3-Beta-Hydroxysteroid Dehydrogenase Deficiency: Background, Pathophysiology, Etiology (original) (raw)
Overview
Background
3-Beta–hydroxysteroid dehydrogenase (3BHSD) deficiency is a rare form of congenital adrenal hyperplasia that results in decreased production of all three groups of adrenal steroids: mineralocorticoids, glucocorticoids, and sex steroids. In severely affected individuals, decreased mineralocorticoid secretion results in varying degrees of salt wasting in both males and females, and deficient androgen production results in ambiguous genitalia in 46,XY males. Although first described in male infants with ambiguous genitalia and severe salt wasting, 3-beta–hydroxysteroid dehydrogenase deficiency also occurs in 46,XX female infants (who may have mild clitoromegaly), as well as in older patients who present with a milder or so-called late-onset variant. [1]
This condition is due to defects in type II 3-beta–hydroxysteroid dehydrogenase, an enzyme that occurs almost exclusively in the gonads and adrenal glands. A variety of mutations in the HSD3B2 gene affect the activity of this enzyme, resulting in the extremely variable, phenotypic presentations of 3-beta–hydroxysteroid dehydrogenase deficiency. [2, 3]
- With severe deficiency, the most common presentation is that of a newborn infant with adrenal insufficiency due to both glucocorticoid and mineralocorticoid deficiency and ambiguous genitalia in 46,XY patients. Infants with less severe (non–salt-wasting) forms may be relatively asymptomatic.
- Older patients with mild defects in 3-beta–hydroxysteroid dehydrogenase activity (late-onset or nonclassic variant) may present with premature pubic hair development, hirsutism, irregular menstrual cycles or primary amenorrhea.
![]()
Pathophysiology
Anatomically, the adrenal gland can be divided into three zones, (1) the zona glomerulosa, which predominately produces mineralocorticoid, (2) the zona fasciculata, which predominately produces glucocorticoid, and (3) the zona reticularis, which predominantly produces androgens. Think of the zona glomerulosa and the zonae fasciculata and reticularis as two separate endocrine organs because they are under separate control. Aldosterone (mineralocorticoid) synthesis and secretion is regulated via the renin-angiotensin system, which is responsive to the state of electrolyte balance and the plasma volume. Aldosterone secretion is also directly stimulated by high serum potassium concentrations. By contrast, cortisol synthesis and secretion is regulated by adrenocorticotropic hormone (ACTH), which stimulates the enzyme P-450scc (20,22 desmolase), with subsequent increased production of all adrenal steroids in both the zona fasciculata and the zona reticularis (see image below).
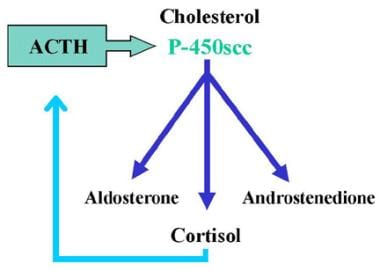
3-Beta-Hydroxysteroid Dehydrogenase Deficiency. Normal adrenal steroid biosynthesis results in 3 products: mineralocorticoid (aldosterone), glucocorticoids (cortisol), and androgens (androstenedione). Cortisol production is regulated by feedback with adrenocorticotropic hormone (ACTH). ACTH stimulates the enzyme P-450scc (20,22 desmolase) with subsequent increased production of all adrenal steroids.
Congenital adrenal hyperplasia (CAH) is a family of autosomal recessive disorders of adrenal steroid biosynthesis [4] in which activity of one of the enzymes necessary for cortisol production is deficient (see image below). Decreased serum cortisol levels stimulate ACTH release via negative feedback. The adrenal glands undergo hypertrophy, apparently because of ACTH-stimulated production of insulinlike growth factor–2 (IGF-2). Increased ACTH secretion also produces overproduction of both the adrenal steroids preceding the missing enzyme and those not requiring the missing enzyme (ie, build-up of compounds both before the block and "sideways" from the block). Treatment with exogenous glucocorticoid results in decreased ACTH secretion and subsequent suppression of the overproduced steroids.
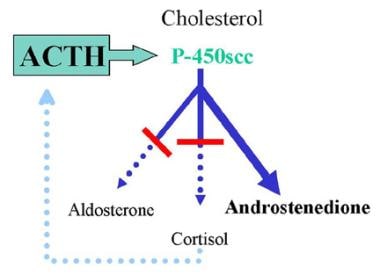
3-Beta-Hydroxysteroid Dehydrogenase Deficiency. Representation of typical congenital adrenal hyperplasia (CAH). In this example, both the mineralocorticoid and glucocorticoid pathways are deficient. Decreased serum cortisol levels stimulate adrenocorticotropic hormone (ACTH) release via negative feedback. Increased ACTH secretion results in overproduction of adrenal steroids preceding the missing enzyme as well as those not requiring the missing enzyme. In this example, a deficiency of 21-hydroxylase results in deficient mineralocorticoid and glucocorticoid production and excessive androgen production.
An 8-kilobase (kb) gene, HSD3B2, located on the p11-13 region of chromosome 1 encodes 3-beta–hydroxysteroid dehydrogenase. [5] Two isoenzymes of 3-beta–hydroxysteroid dehydrogenase have been described, differing by only 23 amino acids.
Type I 3-beta–hydroxysteroid dehydrogenase isoenzyme occurs in the peripheral tissues, primarily the liver. Deficiencies of this isoenzyme primarily effect bile acid metabolism and may present with cholestasis, hepatomegaly, steatorrhea, failure to thrive, and low serum levels of the fat soluble vitamins A, E, and D. [6]
Type II 3-beta–hydroxysteroid dehydrogenase occurs almost exclusively in the gonads and adrenal glands [7] and is the focus of this review. Various mutations in the HSD3B2 gene have been shown to be responsible for the varying phenotypic presentations. [8]
Patients with classic 3-beta–hydroxysteroid dehydrogenase deficiency have various nonconservative missense, nonsense, splicing, and frameshift mutations in the type II 3-beta–hydroxysteroid dehydrogenase gene with no mutation in the type I gene. Such mutations of the type II isoenzyme typically occur in gene domains essential for normal enzyme activity, but in vitro enzyme activity does not necessarily correlate with genital phenotype. [9]
Missense mutations in the type II gene have been described in nonclassic late-onset 3-beta–hydroxysteroid dehydrogenase deficiency. Various mutations have been described in the type II gene, including T259M and G129R/P222Q mutations in female patients and P222Q in a male patient with salt-wasting.
The synthesis of all three groups of adrenal steroids requires 3-beta–hydroxysteroid dehydrogenase. The adrenal steroids are mineralocorticoids, glucocorticoids, and sex steroids. 3-beta–hydroxysteroid dehydrogenase catalyzes the 3-beta-dehydrogenation and isomerization of the double bond of the steroid B ring to the steroid A ring, converting pregnenolone to progesterone (mineralocorticoid pathway), 17-alpha-hydroxypregnenolone to 17-alpha-hydroxyprogesterone (glucocorticoid pathway), and dehydroepiandrosterone (DHEA) to androstenedione (sex steroid pathway). See image below.
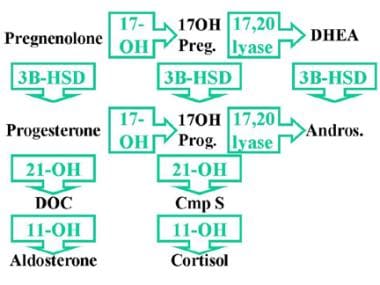
3-Beta-Hydroxysteroid Dehydrogenase Deficiency. 3-beta-hydroxysteroid dehydrogenase (3BHSD) is required for the synthesis of all three groups of adrenal steroids: mineralocorticoids, glucocorticoids, and sex steroids. 3BHSD catalyzes the conversion of pregnenolone to progesterone (mineralocorticoid pathway), 17-alpha-hydroxypregnenolone to 17-alpha-hydroxyprogesterone (glucocorticoid pathway), and dehydroepiandrosterone to androstenedione (sex steroid pathway). Complete absence of this enzyme thus impairs all steroid production. 17OH Preg = 17-alpha-hydroxypregnenolone; DHEA = Dehydroepiandrosterone; 17OH Prog = 17-alpha-hydroxyprogesterone; Andros = Androstenedione; DOC = Deoxycorticosterone; Cmp S = Compound S.
Therefore, absence of this enzyme impairs all steroid production. Low levels of cortisol result in increased ACTH stimulation of steroids prior to the 3-beta–hydroxysteroid dehydrogenase step, producing increased accumulation and secretion of pregnenolone, 17-alpha-hydroxypregnenolone, and DHEA. Adrenal insufficiency occurs secondary to aldosterone and cortisol deficiency. Reduced sex steroid production leads to ambiguous external genitalia in 46,XY individuals; some virilization may occur in 46,XX infants or in older children of either sex because of excessive DHEA production.
Affected 46,XX infants appear normal or may have mild-to-moderate clitoromegaly due to either direct androgen effects of elevated DHEA or peripheral conversion of excess DHEA to testosterone via peripheral type I 3-beta–hydroxysteroid dehydrogenase isoenzyme. Effects of excessive androgen activity in older 46,XX children include acne, premature pubarche, and advanced linear and skeletal growth.
By contrast, 46,XY infants present with varying degrees of ambiguous genitalia due to defective androgen production. 46,XY individuals with milder defects may present as adolescents with ambiguous genitalia, poor virilization, and gynecomastia. Virilization or spontaneous puberty has been reported in occasional male patients secondary to either direct effects of DHEA or to sufficient conversion of DHEA to testosterone via peripheral type I 3-beta–hydroxysteroid dehydrogenase isoenzyme. 3-beta–hydroxysteroid dehydrogenase activity may vary in the gonadal, adrenal, and peripheral tissues within the same individual. [10] At least one patient has been reported with partial 3-beta–hydroxysteroid dehydrogenase activity in the testes coupled with complete absence of adrenal 3-beta–hydroxysteroid dehydrogenase activity.
Finally, a deficiency in the related 3-alpha-hydrozysteroid dehydrogenase may also play a role in hirsutism. 3-alpha HSD is encoded by the AKR1C2 gene and is required for normal metabolism of dihydrotestosterone (DHT) in peripheral tissues. Deficient 3-alpha HSD activity may lead to increased tissue levels of DHT and subsequent hirsutism. [11]
![]()
Etiology
3-beta–hydroxysteroid dehydrogenase deficiency is inherited as an autosomal recessive trait. 3-beta–hydroxysteroid dehydrogenase is encoded by an 8-kb gene located on the p11-13 region of chromosome 1. Two isoenzymes of 3-beta–hydroxysteroid dehydrogenase have been described, [12] differing by only 23 amino acids. Type I 3-beta–hydroxysteroid dehydrogenase isoenzyme occurs in the peripheral tissues, primarily the liver but including the aorta, and type II 3-beta–hydroxysteroid dehydrogenase almost exclusively occurs in the gonads and adrenal glands.
Type I 3-beta–hydroxysteroid dehydrogenase isoenzyme is normal in patients with type II 3-beta–hydroxysteroid dehydrogenase deficiency. At least 31 different mutations in the type II 3-beta–hydroxysteroid dehydrogenase gene have been identified in 32 unrelated families with 3-beta–hydroxysteroid dehydrogenase deficiency.
Patients with classic salt-losing 3-beta–hydroxysteroid dehydrogenase deficiency have been shown to have various mutations, including splicing (1 patient), in-frame (1 patient), nonsense (3 patients), frameshift (4 patients), and missense (22 patients) mutations in the type II 3-beta–hydroxysteroid dehydrogenase gene with no mutation in the type I gene. [12]
No functional 3-beta–hydroxysteroid dehydrogenase type II enzyme is found in the adrenals or gonads of patients with severe salt-losing disease. The non–salt-losing form can occur with a missense mutation causing only partial deficiency in enzyme activity. [13]
Different missense mutations of the type II 3-beta–hydroxysteroid dehydrogenase gene (p.Y190C and p.S218P) have been identified in a female patient with late-onset 3-beta–hydroxysteroid dehydrogenase deficiency. [14]
![]()
Epidemiology
Most individuals worldwide with CAH have 21-hydroxylase deficiency (80-90%). The incidence of classic 21-hydroxylase deficiency varies by population and ranges from 1 case per 5000-15,000 live births to as high as 1 case per 300-700 births in Alaskan Yupik Eskimos. The next most common type of CAH, 11-beta-hydroxylase deficiency, has an incidence of about 1 in 100,000 persons. Approximately 60 unrelated families affected by 3-beta–hydroxysteroid dehydrogenase deficiency have been reported. [15]
In one study of 81 children with ambiguous genitalia, only two were found to have 3-beta–hydroxysteroid dehydrogenase deficiency. [16] The incidence is higher in some populations; 3-beta–hydroxysteroid dehydrogenase deficiency is relatively common in the Old Order Amish in North America associated with a HSD3B2 c.35G, a founder mutation. [17]
Mild 3-beta–hydroxysteroid dehydrogenase defects are probably rare because most children with premature appearance of pubic hair (pubarche) or older women with irregular menstrual cycles and hirsutism and mildly elevated DHEA or 17-hydroxypregnenolone levels only rarely have mutations in the 3-beta–hydroxysteroid dehydrogenase II gene. For example, in 1996, Sakkal-Alkaddour et al reported normal type II 3-beta–hydroxysteroid dehydrogenase gene sequences in 15 infants and children with premature pubarche and mildly elevated DHEA levels. [18] Among 30 women with hirsutism and elevated baseline (unstimulated or random) DHEA levels, none had ACTH-stimulated increases in 17-alpha-hydroxypregnenolone and had DHEA levels consistent with elevations typically observed in genetically proven classic 3-beta–hydroxysteroid dehydrogenase deficiency.
![]()
Prognosis
3-Beta–hydroxysteroid dehydrogenase is required for the synthesis of all three groups of adrenal steroids, which are mineralocorticoids, glucocorticoids, and sex steroids. Therefore, absence of this enzyme impairs all steroid production, and adrenal insufficiency occurs secondary to aldosterone and cortisol deficiency.
A great deal of heterogeneity is observed with 3-beta–hydroxysteroid dehydrogenase deficiency. The most severely affected patients may have fatal salt-losing adrenal crises in infancy. By contrast, some patients with classic 3-beta–hydroxysteroid dehydrogenase deficiency do not have salt-losing crises; milder or late-onset variants have also been described in which patients do not present until later childhood or adolescence.
Prognosis is usually good-to-excellent with adequate replacement glucocorticoid and mineralocorticoid (if needed) therapy and monitoring.
Sex steroid replacement may be necessary for the development of secondary sexual characteristics in both males and females and cyclic menstrual bleeding in females. Decreased spermatogenesis may occur in affected males, [12] although fertility has been reported in men who receive treatment. [15] In postpubertal females with late-onset 3-beta–hydroxysteroid dehydrogenase deficiency, menstrual irregularity and infertility may correct with glucocorticoid replacement alone.
Benign testicular adrenal rest tumors are found in adult men in association with poorly controlled congenital adrenal hyperplasia (CAH). Such men may have gonadal dysfunction and infertility, perhaps due to obstruction of seminiferous tubules. [19] High-resolution ultrasonography has recently been used to estimate the prevalence of testicular adrenal rest tumors in male children with CAH, with a reported incidence ranging from 21-24%. [20] Although the testes are by far the most common location for such rest tumors, ectopic adrenal rest tumors may be present elsewhere. [21]
![]()
Patient Education
Patients with complete 3-beta–hydroxysteroid dehydrogenase deficiency are at risk for acute adrenal insufficiency when ill. Patients and their families should be instructed in the use of stress doses of glucocorticoids for acute illness (eg, temperature ≥101°F) or major trauma. If medication can be taken orally, the patient should double or triple the usual dose of glucocorticoid for 3 days. Mineralocorticoid doses do not need to be increased. If the patient cannot take the medication orally because of vomiting, altered state of consciousness, or surgery, parenteral glucocorticoids, preferably hydrocortisone, should be administered.
Patients should wear MedicAlert identification and be taken to their local health care provider as soon as possible when acutely ill for evaluation.
![]()
- Grumbach MM, Conte FA. Disorders of sex differentiation. Williams Textbook of Endocrinology. 8th ed. Philadelphia, PA: WB Saunders Co; 1992. 853-951.
- Menegatti E, Tessaris D, Barinotti A, Matarazzo P, Einaudi S. Genetic testing for a patient with suspected 3 beta-hydroxysteroid dehydrogenase deficiency: a case of unreported genetic variants. J Clin Med. 2022 Sep 29. 11(19):5767. [QxMD MEDLINE Link]. [Full Text].
- Alkhatib EH, Adams SD, Miller ER. Case of an unreported genetic variant of salt losing 3-β-hydroxysteroid dehydrogenase deficiency. Oxf Med Case Reports. 2021 May. 2021(5):omab021. [QxMD MEDLINE Link]. [Full Text].
- Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009 Apr. 23(2):181-92. [QxMD MEDLINE Link].
- Moisan AM, Ricketts ML, Tardy V, et al. New insight into the molecular basis of 3beta-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3B2 gene eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzym. J Clin Endocrinol Metab. 1999 Dec. 84(12):4410-25. [QxMD MEDLINE Link]. [Full Text].
- Subramaniam P, Clayton PT, Portmann BC, Mieli-Vergani G, Hadzic N. Variable clinical spectrum of the most common inborn error of bile acid metabolism--3beta-hydroxy-Delta 5-C27-steroid dehydrogenase deficiency. J Pediatr Gastroenterol Nutr. 2010 Jan. 50(1):61-6. [QxMD MEDLINE Link].
- Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr Rev. 2005 Jun. 26(4):525-82. [QxMD MEDLINE Link].
- Simard J, Moisan AM, Morel Y. Congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase/Delta(5)-Delta(4) isomerase deficiency. Semin Reprod Med. 2002 Aug. 20(3):255-76. [QxMD MEDLINE Link].
- Welzel M, Wustemann N, Simic-Schleicher G, et al. Carboxyl-terminal mutations in 3beta-hydroxysteroid dehydrogenase type II cause severe salt-wasting congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2008 Apr. 93(4):1418-25. [QxMD MEDLINE Link].
- Schneider G, Genel M, Bongiovanni AM, et al. Persistent testicular delta5-isomerase-3beta-hydroxysteroid dehydrogenase (delta5-3beta-HSD) deficiency in the delta5-3beta-HSD form of congenital adrenal hyperplasia. J Clin Invest. 1975 Apr. 55(4):681-90. [QxMD MEDLINE Link]. [Full Text].
- Steiner AZ, Chang L, Ji Q, et al. 3alpha-Hydroxysteroid dehydrogenase type III deficiency: a novel mechanism for hirsutism. J Clin Endocrinol Metab. 2008 Apr. 93(4):1298-303. [QxMD MEDLINE Link].
- Burckhardt MA, Udhane SS, Marti N, et al. Human 3β-hydroxysteroid dehydrogenase deficiency seems to affect fertility but may not harbor a tumor risk: lesson from an experiment of nature. Eur J Endocrinol. 2015 Nov. 173(5):K1-K12. [QxMD MEDLINE Link].
- Sanchez R, Rheaume E, Laflamme N, et al. Detection and functional characterization of the novel missense mutation Y254D in type II 3 beta-hydroxysteroid dehydrogenase (3 beta HSD) gene of a female patient with nonsalt-losing 3 beta HSD deficiency. J Clin Endocrinol Metab. 1994 Mar. 78(3):561-7. [QxMD MEDLINE Link].
- Takasawa K, Ono M, Hijikata A, et al. Two novel HSD3B2 missense mutations with diverse residual enzymatic activities for Δ5-steroids. Clin Endocrinol (Oxf). 2014 Jun. 80(6):782-9. [QxMD MEDLINE Link].
- Donadille B, Houang M, Netchine I, Siffroi JP, Christin-Maitre S. Human 3beta-hydroxysteroid dehydrogenase deficiency associated with normal spermatic numeration despite a severe enzyme deficit. Endocr Connect. 2018 Mar. 7(3):395-402. [QxMD MEDLINE Link]. [Full Text].
- Al-Jurayyan NA. Ambiguous genitalia: two decades of experience. Ann Saudi Med. 2011 May-Jun. 31(3):284-8. [QxMD MEDLINE Link]. [Full Text].
- Benkert AR, Young M, Robinson D, Hendrickson C, Lee PA, Strauss KA. Severe salt-losing 3β-hydroxysteroid dehydrogenase deficiency: treatment and outcomes of HSD3B2 c.35G>A homozygotes. J Clin Endocrinol Metab. 2015 Aug. 100(8):E1105-15. [QxMD MEDLINE Link]. [Full Text].
- Sakkal-Alkaddour H, Zhang L, Yang X, et al. Studies of 3 beta-hydroxysteroid dehydrogenase genes in infants and children manifesting premature pubarche and increased adrenocorticotropin-stimulated delta 5-steroid levels. J Clin Endocrinol Metab. 1996 Nov. 81(11):3961-5. [QxMD MEDLINE Link].
- Claahsen-van der Grinten HL, Sweep FC, Blickman JG, Hermus AR, Otten BJ. Prevalence of testicular adrenal rest tumours in male children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol. 2007 Sep. 157(3):339-44. [QxMD MEDLINE Link].
- Martinez-Aguayo A, Rocha A, Rojas N, et al. Testicular adrenal rest tumors and Leydig and Sertoli cell function in boys with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2007 Dec. 92(12):4583-9. [QxMD MEDLINE Link].
- Claahsen-van der Grinten HL, Duthoi K, Otten BJ, d'Ancona FC, Hulsbergen-vd Kaa CA, Hermus AR. An adrenal rest tumour in the perirenal region in a patient with congenital adrenal hyperplasia due to congenital 3beta-hydroxysteroid dehydrogenase deficiency. Eur J Endocrinol. 2008 Oct. 159(4):489-91. [QxMD MEDLINE Link].
- Johannsen TH, Mallet D, Dige-Petersen H, et al. Delayed diagnosis of congenital adrenal hyperplasia with salt wasting due to type II 3beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2005 Apr. 90(4):2076-80. [QxMD MEDLINE Link]. [Full Text].
- Nordenstrom A, Forest MG, Wedell A. A case of 3beta-hydroxysteroid dehydrogenase type II (HSD3B2) deficiency picked up by neonatal screening for 21-hydroxylase deficiency: difficulties and delay in etiologic diagnosis. Horm Res. 2007. 68(4):204-8. [QxMD MEDLINE Link].
- Jeandron DD, Sahakitrungruang T. A novel homozygous Q334X mutation in the HSD3B2 gene causing classic 3ß-hydroxysteroid dehydrogenase deficiency: an unexpected diagnosis after a positive newborn screen for 21-hydroxylase deficiency. Horm Res Paediatr. 2012. 77(5):334-8. [QxMD MEDLINE Link].
- Marui S, Castro M, Latronico AC, et al. Mutations in the type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin Endocrinol (Oxf). 2000 Jan. 52(1):67-75. [QxMD MEDLINE Link].
- Mermejo LM, Elias LL, Marui S, Moreira AC, Mendonca BB, de Castro M. Refining hormonal diagnosis of type II 3beta-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2 genotyping. J Clin Endocrinol Metab. 2005 Mar. 90(3):1287-93. [QxMD MEDLINE Link]. [Full Text].
- Penny S, Gridley D and Young J. Congenital adrenal hyperplasia secondary to 3-beta hydroxysteroid dehydrogenase deficiency. Appl Radiol. Nov 2017. 46(11):46-47. [Full Text].
- [Guideline] Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010 Sep. 95(9):4133-60. [QxMD MEDLINE Link]. [Full Text].
- Han TS, Conway GS, Willis DS, Krone N, Rees DA, et al. Relationship between final height and health outcomes in adults with congenital adrenal hyperplasia: United Kingdom congenital adrenal hyperplasia adult study executive (CaHASE). J Clin Endocrinol Metab. 2014 Aug. 99 (8):E1547-55. [QxMD MEDLINE Link].
- Han TS, Krone N, Willis DS, et al, for the United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE). Quality of life in adults with congenital adrenal hyperplasia relates to glucocorticoid treatment, adiposity and insulin resistance: United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE). Eur J Endocrinol. 2013 Jun. 168(6):887-93. [QxMD MEDLINE Link]. [Full Text].
- Ross RJ, Rostami-Hodjegan A. Timing and type of glucocorticoid replacement in adult congenital adrenal hyperplasia. Horm Res. 2005. 64 Suppl 2:67-70. [QxMD MEDLINE Link].
- Bentsen D, Schwartz DS, Carpenter TO. Sonography of congenital adrenal hyperplasia due to partial deficiency of 3beta-hydroxysteroid dehydrogenase: a case report. Pediatr Radiol. 1997 Jul. 27(7):594-5. [QxMD MEDLINE Link].
- Moran C, Potter HD, Reyna R, et al. Prevalence of 3beta-hydroxysteroid dehydrogenase-deficient nonclassic adrenal hyperplasia in hyperandrogenic women with adrenal androgen excess. Am J Obstet Gynecol. 1999 Sep. 181(3):596-600. [QxMD MEDLINE Link].
- Morel Y, Mebarki F, Rheaume E, et al. Structure-function relationships of 3 beta-hydroxysteroid dehydrogenase: contribution made by the molecular genetics of 3 beta-hydroxysteroid dehydrogenase deficiency. Steroids. 1997 Jan. 62(1):176-84. [QxMD MEDLINE Link].
- Nakamura Y, Suzuki T, Inoue T, et al. 3beta-Hydroxysteroid dehydrogenase in human aorta. Endocr J. 2005 Feb. 52(1):111-5. [QxMD MEDLINE Link].
- Pang S, Carbunaru G, Haider A, et al. Carriers for type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) deficiency can only be identified by HSD3B2 genotype study and not by hormone test. Clin Endocrinol (Oxf). 2003 Mar. 58(3):323-31. [QxMD MEDLINE Link].
- Rheaume E, Simard J, Morel Y, et al. Congenital adrenal hyperplasia due to point mutations in the type II 3 beta-hydroxysteroid dehydrogenase gene. Nat Genet. 1992 Jul. 1(4):239-45. [QxMD MEDLINE Link].
- Rosler A, Levine LS, Schneider B, et al. The interrelationship of sodium balance, plasma renin activity and ACTH in congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1977 Sep. 45(3):500-12. [QxMD MEDLINE Link].
- 3-Beta-Hydroxysteroid Dehydrogenase Deficiency. Normal adrenal steroid biosynthesis results in 3 products: mineralocorticoid (aldosterone), glucocorticoids (cortisol), and androgens (androstenedione). Cortisol production is regulated by feedback with adrenocorticotropic hormone (ACTH). ACTH stimulates the enzyme P-450scc (20,22 desmolase) with subsequent increased production of all adrenal steroids.
- 3-Beta-Hydroxysteroid Dehydrogenase Deficiency. Representation of typical congenital adrenal hyperplasia (CAH). In this example, both the mineralocorticoid and glucocorticoid pathways are deficient. Decreased serum cortisol levels stimulate adrenocorticotropic hormone (ACTH) release via negative feedback. Increased ACTH secretion results in overproduction of adrenal steroids preceding the missing enzyme as well as those not requiring the missing enzyme. In this example, a deficiency of 21-hydroxylase results in deficient mineralocorticoid and glucocorticoid production and excessive androgen production.
- 3-Beta-Hydroxysteroid Dehydrogenase Deficiency. 3-beta-hydroxysteroid dehydrogenase (3BHSD) is required for the synthesis of all three groups of adrenal steroids: mineralocorticoids, glucocorticoids, and sex steroids. 3BHSD catalyzes the conversion of pregnenolone to progesterone (mineralocorticoid pathway), 17-alpha-hydroxypregnenolone to 17-alpha-hydroxyprogesterone (glucocorticoid pathway), and dehydroepiandrosterone to androstenedione (sex steroid pathway). Complete absence of this enzyme thus impairs all steroid production. 17OH Preg = 17-alpha-hydroxypregnenolone; DHEA = Dehydroepiandrosterone; 17OH Prog = 17-alpha-hydroxyprogesterone; Andros = Androstenedione; DOC = Deoxycorticosterone; Cmp S = Compound S.

Author
J Paul Frindik, MD, FACE Associate Professor, Department of Pediatrics, University of Arkansas for Medical Sciences College of Medicine
J Paul Frindik, MD, FACE is a member of the following medical societies: American Association of Clinical Endocrinologists
Disclosure: Nothing to disclose.
Specialty Editor Board
Mary L Windle, PharmD Adjunct Associate Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Nothing to disclose.
Barry B Bercu, MD Professor, Departments of Pediatrics, Molecular Pharmacology and Physiology, University of South Florida College of Medicine, All Children's Hospital
Barry B Bercu, MD is a member of the following medical societies: American Academy of Pediatrics, American Association of Clinical Endocrinologists, American Medical Association, American Pediatric Society, Association of Clinical Scientists, Endocrine Society, Florida Medical Association, Pediatric Endocrine Society, Society for Pediatric Research, Southern Society for Pediatric Research, Society for the Study of Reproduction, American Federation for Clinical Research, Pituitary Society
Disclosure: Nothing to disclose.
Chief Editor
Robert P Hoffman, MD Professor and Program Director, Department of Pediatrics, Ohio State University College of Medicine; Pediatric Endocrinologist, Division of Pediatric, Endocrinology, Diabetes, and Metabolism, Nationwide Children's Hospital
Robert P Hoffman, MD is a member of the following medical societies: American College of Pediatricians, American Diabetes Association, American Pediatric Society, Christian Medical and Dental Associations, Endocrine Society, Midwest Society for Pediatric Research, Pediatric Endocrine Society, Society for Pediatric Research
Disclosure: Nothing to disclose.