoprecipitations were carried out from in vitro splicing reactions containing radiolabeled pre-mRNA. RNA was recovered from the immunoprecipitates and analyzed by denaturing PAGE. The anti-PP2Cγ monoclonal antibody efficiently immunoprecipitated the precursor, intermediates, and products of splicing, showing that PP2Cγ is stably associated with the spliceosome at multiple steps (Fig. 7B). The amount of RNA in the anti-PP2Cγ immunoprecipitate is comparable with that of two control antibodies against snRNPs (Sm and trimethyl G). As a negative control, a monoclonal antibody directed against E. coli maltose-binding protein (MalE) was used, and in this case no radiolabeled RNAs were immunoprecipitated. This experiment does not reveal whether the interaction between PP2Cγ and RNA is direct or indirect. UV-cross-linking/immunoprecipitation experiments did not reveal a direct association of PP2Cγ with RNA (data not shown).
Subcellular localization of PP2Cγ
If PP2Cγ is involved in splicing in vivo, then at least some of it should be localized to the nucleus. The intracellular localization of endogenous PP2Cγ was determined by indirect immunofluorescence with anti-PP2Cγ monoclonal antibodies (Fig. 7C). PP2Cγ localized to the nucleoplasm and was excluded from the nucleolus. No staining was detectable in the cytoplasm. All three anti-PP2Cγ monoclonal antibodies gave the same pattern (data not shown). The nucleoplasmic localization is consistent with a role in pre-mRNA splicing, but is in contrast to the speckle pattern observed for a number of other splicing factors, such as SC35 (Fu and Maniatis 1990). On the other hand, the splicing factor U1-70K and some hnRNP proteins that have been implicated in splicing give a staining pattern similar to that observed for PP2Cγ (Misteli and Spector 1998). A similar localization was reported for an epitope-tagged version of a truncated form of mouse PP2Cγ lacking the first 150 amino acids (Guthridge et al. 1997).
Discussion
SCF1 is a new pre-mRNA splicing activity, defined as one of five complementing fractions derived from HeLa nuclear extract. SCF1 was purified, and one of its components is the Ser/Thr phosphatase PP2Cγ. Monoclonal antibodies were used to show that PP2Cγ is a nuclear phosphatase and is physically associated with the spliceosome in vitro. SCF1 is required for efficient A complex assembly, an early event in the splicing pathway. The spliceosome assembly activity is clearly attributable to PP2Cγ, as recombinant PP2Cγ can fully substitute for a partially purified fraction that defines SCF1.
Although SCF1 is required for the first step of splicing, recombinant PP2Cγ only weakly promoted catalysis when substituted for a more active, partially purified SCF1 fraction. Therefore, full activity in splicing catalysis must require another component, in addition to PP2Cγ, or a necessary modification of PP2Cγ is lacking in the recombinant protein. We cannot presently distinguish between these possibilities. This presumptive factor or modification is not required for A complex assembly, but it is required at a later step prior to, or coinciding with, the catalytic steps of splicing. If a factor in addition to PP2Cγ is required for catalysis, then it is likely that such a factor was only partially separated from PP2Cγ at the final purification step but did not cochromatograph with SCF1. We attempted to remove PP2Cγ from nuclear extract by immunodepletion. Although extensive depletion could be obtained, as determined by Western blotting, no effect on splicing was observed (data not shown). However, complete removal of a catalytic activity from an extract is difficult, and a low level of PP2Cγ may be sufficient for splicing.
PP2Cγ and other type 2C Ser/Thr protein phosphatases
The PP2C family is a diverse group of proteins, and type 2C phosphatases contain additional motifs such as EF hands, kinase-interacting, and membrane-spanning domains (see Das et al. 1996). Consistent with this structural diversity, type 2C phosphatases have been shown to be involved in diverse biological processes in both prokaryotes and eukaryotes. In mammals, two other PP2C enzymes have been identified, PP2Cα and PP2Cβ, which lack discernible motifs other than the PP2C homology (Tamura et al. 1989; Mann et al. 1992; Wenk et al. 1992). Although these two phosphatases have been characterized biochemically (e.g., see Marley et al. 1996; Kusuda et al. 1998), little is known about their roles in biological processes. There is evidence that PP2Cα negatively regulates the JNK MAPK pathway (Takekawa et al. 1998), the activity of the cystic fibrosis transmembrane conductance regulator (Travis et al. 1997), and the AMP-activated protein kinase (Moore et al. 1991; Davies et al. 1995).
Mammalian PP2Cγ differs from other type 2C phosphatases by the presence of a large internal domain of acidic character. Characterization of bovine PP2Cγ showed that the substrate specificity in vitro is broad but that it has a preference for basic proteins (Wang et al. 1995). This preference suggests that electrostatic interactions mediated by the acidic domain may be involved in substrate specificity. The mouse ortholog of PP2Cγ, called FIN13, was identified in a screen for cDNAs induced by the fibroblast growth factor FGF-4 (Guthridge et al. 1996). Interestingly, overexpression of FIN13 lacking the first 150 amino acids causes a G1/S arrest, suggesting a direct or indirect role in cell cycle control (Guthridge et al. 1997). No obvious orthologs of PP2Cγ are present among the type 2C phosphatases in Saccharomyces cerevisiae, and the extensive genetic analyses of splicing in this organism have not yet revealed a role for phosphatases in splicing. Therefore, the requirement for PP2Cγ in mammalian splicing may reflect the more complex aspects of fidelity and regulation of splice-site selection, compared with budding yeast.
The role of phosphatases in splicing
PP2Cγ catalyzes at least one specific dephosphorylation event required for A complex formation during spliceosome assembly. In contrast, general inhibitors of PP1 and PP2A activities block the catalytic steps of splicing, but not assembly of the A, B, and C complexes (Mermoud et al. 1992; Tazi et al. 1992). As the inhibitors used do not affect type 2C phosphatases, earlier studies could not address the role of these phosphatases in splicing. Specific inhibitors of type 2C phosphatases have not been described, but PP2Cγ is inhibited by calcium (Wang et al. 1995). The same concentration of calcium that inhibits the phosphatase activity of PP2Cγ also inhibits in vitro splicing in unfractionated nuclear extract (data not shown). This observation is consistent with the requirement for PP2Cγ in splicing.
In addition to phosphatase inhibitor studies, the catalytic steps of splicing, but not spliceosome assembly, are blocked when either of two proteins, the U1 snRNP protein U1-70K and the splicing factor SF2/ASF, are made phosphatase resistant by thiophosphorylation (Tazi et al. 1993; Cao et al. 1997). These data suggested that these factors must be dephosphorylated after formation of the spliceosome but before the first catalytic step. However, this approach did not permit assignment of the dephosphorylation activity to a particular Ser/Thr phosphatase. Even though PP2Cγ has a role in A complex formation early in spliceosome assembly, it may have an additional role later in the splicing reaction. In fact, immunoprecipitation experiments show that PP2Cγ is associated with the spliceosome at all detectable stages, suggesting that PP2Cγ may have a function at later stages of splicing.
Phosphorylation may also play a role in the regulation of alternative splicing, which is affected by the activity and/or levels of general splicing factors, such as SR proteins and hnRNP proteins (Wang et al. 1997). Overexpression of the Clk/Sty kinase, which can phosphorylate SR proteins, changes the alternative splicing of its own pre-mRNA and of an adenovirus pre-mRNA (Duncan et al. 1997). Addition of exogenous PP1 to nuclear extracts changes alternative 5′ splice site use in HeLa nuclear extracts (Cardinali et al. 1994). A splicing silencer within an adenovirus intron can be regulated by the phosphorylation state of SR proteins, and in adenovirus-infected cells, SR protein phosphorylation may be controlled by PP2A and the viral PP2A-regulatory subunit E4-ORF4 (Kanopka et al. 1998). We do not know if PP2Cγ regulates alternative splicing, but it is interesting that the levels of mouse PP2Cγ can be upregulated by the growth factor FGF-4 (Guthridge et al. 1996). The levels of PP2Cγ could either modulate the intrinsic activity of a splicing factor or affect its intracellular location, and hence its local concentration or availability. For example, there is evidence that phosphorylation/dephosphorylation regulates the localization of splicing factors within the subnuclear speckle domain (Gui et al. 1994; Colwill et al. 1996; Misteli and Spector 1996), the recruitment of SR proteins to transcriptionally active sites (Misteli et al. 1998), and the shuttling of SR proteins (Cáceres et al. 1998).
Potential targets of PP2Cγ
Although the targets of PP2Cγ action are not presently known, several of the known mammalian splicing factors are phosphoproteins, and hence, potential substrates of PP2Cγ. In particular, splicing factors known to be required for A complex assembly (see Reed and Palandjian 1997) are good candidates. Among these are the SR proteins, which are essential splicing factors containing serine-phosphorylated carboxy-terminal repeats of arginine/serine dipeptides (for review, see Fu 1995). Phosphorylation of SR proteins appears to be required for splicing in vitro (Mermoud et al. 1994; Cao et al. 1997; Xiao and Manley 1997), and in particular for recruitment of the U4/U6 · U5 tri-snRNP to the A complex (Roscigno and Garcia-Blanco 1995). In addition, the interaction between another phosphoprotein, U1-70K, and the SR protein SF2/ASF, is phosphorylation dependent (Xiao and Manley 1997). This interaction is required for stimulation of U1 snRNP binding to a 5′ splice site by SF2/ASF, which occurs prior to A complex formation (Kohtz et al. 1994). However, the fact that thiophosphorylation of SF2/ASF inhibits catalysis but not spliceosome assembly (Cao et al. 1997) suggests that PP2Cγ does not dephosphorylate SF2/ASF during A complex formation. Another phosphorylated splicing factor required for A complex assembly is SAP155, a component of the 17S U2 snRNP (Wang et al. 1998a). SAP155 has homology to PP2A regulatory subunits, and is phosphorylated after the first catalytic step of splicing. Recently, SAP155 has been shown to interact with the cyclin E–cdk2 kinase (Seghezzi et al. 1998).
The physiological substrate specificity of PP2Cγ is unlikely to be solely an inherent property of its active site. Specificity will also depend on the subcellular localization of the enzyme, its physical association with the spliceosome, and other protein–protein interactions. The fact that PP2Cα cannot function in place of PP2Cγ, despite their similar biochemical properties and strong sequence homology, suggests that the unique acidic domain of PP2Cγ plays a crucial role in determining substrate specificity. Thus, a meaningful assessment of the substrate specificity of PP2Cγ in splicing will likely require the full context of the spliceosome.
Materials and methods
Preparation of HeLa nuclear extract and ammonium sulfate fractionation
HeLa nuclear extracts were prepared as described (Mayeda and Krainer 1999b). For ammonium sulfate fractionation, 2 vol of buffer E [20 mm HEPES–KOH (pH 8), 0.2 mm EDTA, 1 mm DTT] and 0.75 vol of buffer E-AS (saturated with ammonium sulfate at 0°C) was added. The suspension was rocked for 30 min and spun at 14,460_g_max for 15 min. The supernatant was diluted with 0.33 vol of buffer E-AS. The suspension was rocked for 30 min and spun as above. The pellet was resuspended in 40% saturated ammonium sulfate in buffer E and respun for 30 min. The pellet was redissolved in half of the volume of the starting nuclear extract in buffer D [buffer E + 100 mm KCl, 0.5 mm PMSF, 5% (vol/vol) glycerol] and dialyzed against buffer D. The supernatant from the initial 40% cut was adjusted to 90% saturation by the addition of dry ammonium sulfate (0.335 grams/ml solution). The suspension was rocked for 30 min and spun for 15 min. The pellet was redissolved in buffer D and dialyzed against buffer D. All fractions were stored at −70°C.
Purification of SCF1
Frozen HeLa or 293 cells (100 liters) were thawed and collected as described (Krainer et al. 1990). The washed cell pellet was resuspended in an equal volume of buffer A + PI [10 mm HEPES–KOH (pH 8.0), 10 mm KCl, 1.5 mm MgCl2; PI = 0.5 mm PMSF, 3.5 μg/ml pepstatin, 2.5 μg/ml leupeptin, 0.5 μg/ml aprotinin) and incubated for 10 min on ice. Cells were lysed by 30 strokes in a 100-ml Dounce homogenizer (B pestle) followed by the addition of 0.078 vol of 4 m KCl. The homogenate was stirred for 20 min and spun at 4000 rpm in an H6000A rotor for 15 min. The supernatant was diluted with 1 vol of buffer E plus 1.33 vol of buffer E-AS, stirred for 20 min and spun at 14,680_g_max for 30 min. Dry ammonium sulfate (0.335 gram/ml) was added to the supernatant, dissolved by stirring for 30 min and spun as above. The pellet was dissolved in buffer D + PI.
Dry CsCl was added (1 gram/ml) and dissolved by rocking. An equal volume of this solution was layered beneath a 32-ml layer of 30% (wt/vol) CsCl in buffer D in a Ti 45 tube and spun at 235,000_g_max for 24 hr. Gradients were harvested with a gradient fractionator (Haake-Buchler) and fractions were dialyzed against buffer D + PI-500 (500 mm KCl). Active fractions were pooled, diluted fivefold with buffer D + PI-0 (0 m KCl) and loaded onto a 1 × 10 cm Poros 20 HQ column on a Biocad Sprint (Perseptive Biosystems) equilibrated in buffer D. Bound proteins were eluted by stepwise washes of buffer D-400, buffer D-1M, and buffer D-2M (3 column vol) at 10 ml/min. The 2 m eluate was dialyzed against buffer D and loaded onto a 1.6 × 60-cm Sephacryl S-200 HR column (Pharmacia) equilibrated in buffer D at 0.5 ml/min. Active fractions were pooled, diluted with 0.66 vol of buffer D-AS + PI and loaded onto a 1.6 × 100-mm Poros 20 PH column equilibrated in 40% saturated ammonium sulfate in buffer D. A 20-column volume gradient from 40% to 0% saturated ammonium sulfate in buffer D was run at 5 ml/min. Fractions were dialyzed against buffer D + PI.
Splicing assays
In vitro splicing assays were done as described (Mayeda and Krainer 1999a). To assay for SCF1, the following mixture was prepared (amount/reaction): 2 μl of the 20%–40% ammonium sulfate fraction, 0.05 unit of human creatine kinase (1.9 U/μl), 1 μl of purified HeLa SR proteins (0.62 mg/ml), 1 μl of HQFT, 1 μl of HQ1M. To 5 μl of this mixture, up to 4 μl of the fraction to be assayed for SCF1 was added, the volume was adjusted to 9 μl with buffer D, and 6 μl of a splicing premix was added. The reactions were incubated for 2 hr.
Native splicing complex assays were done as described (Konarska 1989), with 0.1 mg/ml heparin.
Immunoprecipitations of splicing reactions were done as described (Blencowe et al. 1994), except that 0.5 mg/ml tRNA was included in the washes.
Peptide sequencing
Fractions 29 and 30 from the Poros 20 PH column were pooled and separated by SDS-PAGE. After Coomassie G staining, the protein bands were excised and subjected to in-gel digestion with Lys-C endopeptidase. The resulting peptides were separated by HPLC and sequenced by automated Edman degradation as described previously (Wang et al. 1996).
Recombinant proteins/antibodies
PP2Cγ was PCR amplified from a human EST clone (IMAGE clone 531667, Research Genetics) using Pfu polymerase (Stratagene) and the primers GTGGATCCACATATGGGTGCCTACCTCTC and CAGGATCCACTAGTCCCTCTTGG. The PCR product was digested with _Nde_I and _Bam_HI and subcloned into pET-19b. The resulting E. coli expression plasmid, pNTPP2Cγ, encodes full-length PP2Cγ with an amino-terminal His tag. The mutant D496A was made from pNTPP2Cγ as described (Deng and Nickoloff 1992) with the primers GGTCATGTTGGCACACCCGGTACCATCCC and CTTCCTTTTTCGATATCATTGAAGCATTT. To express PP2Cγ in Sf9 cells, an _Nco_I– _Bam_HI fragment from pNTPP2Cγ was ligated into the _Nco_I–_Bgl_II sites of the expression vector, pAcSG2. Recombinant virus was made with a Baculogold kit (PharMingen).
To purify rPP2Cγ, 1 liter of E. coli BL21 transformed with pNTPP2Cγ was induced at an OD of 0.4 by addition of 0.4 mm IPTG and shaken at 37°C for 4 hr. Cells were harvested, washed in PBS and lysed by sonication (5–10-sec pulses with 2-min rests) in 30 ml of lysis buffer [20 mm HEPES–KOH (pH 8), 1 m NaCl, 20 mm imidazole, 0.2 mm EGTA, 0.2 mm PMSF, 2 mm MnCl2, 1 mm DTT, 5% glycerol]. The lysate was spun at 32,530_g_max for 15 min. The supernatant was added to 2 ml of NiNTA agarose (Qiagen) equilibrated in lysis buffer without MnCl2 and rocked for 15 min. The resin was pelleted in a table top centrifuge and washed twice with 20 vol of lysis buffer and twice with 20 vol of lysis buffer 100 (100 mm NaCl). Bound proteins were eluted by two washes with 1 vol of elution buffer (lysis buffer 100 + 200 mm imidazole). The eluate was dialyzed against buffer D and loaded onto a 1.6 × 100 mm Poros 20 HQ column equilibrated in buffer D. A 20-column volume gradient from 0.4 m to 1.5 m KCl in buffer D was run at 5 ml/min. Fractions containing PP2Cγ were pooled and dialyzed against buffer D.
rPP2Cγ was purified from 350 ml of baculovirus-infected Sf9 cells (∼106/ml in T-175 flasks) as above, except for the following modifications. Lysis and elution buffers contained protease inhibitors (see above) and lacked MnCl2 and EGTA. Cells were resuspended in 5 ml of lysis buffer and sonicated five times for 5 sec with 1 min rests. Cleared lysate was added to 0.25 ml NiNTA agarose.
To raise monoclonal antibodies, BALB/c mice were immunized with rPP2Cγ from E. coli. Hybridomas were generated and screened by dot blotting at the Cold Spring Harbor Monoclonal Antibody Facility by standard procedures (Harlow and Lane 1988).
Phosphatase assays
Type 2C phosphatase activity was assayed as described (Wang et al. 1995) by use of radiolabeled MBP phosphorylated with protein kinase A.
Immunofluorescence
Immunofluoresence experiments were carried out as described (Misteli and Spector 1996).
Acknowledgments
We thank D.S. Horowitz and T. Misteli for critical reading of the manuscript, C. Bautista and M. Falkowski for technical assistance in the production of monoclonal antibodies, L. Shao for help in constructing mutant rPP2Cγ, D. Barford for the generous gift of rPP2Cα, and members of the laboratory for helpful advice. This work was supported by National Institutes of Health grant GM42699 to A.R.K. and National Cancer Institute training grant T32-CA09311 to M.V.M.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL krainer@cshl.org; FAX (516) 367-8453.
References
- Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997;89:781–787. doi: 10.1016/s0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- Blencowe BJ, Nickerson JA, Issner R, Penman S, Sharp PA. Association of nuclear matrix antigens with exon-containing splicing complexes. J Cell Biol. 1994;127:593–607. doi: 10.1083/jcb.127.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosi R, Hauri HP, Krämer A. Separation of splicing factor SF3 into two components and purification of SF3a activity. J Biol Chem. 1993;268:17640–17646. [PubMed] [Google Scholar]
- Cáceres JF, Screaton GR, Krainer AR. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes & Dev. 1998;12:55–66. doi: 10.1101/gad.12.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA. 1997;3:1456–1467. [PMC free article] [PubMed] [Google Scholar]
- Cardinali B, Cohen PT, Lamond AI. Protein phosphatase 1 can modulate alternative 5′ splice site selection in a HeLa splicing extract. FEBS Lett. 1994;352:276–280. doi: 10.1016/0014-5793(94)00973-2. [DOI] [PubMed] [Google Scholar]
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Das AK, Helps NR, Cohen PT, Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 Å resolution. EMBO J. 1996;15:6798–6809. [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- Deng WP, Nickoloff JA. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- Duncan PI, Stojdl DF, Marius RM, Bell JC. In vivo regulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Mol Cell Biol. 1997;17:5996–6001. doi: 10.1128/mcb.17.10.5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD. The superfamily of arginine/serine-rich splicing factors. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Maniatis T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature. 1990;343:437–441. doi: 10.1038/343437a0. [DOI] [PubMed] [Google Scholar]
- Gozani O, Feld R, Reed R. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes & Dev. 1996;10:233–243. doi: 10.1101/gad.10.2.233. [DOI] [PubMed] [Google Scholar]
- Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- Guthridge MA, Seldin M, Basilico C. Induction of expression of growth-related genes by FGF-4 in mouse fibroblasts. Oncogene. 1996;12:1267–1278. [PubMed] [Google Scholar]
- Guthridge MA, Bellosta P, Tavoloni N, Basilico C. FIN13, a novel growth factor-inducible serine-threonine phosphatase which can inhibit cell cycle progression. Mol Cell Biol. 1997;17:5485–5498. doi: 10.1128/mcb.17.9.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- Kanopka A, Mühlemann O, Petersen-Mahrt S, Estmer C, Öhrmalm C, Aküsjarvi G. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature. 1998;393:185–187. doi: 10.1038/30277. [DOI] [PubMed] [Google Scholar]
- Kohtz JD, Jamison SF, Will CL, Zuo P, Lührmann R, Garcia-Blanco MA, Manley JL. Protein–protein interactions and 5′ splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- Konarska MM. Analysis of splicing complexes and small nuclear ribonucleoprotein particles by native gel electrophoresis. Methods Enzymol. 1989;180:442–453. doi: 10.1016/0076-6879(89)80116-8. [DOI] [PubMed] [Google Scholar]
- Krainer AR, Conway GC, Kozak D. Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes & Dev. 1990;4:1158–1171. doi: 10.1101/gad.4.7.1158. [DOI] [PubMed] [Google Scholar]
- Krämer A. Purification of splicing factor SF1, a heat-stable protein that functions in the assembly of a presplicing complex. Mol Cell Biol. 1992;12:4545–4552. doi: 10.1128/mcb.12.10.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- Kuroyanagi N, Onogi H, Wakabayashi T, Hagiwara M. Novel SR-protein-specific kinase, SRPK2, disassembles nuclear speckles. Biochem Biophys Res Commun. 1998;242:357–364. doi: 10.1006/bbrc.1997.7913. [DOI] [PubMed] [Google Scholar]
- Kusuda K, Kobayashi T, Ikeda S, Ohnishi M, Chida N, Yanagawa Y, Shineha R, Nishihira T, Satomi S, Hiraga A, Tamura S. Mutational analysis of the domain structure of mouse protein phosphatase 2Cβ. Biochem J. 1998;332:243–250. doi: 10.1042/bj3320243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DJ, Campbell DG, McGowan CH, Cohen PT. Mammalian protein serine/threonine phosphatase 2C: cDNA cloning and comparative analysis of amino acid sequences. Biochim Biophys Acta. 1992;1130:100–104. doi: 10.1016/0167-4781(92)90471-b. [DOI] [PubMed] [Google Scholar]
- Marley AE, Sullivan JE, Carling D, Abbott WM, Smith GJ, Taylor IW, Carey F, Beri RK. Biochemical characterization and deletion analysis of recombinant human protein phosphatase 2Cα. Biochem J. 1996;320:801–806. doi: 10.1042/bj3200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A, Krainer AR. Mammalian in vitro splicing assays. In: Haynes SR, editor. Methods in molecular biology: RNA-protein interaction protocols. Totowa, NJ: Humana Press; 1999a. . (In press.) [DOI] [PubMed] [Google Scholar]
- ————— . Preparation of Hela cell nuclear and cytosolic S100 extracts for in vitro splicing. In: Haynes SR, editor. Methods in molecular biology: RNA-protein interaction protocols. Totowa, NJ: Humana Press; 1999b. . (In press.) [DOI] [PubMed] [Google Scholar]
- Mermoud JE, Cohen P, Lamond AI. Ser/Thr-specific protein phosphatases are required for both catalytic steps of pre-mRNA splicing. Nucleic Acids Res. 1992;20:5263–5269. doi: 10.1093/nar/20.20.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermoud JE, Cohen PT, Lamond AI. Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. EMBO J. 1994;13:5679–5688. doi: 10.1002/j.1460-2075.1994.tb06906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Spector DL. Serine/threonine phosphatase 1 modulates the subnuclear distribution of pre-mRNA splicing factors. Mol Biol Cell. 1996;7:1559–1572. doi: 10.1091/mbc.7.10.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The cellular organization of gene expression. Curr Opin Cell Biol. 1998;10:323–331. doi: 10.1016/s0955-0674(98)80007-0. [DOI] [PubMed] [Google Scholar]
- Misteli T, Cáceres JF, Clement JQ, Krainer AR, Wilkinson MF, Spector DL. Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J Cell Biol. 1998;143:297–307. doi: 10.1083/jcb.143.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore F, Weekes J, Hardie DG. Evidence that AMP triggers phosphorylation as well as direct allosteric activation of rat liver AMP-activated protein kinase. A sensitive mechanism to protect the cell against ATP depletion. Eur J Biochem. 1991;199:691–697. doi: 10.1111/j.1432-1033.1991.tb16172.x. [DOI] [PubMed] [Google Scholar]
- Parker AR, Steitz JA. Inhibition of mammalian spliceosome assembly and pre-mRNA splicing by peptide inhibitors of protein kinases. RNA. 1997;3:1301–1312. [PMC free article] [PubMed] [Google Scholar]
- Reed R, Palandjian L. Spliceosome assembly. In: Krainer AR, editor. Eukaryotic mRNA processing. New York, NY: Oxford University Press; 1997. pp. 130–173. [Google Scholar]
- Roscigno RF, Garcia-Blanco MA. SR proteins escort the U4/U6.U5 tri-snRNP to the spliceosome. RNA. 1995;1:692–706. [PMC free article] [PubMed] [Google Scholar]
- Rossi F, Labourier E, Forne T, Divita G, Derancourt J, Riou JF, Antoine E, Cathala G, Brunel C, Tazi J. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature. 1996;381:80–82. doi: 10.1038/381080a0. [DOI] [PubMed] [Google Scholar]
- Seghezzi W, Chua K, Shanahan F, Gozani O, Reed R, Lees E. Cyclin E associates with components of the pre-mRNA splicing machinery in mammalian cells. Mol Cell Biol. 1998;18:4526–4536. doi: 10.1128/mcb.18.8.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenolikar S. Protein serine/threonine phosphatases—new avenues for cell regulation. Annu Rev Cell Biol. 1994;10:55–86. doi: 10.1146/annurev.cb.10.110194.000415. [DOI] [PubMed] [Google Scholar]
- Tacke R, Chen Y, Manley JL. Sequence-specific RNA binding by an SR protein requires RS domain phosphorylation: Creation of an SRp40-specific splicing enhancer. Proc Natl Acad Sci. 1997;94:1148–1153. doi: 10.1073/pnas.94.4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa M, Maeda T, Saito H. Protein phosphatase 2Cα inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J. 1998;17:4744–4752. doi: 10.1093/emboj/17.16.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura S, Lynch KR, Larner J, Fox J, Yasui A, Kikuchi K, Suzuki Y, Tsuiki S. Molecular cloning of rat type 2C (IA) protein phosphatase mRNA. Proc Natl Acad Sci. 1989;86:1796–1800. doi: 10.1073/pnas.86.6.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazi J, Daugeron MC, Cathala G, Brunel C, Jeanteur P. Adenosine phosphorothioates (ATP α S and ATP τ S) differentially affect the two steps of mammalian pre-mRNA splicing. J Biol Chem. 1992;267:4322–4326. [PubMed] [Google Scholar]
- Tazi J, Kornstadt U, Rossi F, Jeanteur P, Cathala G, Brunel C, Lührmann R. Thiophosphorylation of U1-70K protein inhibits pre-mRNA splicing. Nature. 1993;363:283–286. doi: 10.1038/363283a0. [DOI] [PubMed] [Google Scholar]
- Travis SM, Welsh MJ. PP2Cγ: A human protein phosphatase with a unique acidic domain. FEBS Lett. 1997;412:415–419. doi: 10.1016/s0014-5793(97)00837-5. [DOI] [PubMed] [Google Scholar]
- Travis SM, Berger HA, Welsh MJ. Protein phosphatase 2C dephosphorylates and inactivates cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci. 1997;94:11055–11060. doi: 10.1073/pnas.94.20.11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chua K, Seghezzi W, Lees E, Gozani O, Reed R. Phosphorylation of spliceosomal protein SAP 155 coupled with splicing catalysis. Genes & Dev. 1998a;12:1409–1414. doi: 10.1101/gad.12.10.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, Fu XD. SRPK2: A differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol. 1998b;140:737–750. doi: 10.1083/jcb.140.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Kobayashi R, Bishop JM. Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc Natl Acad Sci. 1996;93:8425–8430. doi: 10.1073/pnas.93.16.8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Santini F, Qin K, Huang CY. A Mg(2+)-dependent, Ca(2+)-inhibitable serine/threonine protein phosphatase from bovine brain. J Biol Chem. 1995;270:25607–25612. doi: 10.1074/jbc.270.43.25607. [DOI] [PubMed] [Google Scholar]
- Wang Y-C, Selvakumar M, Helfman DH. Alternative pre-mRNA splicing. In: Krainer AR, editor. Eukaryotic mRNA processing. New York, NY: Oxford University Press; 1997. pp. 242–279. [Google Scholar]
- Wenk J, Trompeter HI, Pettrich KG, Cohen PT, Campbell DG, Mieskes G. Molecular cloning and primary structure of a protein phosphatase 2C isoform. FEBS Lett. 1992;297:135–138. doi: 10.1016/0014-5793(92)80344-g. [DOI] [PubMed] [Google Scholar]
- Will CL, Lührmann R. snRNP structure and function. In: Krainer AR, editor. Eukaryotic mRNA processing. New York, NY: Oxford University Press; 1997. pp. 130–173. [Google Scholar]
- Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS domain affects both protein–protein and protein–RNA interactions and is necessary for splicing. Genes & Dev. 1997;11:334–344. doi: 10.1101/gad.11.3.334. [DOI] [PubMed] [Google Scholar]
- Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: A conserved family of pre-mRNA splicing factors. Genes & Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]