Mature Dendritic Cells Infected with Herpes Simplex Virus Type 1 Exhibit Inhibited T-Cell Stimulatory Capacity (original) (raw)
Abstract
Mature dendritic cells (DC) are the most potent antigen-presenting cells within the entire immune system. Interference with the function of these cells therefore constitutes a very powerful mechanism for viruses to escape immune responses. Several members of the Herpesviridae family have provided examples of such escape strategies, including interference with antigen presentation and production of homologous cytokines. In this study we investigated the infection of mature DC with herpes simplex virus type 1 (HSV-1) and the way in which infection alters the phenotype and function of mature DC. Interestingly, the T-cell-stimulatory capacity of these DC was strongly impaired. Furthermore, we demonstrated that HSV-1 leads to the specific degradation of CD83, a cell surface molecule which is specifically upregulated during DC maturation. These data indicate that HSV-1 has developed yet another novel mechanism to escape immune responses.
Herpes simplex virus type 1 (HSV-1) belongs to the Herpesviridae, a large and diverse family of vertebrate pathogens including several human pathogens such as HSV-1 and HSV-2 (23). Members of the Herpesviridae characteristically have a large double-stranded DNA genome, and virions consist of an icosahedral nucleocapsid surrounded by a lipid layer envelope. After the initial acute infection of the host, HSV-1 establishes a persistent infection. Such persistent infections are established when host immune responses fail to eliminate the virus which is produced during the acute stage of infection.
Dendritic cells (DC) are the most potent antigen-presenting cells and are also capable of stimulating immunologically naive T cells (4, 8). DC have to mature in order to become potent T-cell stimulators. As immature DC, they acquire antigens in peripheral tissues and migrate to the secondary lymphoid organs, where, as mature DC, they present processed peptides very efficiently to rare antigen-specific T cells. During this maturation, a variety of different DC-specific genes are induced and expressed. In particular, costimulatory and accessory molecules, as well as major histocompatibility complex (MHC) class I and class II molecules are upregulated during DC maturation (4, 8, 30, 37, 38).
In addition to these gene products, CD83, a cell surface protein representing one of the best markers for mature DC, is specifically upregulated during maturation (35, 39, 40). Although the precise function of CD83 remains elucidated, its upregulation during maturation, together with the upregulation of the costimulatory molecules, points to an important function. Moreover, we recently demonstrated that inhibition of CD83 cell surface expression on mature DC leads to a dramatic reduction of their T-cell stimulatory capacity (19).
DC are essential stimulators of antiviral immune responses. In the murine system, for instance, DC are the most effective antigen-presenting cells in stimulating recall cytotoxic T-lymphocyte (CTL) responses to Sendai virus, Moloney murine leukemia virus (17), HSV (14), and influenza virus (22). The ability of DC to also induce a protective immune response to viral infection has recently been demonstrated by adoptive transfer with lymphocytic choriomeningitis virus peptide-pulsed DC and with DC which constitutively expressed the same lymphocytic choriomeningitis virus epitope (20). DC were also found to be pivotal for the initiation of anti-influenza virus responses at the CTL level (13).
The exposure of human DC to influenza virus leads to an efficient infection in vitro, as shown by the expression of the viral antigens hemagglutinin (HA) and nonstructural protein 1 (NS1) (7). However, this infection does not lead to rapid cell death and only small amounts of infectious virus particles are produced. Interestingly, infected DC, but not macrophages, can induce CTL responses from purified blood CD8+ cells in the absence of exogenous cytokines (5).
For HSV-1, it has been shown that viral proteins are expressed which can interfere with the immune response. For instance, ICP47, an immediate-early protein, inhibits TAP function in human fibroblasts and keratinocytes (18) by binding via its N terminus to the cytosolic peptide binding domain of human transporter associated with antigen presentation (TAP) (1, 33, 34) and thereby inhibits MHC class I-mediated peptide presentation (36).
In this study we show that HSV-1-infected mature DC do not propagate infectious virus particles and, interestingly, that these infected DC have a dramatically reduced T-cell-stimulatory capacity. Furthermore, we demonstrate that HSV-1 leads to the specific degradation of CD83, a cell surface molecule which is specifically upregulated on mature DC, indicating a new strategy for viral immune evasion.
MATERIALS AND METHODS
Cells and culture.
Peripheral blood mononuclear cells (5 × 107) were isolated from healthy donors by sedimentation in Ficoll-Hypaque (Pharmacia Biotech, Uppsala, Sweden) and cultured in RPMI 1640 (BioWhittaker, Walkersville, Md.) supplemented with 1% human plasma from a single AB donor, glutamine (300 μg/ml), penicillin-streptomycin (20 μg/ml each), and 10 mM HEPES (pH 7.5) (Sigma, Deisenhofen, Germany). DC precursors were seeded onto immunoglobulin G (IgG)-coated (10 μg of Ig-globulin per ml from the Cohn fraction [Sigma]) culture dishes for 1 h. A first nonadherent fraction was harvested after 1 h, and a second was harvested after a further 7 h. Immature DC were generated using the cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF) (800 U/ml [Novartis Research Institute, Vienna, Austria]) and interleukin-4 (IL-4) (1,000 U/ml [Genzyme Corp., Cambridge, Mass.]). Fresh medium (3 ml) containing 4,000 U of GM-CSF and 5,000 U of IL-4 was added to the cell culture on day 3. On day 5, nonadherent cells were collected, counted, and transferred into new dishes and the final DC maturation was induced by adding tumor necrosis factor alpha (2.5 ng/ml [Boehringer Ingelheim, Vienna, Austria]), prostaglandin E2 (1 ng/ml [Cayman Chemical, Ann Arbor, Mich.]), GM-CSF (200 U/ml), and IL-4 (250 U/ml) to the medium.
HSV-1 infection of mature DC.
Mature DC were prepared from healthy donors, washed, and resuspended in RPMI 1640 at a density of 0.5 × 106 to 1 × 106 cells/ml. The cells were inoculated either with infectious HSV-1 (strain ang) (21) at a multiplicity of infection (MOI) of 0.01, 0.1, or 1 or with UV-inactivated HSV-1 for 1 h at 37°C. Following three washes, 0.5 × 106 cells were transferred into six-well plates and cultivated for a further 24 h. Control experiments were performed in the presence of cycloheximide (CHI) (Sigma) at a final concentration of 10 μg/ml.
FACS analyses.
The mature DC phenotype was analyzed by fluorescence-activated cell sorting (FACS) using the following monoclonal antibodies: CD83 (Immunotech, Marseilles, France); CD25, CD80, and CD86 (Becton-Dickinson, Heidelberg, Germany); and CD40, CD95, MHC class I, and MHC class II (PharMingen, Hamburg, Germany). As isotype controls, IgG1a, IgG2a and IgG2b were used (Becton-Dickinson) and run in parallel. Then 10 h postinfection the HSV-1 infection efficacy was determined using a VP16-specific antibody in combination with intracellular FACS staining (3).
Reverse transcription-PCR.
Total cellular RNA was isolated from DC (106) infected with HSV-1 at a MOI of 0.01, 0.1, or 1, using TRIzol (Gibco-Life Technologies, Eggenstein, Germany). Subsequently, the RNA was reverse transcribed into single-stranded cDNA by using avian myeloblastosis virus reverse transcriptase as specified by the manufacturer (Roche Molecular Biochemicals, Mannheim, Germany). These cDNAs were used as PCR templates to amplify the viral transcripts ICP27, ICP8, and glycoprotein G (gG), as well as CD83- and S14-specific fragments.
The following PCR primers were used: ICP27 sense (5′-CGAGACCAGACGGGTCTCCTGG-3′) and antisense (5′-GCAGACACGACTCGAACACTCCTG-3′), ICP8 sense (5′-CTCGGTAACGACCAGATACAGGAGG-3′) and antisense (5′-CCAAGACGGCAACCACCATCAAGG-3′), gG sense (5′-CATGCCAAGTATTGGACTGGAGGAG-3′) and antisense (5′-CACAGGTGTGTCGCCATCGCAC-3′), CD83 sense (5′-GTTATTGGAGGGTGGTGAAGAGAGG-3′) and antisense (5′-GTGAGGAGTCACTAGCCCTAAATGC-3′), and S14 sense (5′-GGCAGACCGAGATGAATCCTCA-3′) and antisense (5′-CAGGTCCAGGGGTCTTGGTCC-3′). The following PCR cycling profile (30 cycles) was used: 95°C for 60 s, 60°C for 60 s, and 72°C for 90 s. The reaction products were analyzed on 2% agarose gels and visualized by ethidium bromide staining.
Determination of viral particle numbers using a plaque test.
Mature DC were infected with HSV-1 for 24 h at an MOI of 0.01, 0.1, or 1. The cells were subsequently washed twice with phosphate-buffered saline (PBS) and lysed by three freeze-thaw steps. Cell debris was removed by centrifugation, and the viral titer produced by the infected DC was determined in the supernatants. Cell supernatants were diluted six times (10−1 to 10−6), and 100 μl of each dilution was added for 1 h at 37°C to confluent Vero cells (5 × 106 cells/well) in 24-well plates. After virus particle adsorption, 1 ml of minimal essential medium (Gibco-BRL) containing 10% fetal calf serum (Gibco-BRL) was added. After 48 h, the medium was removed and cells were fixed for 2 min using 5% (vol/vol) formalin in water. Then cells were washed five times with water, incubated with 1% (vol/vol) crystal violet in water for 2 min, and washed again five times with water. The viral titers were determined by plaque counting.
Allogeneic T-cell proliferation.
Human PBMC were isolated from buffy coats, and the T-cell fraction was purified by rosetting with neuraminidase-treated sheep red blood cells as described by Bender et al. (6). T cells (2 × 105/well) were cocultured with DC, which had been inoculated with infectious virus or with UV-inactivated virus or left untreated, for 4 days in 200 μl of RPMI 1640 supplemented with 5% human serum from a single AB donor in 96-well plates. In addition, the allogeneic mixed leukocyte reaction (MLR) assay was performed using a mixture of uninfected and infected DC as follows. A total of 200, 900, 2,900, or 9,900 uninfected or HSV-1-infected DC were added to 100 HSV-1-infected DC and cocultured with T cells as described above. Then the cells were pulsed with [3H]thymidine (1 μCi/well) (Amersham, Braunschweig, Germany) for 8 to 16 h. The culture supernatants were harvested onto glass fiber filters (Printed Filtermat A; Wallac, Turku, Finland) by using an ICH-110 harvester (Inotech, Dottikon, Switzerland), and [3H]thymidine incorporation was determined using a microplate counter (Wallac).
Protein gel electrophoresis and immunoblotting.
Mature DC (0.5 × 106) were harvested, washed twice with PBS, and then inoculated with infectious virus or with UV-inactivated virus, treated with CHI, or left untreated. The cells were collected by centrifugation and solubilized in gel-loading buffer (50 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 5% β-mercaptoethanol, 0.1% bromphenol blue), cell extracts were separated by SDS-polyacrylamide gel electrophoresis, and the proteins were transferred to nitrocellulose membranes (Amersham). The membranes were blocked with 10% dry milk (Nestlé, Glendale, Calif.) in TBST (150 mM NaCl, 10 mM Tris-HCl, 0.05% Tween 20 [pH 8.0]) at 4°C overnight, incubated for 2 h at 25°C with a monoclonal anti-CD83 antibody (Dianova, Hamburg, Germany), washed five times in TBST, and incubated with the secondary antibody coupled to peroxidase (Dianova) at a dilution of 1:10,000 in TBST plus 5% dry milk for 1 h at 25°C. Specific bands were visualized using an enhanced chemiluminescence detection system (Amersham).
Immunofluorescence microscopy.
The expression plasmid p3CD83, coding for the entire CD83 molecule, was microinjected into the nuclei of Vero cells, cultured for 20 h. Subsequently, cells were inoculated with infectious HSV-1 at an MOI of 1 for a further 7 h. Then indirect immunofluorescence studies were performed. Cells were fixed with 2% paraformaldehyde (Merck, Darmstadt, Germany) and subsequently permeabilized using 0.1% Triton X-100 (Sigma) for 4 min and blocked with 1% bovine serum albumin (Sigma) for 30 min. The cells were then stained for 30 min with a monoclonal anti-CD83 antibody (Immunotech), an anti-cathepsin D antibody (kindly provided by A. Hille, Göttingen, Germany), or an anti-VP16 antibody. Following extensive washing steps in PBS, cells were incubated for 30 min with appropriate secondary antibodies conjugated to Cy2- or Cy3-conjugated fluorophores (Rockland, Gilbertsville, Pa.). Samples were washed in PBS, mounted in Mowiol (Calbiochem, Bad Soden, Germany), and analyzed using an Axiovert-135 microscope (Zeiss, Jena, Germany). Images were recorded with a cooled MicroMax charge-coupled device camera (Princeton Instruments, Stanford, Calif.) and processed using IPLap Spectrum and Adobe Photoshop software packages. All reactions were performed at room temperature.
DC which were inoculated as described above were also centrifuged onto polylysine-coated microslides (Menzel-Gläser, Mainz, Germany) for 30 s at 400 rpm using a Cytospin 3 centrifuge (Shandon, Pittsburgh, Pa.). CD83 expression was determined using indirect immunofluorescence as described above.
RESULTS
Phenotypic characterization of mature DC infected with HSV-1.
Mature DC were generated from nonproliferating blood progenitors by using an improved two-step protocol (6, 24). Briefly, DC precursors were generated from adherent monocytes cultured in medium containing 1% human plasma as well as GM-CSF and IL-4. A cytokine cocktail composed of tumor necrosis factor alpha, prostaglandin E2, GM-CSF, and IL-4 (15; M. Kruse et al., unpublished data) was then used to generate stable mature DC. These DC showed the characteristic features of mature DC, e.g., stellate cell shape; nonadherence to plastic; high expression of MHC class I and class II molecules, adhesins, and costimulatory molecules; and a very potent T-cell-stimulatory activity (data not shown).
Using these cells, we wanted in particular to investigate whether HSV-1 interferes with the phenotype and/or function of DC as observed with other viruses such as vaccinia virus (9) and measles virus (28). FACS analyses of the cell surface expression of typical DC markers were performed to monitor the DC phenotype. Strikingly, 24 h after HSV-1 infection, the surface expression of CD83 was drastically reduced (Fig. 1A, left panel). In sharp contrast, untreated DC or those which were inoculated with UV-inactivated HSV-1 showed no CD83 downregulation (Fig. 1A, left panel). On the other hand, HSV-1 had only a limited effect on the cell surface expression of MHC class II molecules (Fig. 1A, right panel). Furthermore, other investigated molecules, including CD25, CD40, CD80, CD86, CD95 and MHC class I, revealed no significant changes in their cell surface expression (data not shown).
FIG. 1.

(A) HSV-1 infection induces CD83 downregulation on mature DC. Mature DC were inoculated either with infectious HSV-1 at a MOI of 1 or with UV-inactivated virus and analyzed by FACS analysis after 0, 4, and 24 h. A strong downregulation of cell surface expression of CD83 was observed (left panel). No such effects could be observed when DC were left untreated or inoculated with UV-inactivated HSV-1. In contrast, the expression of MHC class II molecules was not dramatically affected by the HSV-1 infection (right panel). (B) Time course of HSV-1-induced CD83 downregulation. At 10 h after the HSV-1 infection (MOI = 1), a dramatic reduction of the CD83 cell surface expression is already observed. (C) CD83 downregulation is HSV-1 titer dependent. Mature DC were infected with HSV-1 at different MOIs (0.01, 0.1, and 1) and analyzed after 24 h by FACS analysis. The viral infection rate significantly influences the CD83 expression.
To expand our studies on the CD83 downregulation, more elaborate time course experiments were conducted. This analysis clearly demonstrated that CD83 downregulation occurs between 4 and 10 h after virus infection (Fig. 1B). Next we tested if this effect was MOI dependent. No downregulation of CD83 was visible at an MOI of 0.01. Only a slight effect was observed at an MOI of 0.1, while a dramatic CD83 downregulation was observed at an MOI of 1 (Fig. 1C). Furthermore, there was no increase in toxicity upon HSV-1 infection during the first 24 h, as monitored by propidium iodide staining and FACS analysis (data not shown).
HSV-1 infects mature DC very efficiently.
To determine the viral infection level, mature DC were inoculated with HSV-1 at an MOI of 1 and analyzed 10 h postinfection. Using a VP16-specific antibody in combination with indirect immunofluorescence microscopy, we demonstrated that most of the DC were infected (Fig. 2A); intracellular FACS analyses showed that over 90% of the DC were infected (Fig. 2C).
FIG. 2.

Efficient infection of mature DC by HSV-1. Mature DC were infected with HSV-1 at an MOI of 1 and analyzed after 10 h by intracellular FACS staining or by indirect immunofluorescence using HSV-1 VP16-specific antibody. (A and B) VP16 staining (A) and phase-contrast microscopy (B) of infected DC. (C) More than 90% of the mature DC were infected after this time.
Analyses of HSV-1 transcripts in infected mature DC.
On the RNA level, we next investigated whether HSV-1 was able to express various classes of viral mRNAs in mature DC. Using specific PCR primers, we amplified fragments of prototypical immediate-early (ICP27), early (ICP8), and late (gG) transcripts. As shown in Fig. 3A, the immediate-early ICP27 and early ICP8 gene products were transcribed after 24 h at an MOI of 1. Interestingly, in contrast to ICP27 and ICP8, only very small amounts of the late gG transcript could be amplified at this MOI (Fig. 3A, right lanes). These data indicate that mainly immediate-early and early HSV-1 transcripts are synthesized at larger amounts in mature DC. Next, the levels of CD83-specific mRNA were investigated. As shown in Fig. 3B, there was no difference in CD83 transcription after 4 and 10 h when HSV-1-infected (MOI = 1) and uninfected samples were compared. These data clearly show that the downregulation of CD83 from the cell surface, which was already observed 10 h after HSV-1 infection (Fig. 1B), was not due to an inhibition of CD83 transcription.
FIG. 3.

RT-PCR analyses of HSV-1- and CD83-specific mRNA. (A) Total cellular RNA was isolated after 24 h and reverse transcribed from uninfected and HSV-1 (MOI = 1)-infected mature DC. HSV-1-specific PCR primers were used to amplify transcripts from immediate-early (ICP27), early (ICP8), and late (gG) gene transcripts. Amplification of ribosomal protein S14-specific sequences served as an internal control. (B) RT-PCR of CD83-specific mRNA. No difference in the CD83 signals between mock- and HSV-1-infected samples after 4 and 10 h postinfection was detected.
Mature DC do not support the generation of infectious HSV-1 particles.
Next we checked whether infectious virus particles were generated in infected cells. Mature DC were thus infected for 1 h at different MOIs (0.01, 0.1, and 1), and 24 h later the viral titer present in the supernatants was analyzed using the plaque test. Supernatants of infected mature DC were added to HSV-1-permissive Vero cells. The cells were stained with crystal violet after a 48-h incubation, and the numbers of lysed cells were determined. Supernatants from HSV-1-infected Vero cells, inoculated at an MOI of 0.01, were included as a control and revealed high viral titers (106 viral particles/ml) (Fig. 4, control).
FIG. 4.

HSV-1-infected mature DC do not produce infectious virus. Mature DC were infected with HSV-1 at a MOI of 0.01, 0.1, or 1. Cell supernatants were analyzed using the RITA-plaque test after 24 h to determine the amount of infectious particles generated during the infection. No or only low levels of viral particles were detected in supernatant derived from HSV-1-infected DC. In sharp contrast, Vero cells inoculated at a MOI of 0.01 generated 106 virus particles per ml (control). Note the logarithmic scale of the y axis.
In contrast, we were not able to detect any infectious viral particles in supernatants from DC which were infected at an MOI of 0.01, and only very low levels of infectious particles were detected in the supernatant from DC infected at an MOI of 0.1 (approximately 300 particles/ml). Even at an MOI of 1, only 103 virus particles per ml were generated. These 103 virus particles could theoretically also be derived from the stock initially used to infect the DC. These data indicate that HSV-1 does not productively replicate at high levels in mature DC.
HSV-1 affects the allogeneic T-cell stimulatory capacity of DC.
The most distinctive functional characteristic of mature DC is their ability to stimulate T cells very potently (4, 8, 30, 32). Therefore, we assessed whether HSV-1 interferes with this stimulatory capacity. Mature DC were either left untreated or inoculated with infectious HSV-1 or with UV-inactivated HSV-1. These cultures were then compared in an allogeneic MLR assay. DC treated with UV-inactivated HSV-1 and untreated DC showed a very high stimulatory capacity (Fig. 5A). In contrast, DC inoculated with infectious HSV-1 showed a clear reduction in their allostimulatory capacity. This effect was particularly striking at a T-cell-to-DC ratio of ≤200:1. These data clearly show that HSV-1 infection interferes with DC function, where the downregulation of CD83 might play a role in this process.
FIG. 5.

HSV-1 infection decreases the ability of mature DC to induce an allogeneic T-cell response. (A) DC were inoculated with either infectious virus or UV-inactivated virus or left untreated, cocultured with allogeneic T cells for 4 days, and pulsed with [3H]thymidine for 8 h, and the cell supernatants were analyzed. DC derived from noninfected (□) or UV-inactivated HSV-1 (●) induced a strong allostimulatory reaction in the primary allogeneic MLR. In contrast, cells derived from HSV-1-infected DC (■) showed a strongly reduced ability to induce T-cell proliferation, particularly at a T-cell-to-DC ratio of ≤200:1 (MOI = 1). (B) Increasing numbers of uninfected or HSV-1-infected DC were added to 100 HSV-1-infected DC and subsequently analyzed for their allostimulatory capacity. Only when a large excess of infected DC were added (ratio of infected to uninfected DC = 66:1) was an interference with the allostimulation detectable.
Next we determined if a mixture of HSV-1-infected and uninfected DC would interfere with the alloresponse. Thus, increasing numbers of uninfected or HSV-1-infected DC (MOI of 1) were cocultured with 100 HSV-1-infected DC and their T-cell-stimulatory capacity was analyzed. Only when a large excess of infected DC was added (ratio of infected to uninfected DC = 66:1) was an interference with the allostimulation detectable (Fig. 5B). This strongly suggests that DC must be infected to downregulate their allostimulatory function.
HSV-1 induces downregulation of CD83.
Next we addressed whether the CD83 downregulation was due to molecule uptake into the cell or to protein degradation. To answer this question, cell extracts from DC which were inoculated with replication-competent virus or with UV-inactivated virus, treated with CHI, or left untreated were separated by SDS-gel electrophoresis. Samples taken after 2 and 24 h were compared. Proteins were transferred to nitrocellulose and analyzed by Western blotting. Strikingly, cell extracts derived from HSV-1-infected DC showed an almost complete absence of CD83 after 24 h (Fig. 6A, lane 6). In contrast, no effects were observed in any of the other analyzed samples. As expected, the CHI treatment induced a slight reduction of CD83 expression after 24 h (lane 4). The same blot was reprobed with an anti-CD86 antibody, and, as shown in Fig. 6B, no major differences were observed, indicating that the HSV-1-induced CD83 degradation is specific and is not due to a general protein degradation phenomenon. Furthermore, 10 h after the HSV-1 infection, no CD83 protein was detectable (data not shown). Taken together, these results and the FACS analyses clearly support the notion that CD83 degradation occurs during the first 10 h of viral infection.
FIG. 6.

HSV-1 infection induces CD83 protein degradation in mature DC. (A) Total cellular protein extracts (2- and 24-h samples) from DC which were inoculated either with infectious virus (MOI = 1) or with UV-inactivated virus, treated with CHI, or left untreated were resolved using SDS-polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes, and probed with a CD83-specific monoclonal antibody. (B) The blot shown in panel A was reprobed using a CD86-specific antibody.
HSV-1 infection induces a redistribution of CD83 molecules in mature DC.
To analyze the data described above, we performed immunofluorescence studies on a single-cell basis. Mature DC were either inoculated with virus or left untreated and analyzed after 3 and 23 h, respectively. After fixation, the cells were incubated with a monoclonal antibody specific for CD83 or for MHC class I (Fig. 7). When noninfected DC were analyzed, there was no difference in the expression of CD83 after 3 and 23 h (Fig. 7A and B). The same was true for the expression of MHC class I molecules (Fig. 7E and F). In sharp contrast, a strong reduction of the CD83 signal was observed in HSV-1-infected DC after 23 h (Fig. 7D). No effect of MHC class I expression was observed (Fig. 7G and H). These data further support the notion that CD83 is specifically degraded upon HSV-1 infection.
FIG. 7.

HSV-1 infection strongly affects the expression of CD83 in mature DC. Mature DC were either inoculated for 1 h with HSV-1 (MOI = 1) or left untreated. After an incubation period of 3 h (A and C) or 23 h (B and D), the cells were fixed and analyzed by indirect CD83 immunofluorescence. After 3 h, the infected DC showed only a slight change in their CD83 surface expression (C), but a dramatic reduction of the CD83 signal intensity was observed after 23 h (D). The same cell population was also analyzed using an MHC class I-specific antibody (E to H). These analyses did not reveal major differences between the different cell populations. Bar, 20 μm.
HSV-1 leads to CD83 degradation in lysosomes.
To further investigate the CD83 downregulation phenomenon, we used HSV-1-permissive Vero cells. In contrast to the nonadherent DC, these adherent Vero cells have the advantage that their intracellular morphology can be studied more easily using immunofluorescence microscopy. Therefore, Vero cell nuclei were microinjected with an expression plasmid encoding CD83 and cultured for 20 h before being inoculated with HSV-1 at an MOI of 1. The cells were then analyzed by immunofluorescence using monoclonal antibodies specific for CD83 (Fig. 8A and C) or the viral protein VP16 (Fig. 8B and D). Noninfected, microinjected cells showed a high expression and even distribution of CD83 (Fig. 8A). As expected, no VP16 staining was observed in these noninfected cells (Fig. 8B). In sharp contrast, HSV-1-infected cells showed a dramatic reduction of CD83 expression and a change in the cellular distribution of CD83 (Fig. 8C). Some fluorescent dot structures were visible in the cytoplasm, possibly reflecting the loci of CD83 degradation. The VP16 staining, demonstrating that the cells were indeed infected, is shown in Fig. 8D. These experiments revealed that the HSV-1-induced downregulation of CD83 in Vero cells was comparable to the downregulation observed in DC, indicating that it is feasible to use Vero cells to study HSV-1-induced effects.
FIG. 8.

HSV-1 leads to CD83 degradation. The p3CD83 expression plasmid DNA was microinjected into the nuclei of Vero cells, and the cells were cultured for further 20 h. They were then either left untreated or infected with HSV-1 (MOI = 1) prior to a further incubation period of 7 h. The cells were subsequently analyzed by double immunofluorescence microscopy using antibodies directed against CD83 (A and C) and VP16 (B and D). (A) Noninfected Vero cells showed a characteristic CD83 staining. (C) In contrast, HSV-1 infection affected the normal CD83 distribution, leading to accumulated intracellular structures. (D) Indirect VP16 immune fluorescence served as control for the successful virus infection. More infected cells show less CD83 expression. Compare cells labeled # and ∗ in panels C and D. Bar, 20 μm.
Next we wanted to determine the cellular compartment in which CD83 might be localized and subsequently degraded. Vero cells were therefore microinjected with the CD83-expressing plasmid, infected with HSV-1, and stained with anti-CD83 antibody (Fig. 9A) or with an antibody specific for the lysosomal protein cathepsin D (Fig. 9B). The colocalization of CD83 and cathepsin D became particularly obvious when the individual images were merged (Fig. 9C). These data strongly suggest that after HSV-1 infection, CD83 is downregulated from the cell surface and localized to intracellular lysosomal organelles, where degradation takes place.
FIG. 9.
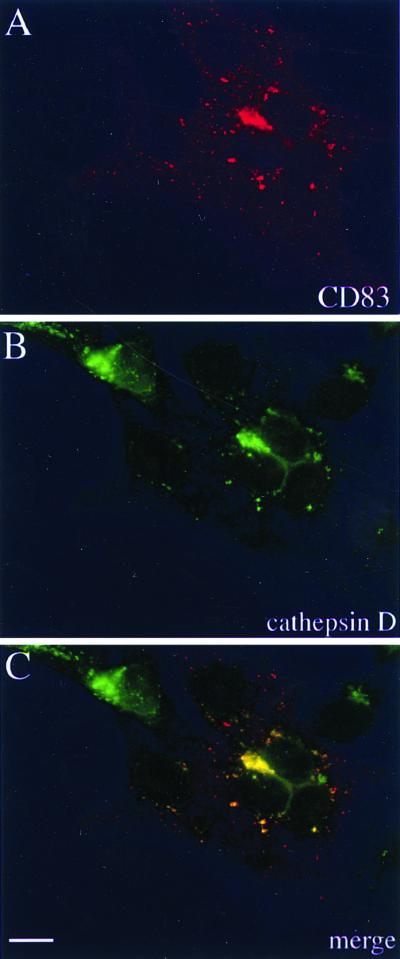
Subcellular localization of CD83. (A and B) To define the cellular CD83 localization after HSV-1 infection, cells were stained with antibodies against CD83 (A) and cathepsin D (B), which showed a similar intracellular distribution. (C) Merging of the images in panels A and B indicates that CD83 accumulates in lysosomal compartments. Bar, 20 μm.
DISCUSSION
Viruses have developed a broad range of mechanisms to escape the host immune response. These include reduced expression of critical antigen epitopes, genetic variations of MHC epitopes, clonal exhaustion, production of immunosuppressive cytokines and homologous cytokine receptors, and downregulation of critical cytokines. From this list, it has recently become evident that many of the viral escape mechanisms specifically target DC function. Measles virus infection, for example, causes apoptosis and syncytium formation in DC, inhibits IL-12 production, and blocks the T-cell stimulatory capacity of DC (10, 12, 28). These data may explain why measles virus infections are often accompanied by a dramatic suppression of the immune system.
Interestingly, vaccinia virus also inhibits DC maturation and leads to impaired allostimulatory properties of the infected DC (9a). Although vaccinia virus has developed so many immune escape mechanisms (reviewed in reference 29), it induces a strong cellular and antibody-mediated immune response. However, the precise mechanism for the induction of this immune response still requires elucidation. A possible explanation may be provided by so-called cross-presentation. Here, apoptotic vaccinia virus-infected DC may be taken up by uninfected bystander DC, and the bystander cells could then present viral antigens to virus-specific cytotoxic T cells. This has already been described for influenza virus-infected apoptotic cells (2). These findings clearly demonstrate the dual role of DC during viral infections. On the one hand, they are able to induce a specific immune response, while on the other hand, they contribute to pathogenic effects and immune suppression.
This dual role has also been reported for human immunodeficiency virus type 1 infections. In this case, DC not only elicit virus-specific CTL responses but also transport the virus from the periphery to the T cells in the lymph nodes, where human immunodeficiency virus type 1 replicates (11). Furthermore, hepatitis C virus seems to impair the allostimulatory capacity of DC, as assessed by examination of peripheral blood DC isolated from hepatitis C virus-infected patients (16). These reports all clearly demonstrate that DC are very important targets for viruses in their struggle for survival against the immune response.
Here we report that HSV-1-infected mature DC are inhibited in their T-cell-stimulatory capacity. Furthermore, we show that mature DC do not generate infectious virus particles. On the transcriptional level, mainly immediate-early and early viral transcripts were detected. Therefore, we speculate that proteins encoded by these gene products might be responsible for the interference with DC function. However, we have not yet identified the molecular mode of action of this HSV-1-mediated inhibitory effect. In contrast to mature DC, immature DC can also be infected efficiently at lower MOI, i.e., 0.01; strikingly, these cells never reach the final state of maturation and are therefore very poor T-cell stimulators (M. Kruse, unpublished results). In support of these findings, a study published during the final preparation of this paper reported that HSV-1 interferes with the maturation of immature DC (27).
Interestingly, when we investigated the phenotype of mature DC infected with HSV-1, we observed a very striking feature, namely, the loss of CD83 cell surface expression. However, the expression of other typical DC molecules, including CD25, CD40, CD80, CD86, CD95, and MHC class I and class II, was not influenced. CD83 expression is induced during DC maturation and represents one of the best-known markers for mature DC (35, 39, 40). Although the function of CD83 is still unknown, the fact that its surface expression is strongly upregulated during DC maturation, along with costimulatory molecules, indicates that it has an important function during the immune response. Noteworthy is the finding that inhibition of CD83 cell surface expression leads to a dramatic reduction of DC-mediated T-cell stimulation (19). Using a specific inhibitor of the eukaryotic initiation factor 5A, a protein which is involved in the nuclear export pathway of specific RNAs (25, 26), we showed that the CD83 cell surface expression on DC could be prevented and that their allostimulatory capacity was strongly reduced (19).
A typical feature of HSV-1 infection is the loss of protein synthesis due to disruption of polysomes and degradation of mRNA (31). However, the loss of CD83 cell surface expression was shown not to be due to CD83 mRNA degradation. The dramatic downregulation of the CD83 protein was already observed 10 h after infection. Nevertheless, at this time point there was no difference between the uninfected and infected samples when CD83 mRNA was analyzed. In contrast, HSV-1 led to a complete and specific degradation of CD83 protein in mature DC. Since we observed a colocalization of CD83 and cathepsin D, this degradation is probably lysosome mediated. Since mainly immediate-early and early viral gene transcripts are generated in mature DC, we speculate that these viral gene products might be involved in the degradation of CD83. A detailed analysis of this hypothesis will be performed in a future study. Although the inhibitory effect of HSV-1 on mature DC function cannot be exclusively attributed to the degradation of CD83, this inhibition represents an additional new viral immune system escape mechanism for HSV-1.
Peripheral DC and Langerhans cells reside close to keratinocytes, fibroblasts, and epithelial cells, the major cells in which HSV-1 replication occurs. Therefore, it is conceivable that DC and Langerhans cells encounter HSV-1, either directly through infection or indirectly via uptake of infected cells, which could lead to an antiviral immune response in both cases. Our data suggest that HSV-1 interferes with the function of mature DC by preventing the T-cell activation by these most powerful antigen-presenting cells. Consequently, caution should be applied to the use of HSV-1-derived vectors currently being developed as tools for vaccination strategies to be implemented in human clinical trials.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 466) and the Wilhelm Sander-Stiftung (96.042.2).
We thank Heidi C. Joao for critical reading of the manuscript.
REFERENCES
- 1.Ahn K, Meyer T H, Uebel S, Sempe P, Djaballah H, Yang Y, Peterson P A, Fruh K, Tampe R. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 1996;13:3247–3255. [PMC free article] [PubMed] [Google Scholar]
- 2.Albert M L, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 3.Assenmacher M, Schmitz J, Radbruch A. Flow cytometric determination of cytokines in activated murine T helper lymphocytes: expression of IL-10 in interferon-gamma and in interleukin-4 expressing cells. Eur J Immunol. 1994;24:1097–1101. doi: 10.1002/eji.1830240513. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau J, Steinman R M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 5.Bender A, Bui L K, Feldman M A, Larsson M, Bhardwaj N. Inactivated influenza virus, when presented on dendritic cells, elicits human CD8+ cytolytic T cell responses. J Exp Med. 1995;182:1663–1671. doi: 10.1084/jem.182.6.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bender A, Sapp M, Schuler G, Steinman R M, Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J Immunol Methods. 1996;196:121–135. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- 7.Bender A, Albert M, Reddy A, Feldman M, Sauter B, Kaplan G, Hellman W, Bhardwaj N. The distinctive features of influenza virus infection of dendritic cells. Immunobiology. 1998;198:552–567. doi: 10.1016/S0171-2985(98)80078-8. [DOI] [PubMed] [Google Scholar]
- 8.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 9.Di Nicola M, Siena S, Bregni M, Longoni P, Magni M, Milanesi M, Matteucci P, Mortarini R, Anichini A, Parmiani G, Drexler I, Erfle V, Sutter G, Gianni A M. Gene transfer into human dendritic antigen-presenting cells by vaccinia virus and adenovirus vectors. Cancer Gene Ther. 1998;5:350–356. [PubMed] [Google Scholar]
- 9a.Engelmayer J, Larsson M, Subklewe M, Chahroudi A, Cox W I, Steinman R M, Bhardwaj N. Vaccinia virus inhibits the maturation of human dendridic cells: a novel mechanism of immune evasion. J Immunol. 1999;163:6762–6768. [PubMed] [Google Scholar]
- 10.Fugier-Vivier I, Servet-Delprat C, Rivailler P, Rissoan M C, Liu Y J, Rabourdin-Combe C. Measles virus suppresses cell-mediated immunity by interfering with the survival and functions of dendritic and T cells. J Exp Med. 1997;186:813–823. doi: 10.1084/jem.186.6.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geijtenbeek T, Kwon D, Torensma R, van Vliet S, van Duijnhoven G, Middel J, Cornelissen I, Nottet H, KewalRamani V, Littam D, Figdor C, van Kook Y. DC-SIGN, a dendritic cell-specific HIV-1 binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 12.Grosjean I, Caux C, Bella C, Berger I, Wild F, Banchereau J, Kaiserlian D. Measles virus infects human dendritic cells and blocks their allostimulatory properties for CD4+ T cells. J Exp Med. 1997;186:801–812. doi: 10.1084/jem.186.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamilton-Easton A, Eichelberger M. Virus-specific antigen presentation by different subsets of cells from lung and mediastinal lymph node tissues of influenza virus-infected mice. J Virol. 1995;69:6359–6366. doi: 10.1128/jvi.69.10.6359-6366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hengel H, Lindner M, Wagner H, Heeg K. Frequency of herpes simplex virus-specific murine cytotoxic T lymphocyte precursors in mitogen- and antigen primary in vitro T cell responses. J Immunol. 1987;139:4196–4202. [PubMed] [Google Scholar]
- 15.Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk A H. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 16.Kanto T, Hayashi N, Takehara T, Tatsumi T, Kuzushita N, Ito A, Sasaki Y, Kasahara A, Hori M. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol. 1999;162:5584–5591. [PubMed] [Google Scholar]
- 17.Kast W M, Boog C J, Roep B O, Voordouv A C, Melief C J. Failure or success in the restoration of virus-specific cytotoxic T lymphocyte response defects by dendritic cells. J Immunol. 1988;140:3186–3193. [PubMed] [Google Scholar]
- 18.Koelle D M, Tigges M A, Burke R L, Symington F W, Riddell S R, Abbo H, Corey L. Herpes simplex virus infection of human fibroblasts and keratinocytes inhibits recognition by cloned CD8+ cytotoxic T lymphocytes. J Clin Investig. 1993;91:961–968. doi: 10.1172/JCI116317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruse M, Rosorius O, Krätzer F, Bevec D, Kuhnt C, Steinkasserer A, Schuler G, Hauber J. Inhibition of CD83 cell surface expression during dendritic cell maturation by interference with nuclear export of CD83 mRNA. J Exp Med. 2000;191:1581–1590. doi: 10.1084/jem.191.9.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludewig B, Ehl S, Karrer U, Odermatt B, Hengartner H, Zinkernagel R M. Dendritic cells efficiently induce protective antiviral immunity. J Virol. 1998;72:3812–3818. doi: 10.1128/jvi.72.5.3812-3818.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munk K, Ludwig G. Properties of plaque variants of herpes virus hominis strains of genital origin. Arch Gesamte Virusforsch. 1972;37:308–315. doi: 10.1007/BF01241453. [DOI] [PubMed] [Google Scholar]
- 22.Nonacs R, Humborg C, Tam J P, Steinman R M. Mechanisms of mouse spleen dendritic cell function in the generation of influenza-specific, cytolytic T lymphocytes. J Exp Med. 1992;176:519–529. doi: 10.1084/jem.176.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roizman B. Whither herpesviruses? Adv Exp Med Biol. 1990;278:285–291. doi: 10.1007/978-1-4684-5853-4_29. [DOI] [PubMed] [Google Scholar]
- 24.Romani N, Reider D, Heuer M, Ebner S, Kampgen E, Eibl B, Niederwieser D, Schuler G. Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J Immunol Methods. 1996;196:137. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- 25.Rosorius O, Reichart B, Krätzer F, Heger P, Dabauvalle M C, Hauber J. Nuclear pore localization and nucleocytoplasmic transport of eIF-5A: evidence for direct interaction with the export receptor CRM1. J Cell Sci. 1999;112:2369–2380. doi: 10.1242/jcs.112.14.2369. [DOI] [PubMed] [Google Scholar]
- 26.Ruhl M, Himmelspach M, Bahr G M, Hammerschmid F, Jaksche H, Wolff B, Aschauer H, Farrington G K, Probst H, Bevec D, Hauber J. Eukaryotic initiation factor 5A is a cellular target of the human immunodeficiency virus type 1 Rev activation domain mediating trans-activation. J Cell Biol. 1993;123:1309–1320. doi: 10.1083/jcb.123.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salio M, Cella M, Suter M, Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol. 1999;29:3245–3253. doi: 10.1002/(SICI)1521-4141(199910)29:10<3245::AID-IMMU3245>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 28.Schnorr J J, Xanthakos S, Keikavoussi P, Kampgen E, ter Meulen V, Schneider-Schaulies S. Induction of maturation of human blood dendritic cell precursors by measles virus is associated with immunosuppression. Proc Natl Acad Sci USA. 1997;94:5326–5331. doi: 10.1073/pnas.94.10.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith G L, Symons J A, Alcami A. Immune modulation by proteins secreted from cells infected by vaccinia virus. Arch Virol Suppl. 1999;15:111–129. doi: 10.1007/978-3-7091-6425-9_8. [DOI] [PubMed] [Google Scholar]
- 30.Steinman R M. Dendritic cells. In: Paul W E, editor. Fundamental immunology. 4th ed. Philadelphia, Pa: Lippincott Raven Publishers; 1999. pp. 547–573. [Google Scholar]
- 31.Sydiskis R J, Roizman B. Polysomes and protein synthesis in cells infected with a DNA virus. Science. 1966;153:76–78. doi: 10.1126/science.153.3731.76. [DOI] [PubMed] [Google Scholar]
- 32.Thomas R, Davis L S, Lipsky P E. Comparative accessory cell function of human peripheral blood dendritic cells and monocytes. J Immunol. 1993;151:6840. [PubMed] [Google Scholar]
- 33.Tomazin R, Hill A B, Jugovic P, York I, van Endert P, Ploegh H L, Andrews D W, Johnson D C. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 1996;15:3256–3266. [PMC free article] [PubMed] [Google Scholar]
- 34.Tomazin R, van Schoot N E, Goldsmith K, Jugovic P, Sempe P, Fruh K, Johnson D C. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol. 1998;72:2560–2563. doi: 10.1128/jvi.72.3.2560-2563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weissman D, Li Y, Orenstein J M, Fauci A S. Both a precursor and a mature population of dendritic cells can bind HIV. However, only the mature population that expresses CD80 can pass infection to unstimulated CD4+ T cells. J Immunol. 1995;155:4111–4117. [PubMed] [Google Scholar]
- 36.York I A, Roop C, Andrews D W, Riddell S R, Graham F L, Johnson D C. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 1994;77:525–535. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 37.Young J W, Steinman R M. Dendritic cells stimulate primary human cytolytic lymphocyte responses in the absence of CD4+ helper T cells. J Exp Med. 1990;171:1315–1332. doi: 10.1084/jem.171.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young J W, Koulova L, Soergel S A, Clark E A, Steinman R M, Dupont B. The B7/BB1 antigen provides one of several costimulatory signals for the activation of CD4+ T lymphocytes by human blood dendritic cells in vitro. J Clin Investig. 1992;90:229–237. doi: 10.1172/JCI115840. . (Erratum, 91:1853, 1993.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou L J, Schwarting R, Smith H M, Tedder T F. A novel cell-surface molecule expressed by human interdigitating reticulum cells, Langerhans cells, and activated lymphocytes is a new member of the Ig superfamily. J Immunol. 1992;149:735–742. [PubMed] [Google Scholar]
- 40.Zhou L J, Tedder T F. Human blood dendritic cells selectively express CD83, a member of the immunoglobulin superfamily. J Immunol. 1995;154:3821–3835. [PubMed] [Google Scholar]