Chapter 2 Linear models | Omics Data Analysis (original) (raw)
library("tidyverse")
## Prepare the data
x <-
data.frame(lmdata) |>
rownames_to_column() |>
pivot_longer(names_to = "sample", values_to = "expression", -rowname) |>
full_join(lmannot) |>
mutate(sample = sub("sample_", "", sample))
## Analyse gene 1
x_i <- filter(x, rowname == "gene_1")
x_i
## # A tibble: 12 × 5
## rowname sample expression condition cell_type
## <chr> <chr> <dbl> <chr> <chr>
## 1 gene_1 1 4.44 ctrl A
## 2 gene_1 2 4.77 ctrl B
## 3 gene_1 3 6.56 ctrl A
## 4 gene_1 4 5.07 ctrl B
## 5 gene_1 5 5.13 ctrl A
## 6 gene_1 6 6.72 ctrl B
## 7 gene_1 7 5.46 cond A
## 8 gene_1 8 3.73 cond B
## 9 gene_1 9 4.31 cond A
## 10 gene_1 10 4.55 cond B
## 11 gene_1 11 6.22 cond A
## 12 gene_1 12 5.36 cond B
mod_i <- lm(expression ~ condition * cell_type, data = x_i)
summary(mod_i)
##
## Call:
## lm(formula = expression ~ condition * cell_type, data = x_i)
##
## Residuals:
## Min 1Q Median 3Q Max
## -1.0196 -0.7652 -0.1210 0.8304 1.1966
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 5.33272 0.56643 9.415 1.33e-05 ***
## conditionctrl 0.04312 0.80105 0.054 0.958
## cell_typeB -0.78302 0.80105 -0.977 0.357
## conditionctrl:cell_typeB 0.92564 1.13286 0.817 0.438
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.9811 on 8 degrees of freedom
## Multiple R-squared: 0.1824, Adjusted R-squared: -0.1242
## F-statistic: 0.595 on 3 and 8 DF, p-value: 0.6357
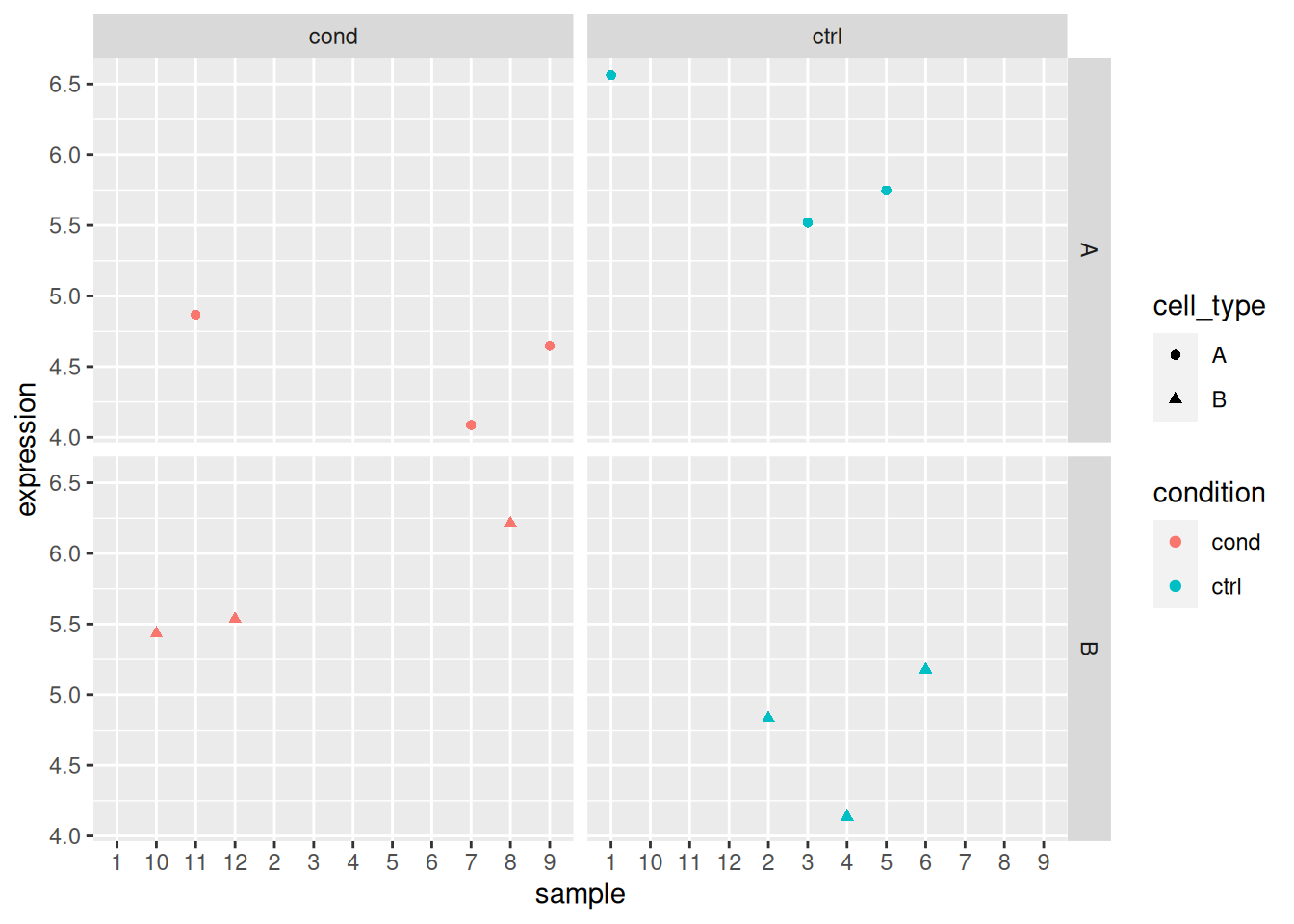
ggplot(x_i, aes(x = sample, y = expression,
colour = condition,
shape = cell_type)) +
geom_point()
## Analyse gene 2
x_i <- filter(x, rowname == "gene_2")
x_i
## # A tibble: 12 × 5
## rowname sample expression condition cell_type
## <chr> <chr> <dbl> <chr> <chr>
## 1 gene_2 1 6.16 ctrl A
## 2 gene_2 2 6.04 ctrl B
## 3 gene_2 3 5.78 ctrl A
## 4 gene_2 4 6.71 ctrl B
## 5 gene_2 5 6.20 ctrl A
## 6 gene_2 6 5.21 ctrl B
## 7 gene_2 7 4.70 cond A
## 8 gene_2 8 3.53 cond B
## 9 gene_2 9 2.93 cond A
## 10 gene_2 10 3.78 cond B
## 11 gene_2 11 2.97 cond A
## 12 gene_2 12 3.27 cond B
mod_i <- lm(expression ~ condition * cell_type, data = x_i)
summary(mod_i)
##
## Call:
## lm(formula = expression ~ condition * cell_type, data = x_i)
##
## Residuals:
## Min 1Q Median 3Q Max
## -0.77745 -0.34149 0.02695 0.17889 1.16551
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 3.535847 0.376883 9.382 1.36e-05 ***
## conditionctrl 2.509857 0.532992 4.709 0.00152 **
## cell_typeB -0.009063 0.532992 -0.017 0.98685
## conditionctrl:cell_typeB -0.045843 0.753765 -0.061 0.95300
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.6528 on 8 degrees of freedom
## Multiple R-squared: 0.8448, Adjusted R-squared: 0.7866
## F-statistic: 14.52 on 3 and 8 DF, p-value: 0.001335
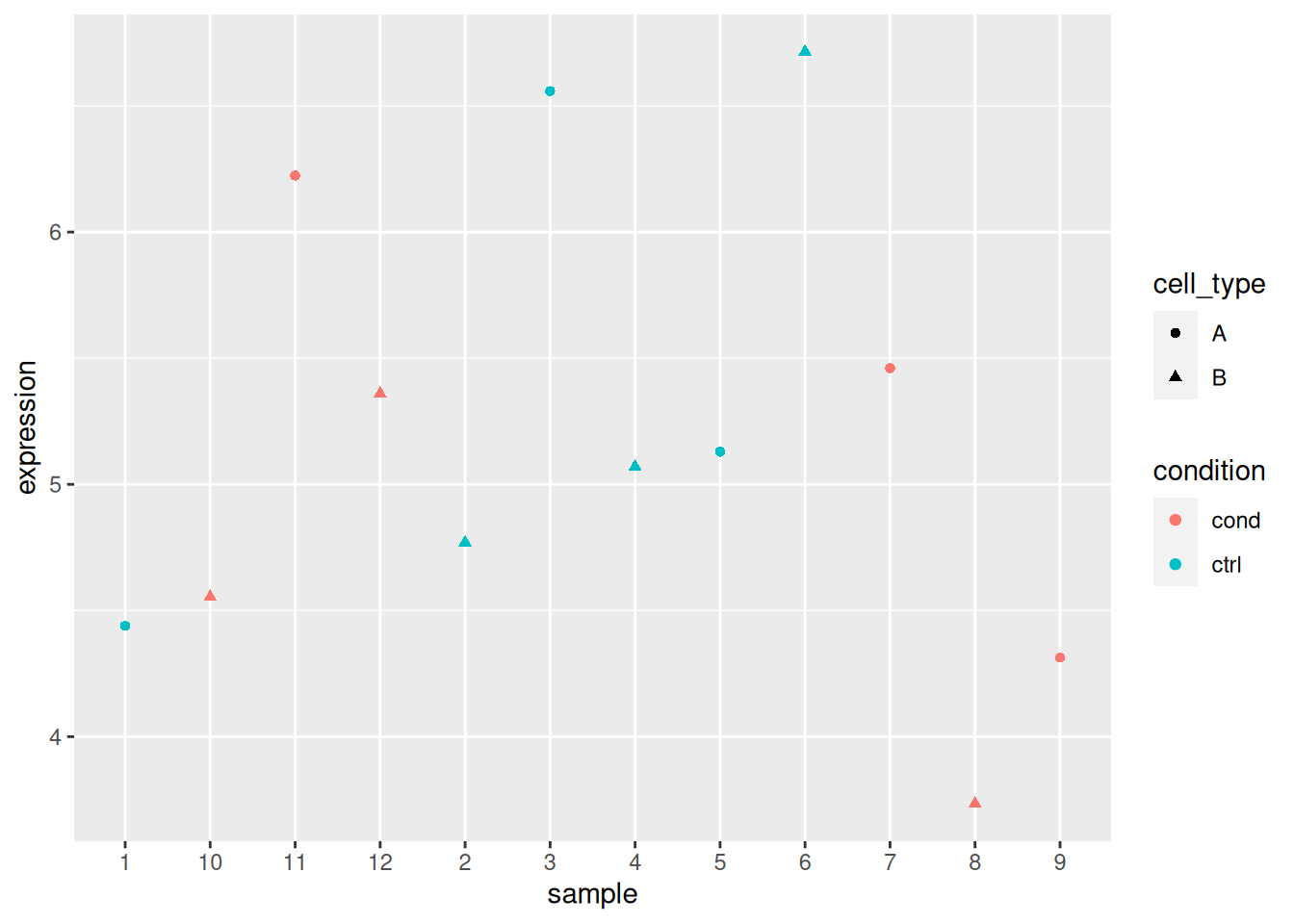
ggplot(x_i, aes(x = sample, y = expression, colour = cell_type)) +
geom_point() +
facet_wrap(~ condition)
## Analyse gene 3
x_i <- filter(x, rowname == "gene_3")
x_i
## # A tibble: 12 × 5
## rowname sample expression condition cell_type
## <chr> <chr> <dbl> <chr> <chr>
## 1 gene_3 1 2.69 ctrl A
## 2 gene_3 2 3.31 ctrl B
## 3 gene_3 3 3.42 ctrl A
## 4 gene_3 4 5.15 ctrl B
## 5 gene_3 5 2.43 ctrl A
## 6 gene_3 6 6.25 ctrl B
## 7 gene_3 7 3.21 cond A
## 8 gene_3 8 4.70 cond B
## 9 gene_3 9 3.45 cond A
## 10 gene_3 10 5.88 cond B
## 11 gene_3 11 3.41 cond A
## 12 gene_3 12 5.69 cond B
mod_i <- lm(expression ~ condition * cell_type, data = x_i)
summary(mod_i)
##
## Call:
## lm(formula = expression ~ condition * cell_type, data = x_i)
##
## Residuals:
## Min 1Q Median 3Q Max
## -1.59352 -0.22242 0.07198 0.31211 1.34698
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 3.357193 0.490117 6.850 0.000131 ***
## conditionctrl -0.511427 0.693130 -0.738 0.481685
## cell_typeB 2.066707 0.693130 2.982 0.017555 *
## conditionctrl:cell_typeB -0.005643 0.980234 -0.006 0.995547
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.8489 on 8 degrees of freedom
## Multiple R-squared: 0.7019, Adjusted R-squared: 0.5901
## F-statistic: 6.278 on 3 and 8 DF, p-value: 0.01696
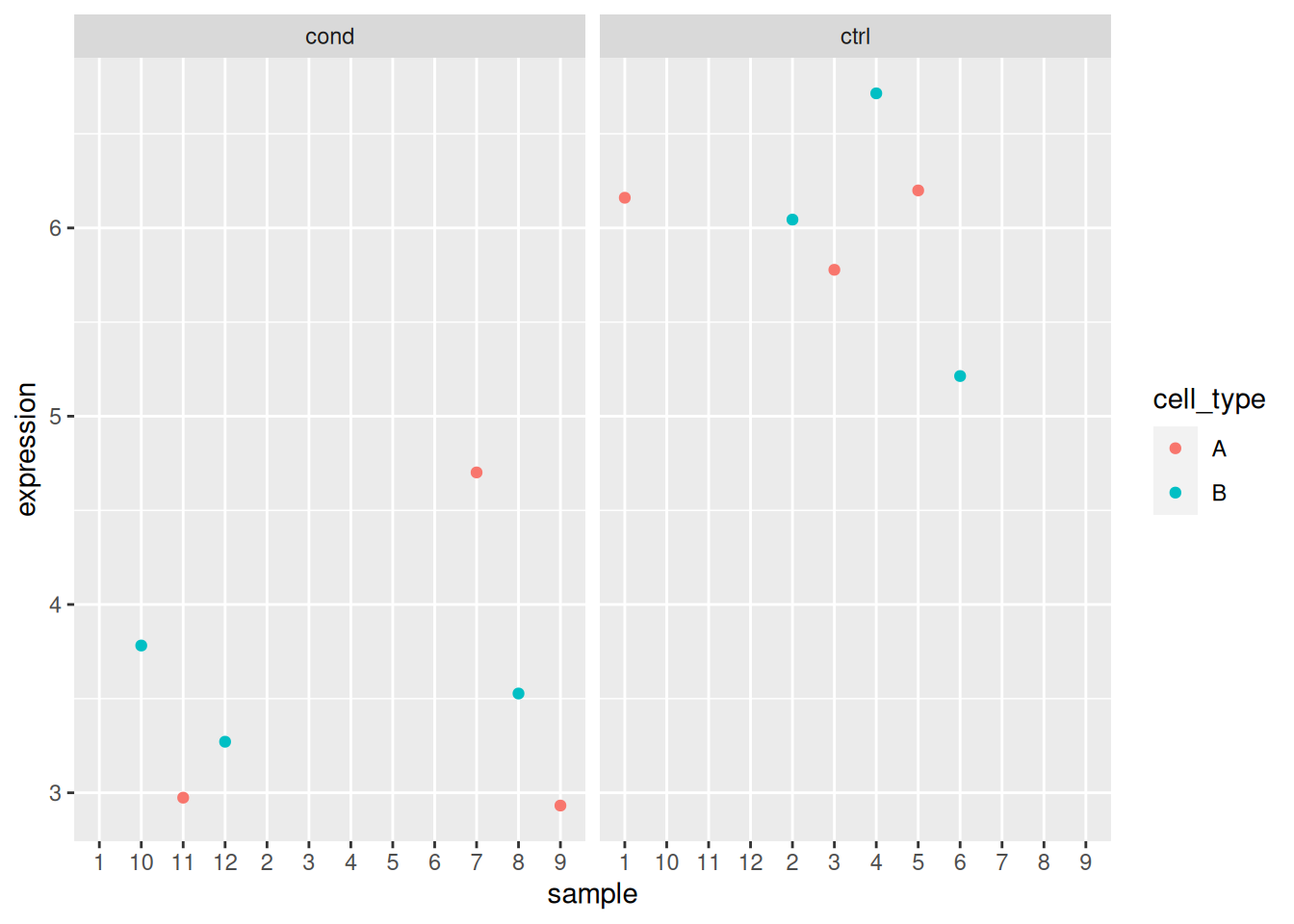
ggplot(x_i, aes(x = sample, y = expression, colour = condition)) +
geom_point() +
facet_wrap(~ cell_type)
## Analyse gene 4
x_i <- filter(x, rowname == "gene_4")
x_i
## # A tibble: 12 × 5
## rowname sample expression condition cell_type
## <chr> <chr> <dbl> <chr> <chr>
## 1 gene_4 1 7.11 ctrl A
## 2 gene_4 2 5.88 ctrl B
## 3 gene_4 3 5.39 ctrl A
## 4 gene_4 4 5.24 ctrl B
## 5 gene_4 5 4.61 ctrl A
## 6 gene_4 6 5.58 ctrl B
## 7 gene_4 7 3.47 cond A
## 8 gene_4 8 10.3 cond B
## 9 gene_4 9 8.42 cond A
## 10 gene_4 10 3.75 cond B
## 11 gene_4 11 5.19 cond A
## 12 gene_4 12 5.07 cond B
mod_i <- lm(expression ~ condition * cell_type, data = x_i)
summary(mod_i)
##
## Call:
## lm(formula = expression ~ condition * cell_type, data = x_i)
##
## Residuals:
## Min 1Q Median 3Q Max
## -2.6323 -1.1485 -0.3208 0.5837 3.9518
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 5.693120 1.296865 4.390 0.00232 **
## conditionctrl 0.009047 1.834044 0.005 0.99619
## cell_typeB 0.693007 1.834044 0.378 0.71537
## conditionctrl:cell_typeB -0.828703 2.593730 -0.320 0.75753
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 2.246 on 8 degrees of freedom
## Multiple R-squared: 0.02982, Adjusted R-squared: -0.334
## F-statistic: 0.08197 on 3 and 8 DF, p-value: 0.968
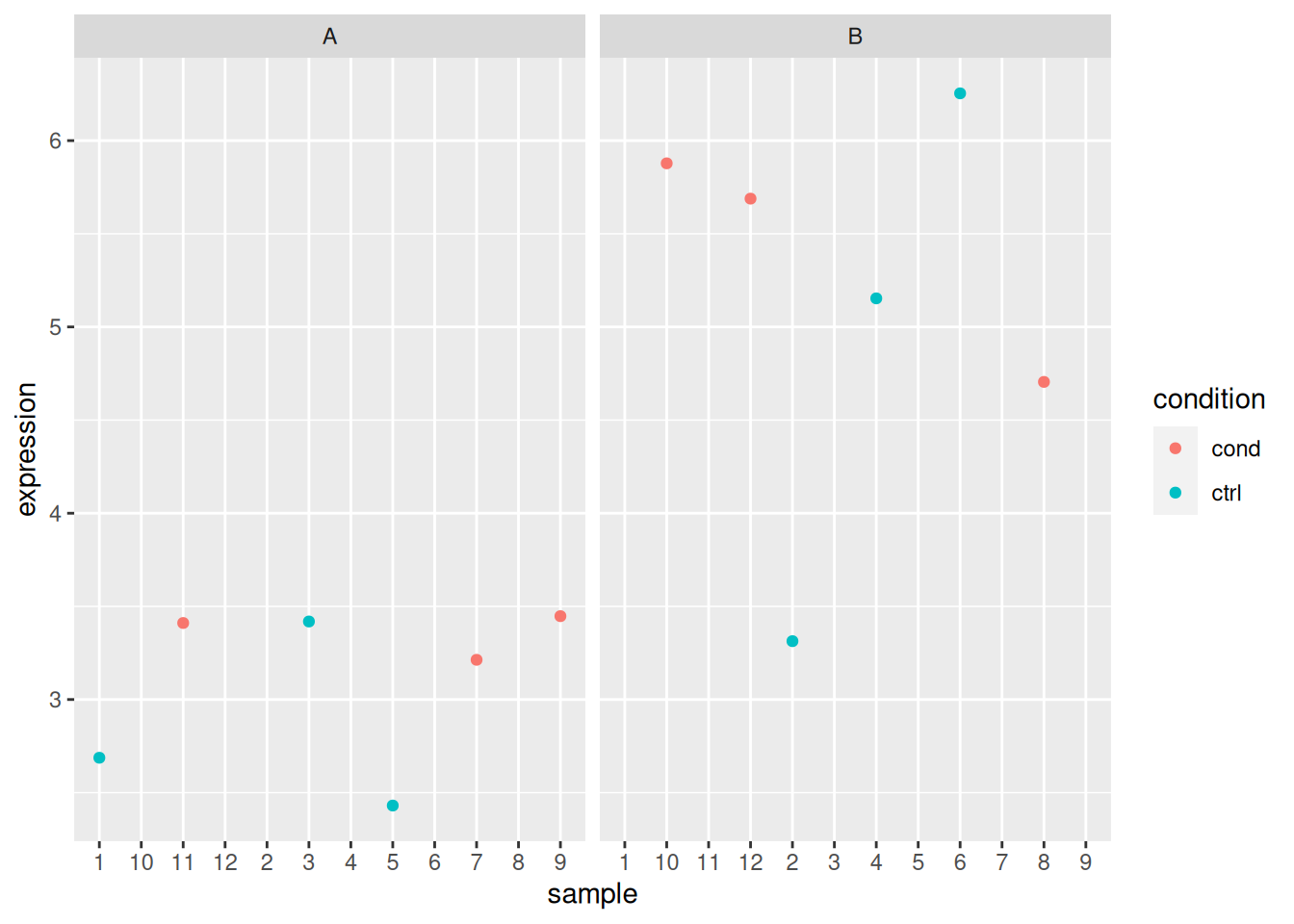
ggplot(x_i, aes(x = sample, y = expression,
colour = condition,
shape = cell_type)) +
geom_point()
## Analyse gene 5
x_i <- filter(x, rowname == "gene_5")
x_i
## # A tibble: 12 × 5
## rowname sample expression condition cell_type
## <chr> <chr> <dbl> <chr> <chr>
## 1 gene_5 1 6.56 ctrl A
## 2 gene_5 2 4.83 ctrl B
## 3 gene_5 3 5.52 ctrl A
## 4 gene_5 4 4.14 ctrl B
## 5 gene_5 5 5.75 ctrl A
## 6 gene_5 6 5.18 ctrl B
## 7 gene_5 7 4.09 cond A
## 8 gene_5 8 6.21 cond B
## 9 gene_5 9 4.65 cond A
## 10 gene_5 10 5.43 cond B
## 11 gene_5 11 4.87 cond A
## 12 gene_5 12 5.54 cond B
mod_i <- lm(expression ~ condition * cell_type, data = x_i)
summary(mod_i)
##
## Call:
## lm(formula = expression ~ condition * cell_type, data = x_i)
##
## Residuals:
## Min 1Q Median 3Q Max
## -0.57907 -0.32745 -0.03921 0.36578 0.61935
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 4.5338 0.2775 16.338 1.98e-07 ***
## conditionctrl 1.4092 0.3924 3.591 0.00708 **
## cell_typeB 1.1929 0.3924 3.040 0.01607 *
## conditionctrl:cell_typeB -2.4201 0.5550 -4.361 0.00241 **
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.4806 on 8 degrees of freedom
## Multiple R-squared: 0.7094, Adjusted R-squared: 0.6005
## F-statistic: 6.511 on 3 and 8 DF, p-value: 0.01536
group_by(x_i, cell_type, condition) |> summarise(mean = mean(expression))
## `summarise()` has grouped output by 'cell_type'. You can override using the
## `.groups` argument.
## # A tibble: 4 × 3
## # Groups: cell_type [2]
## cell_type condition mean
## <chr> <chr> <dbl>
## 1 A cond 4.53
## 2 A ctrl 5.94
## 3 B cond 5.73
## 4 B ctrl 4.72
group_by(x_i, cell_type) |> summarise(mean = mean(expression))
## # A tibble: 2 × 2
## cell_type mean
## <chr> <dbl>
## 1 A 5.24
## 2 B 5.22
group_by(x_i, condition) |> summarise(mean = mean(expression))
## # A tibble: 2 × 2
## condition mean
## <chr> <dbl>
## 1 cond 5.13
## 2 ctrl 5.33
ggplot(x_i, aes(x = sample, y = expression,
colour = condition,
shape = cell_type)) +
geom_point() +
facet_grid(cell_type ~ condition)