H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB (original) (raw)
Abstract
The ABC transporter HlyB is a central element of the HlyA secretion machinery, a paradigm of Type I secretion. Here, we describe the crystal structure of the HlyB-NBD (nucleotide-binding domain) with H662 replaced by Ala in complex with ATP/Mg2+. The dimer shows a composite architecture, in which two intact ATP molecules are bound at the interface of the Walker A motif and the C-loop, provided by the two monomers. ATPase measurements confirm that H662 is essential for activity. Based on these data, we propose a model in which E631 and H662, highly conserved among ABC transporters, form a catalytic dyad. Here, H662 acts as a ‘linchpin', holding together all required parts of a complicated network of interactions between ATP, water molecules, Mg2+, and amino acids both in cis and trans, necessary for intermonomer communication. Based on biochemical experiments, we discuss the hypothesis that substrate-assisted catalysis, rather than general base catalysis might operate in ABC-ATPases.
Keywords: ATPase activity, catalysis, membrane transporter, NBD-dimer, X-ray crystallography
Introduction
In Gram-negative bacteria, different pathways exist to translocate proteins across the inner and outer membranes. Type I secretion is a Sec-independent mechanism that shuttles toxins, proteases, lipases, heme-binding proteins, or lantibiotics in one step across both membranes of Gram-negative bacteria such as Escherichia coli (Holland et al, 2003). All information necessary and sufficient for translocation is encoded at the C-terminus of the transported substrate (allocrite). This secretion signal normally comprises the last 50–60 C-terminal amino acids of the allocrite (Jarchau et al, 1994; Kenny et al, 1994). The first identified allocrite secreted by the Type I pathway was haemolysin (Hly) A, a 107 kDa pore-forming toxin secreted by certain uropathogenic E. coli strains and member of the family of RTX toxins (Welch, 1991).
The Type I secretion apparatus is made up of three components, an ABC transporter, a membrane fusion protein (MFP), and an outer membrane protein (OMP) (Holland et al, 2003). For the HlyA transporter complex, HlyB (ABC transporter) and HlyD (MFP) reside in the inner membrane of E. coli. The OMP component is TolC (Koronakis et al, 2000), which is thought to interact with the MFP to form a continuous channel across the periplasm, from the cytoplasm to the exterior (Holland et al, 2003).
HlyB belongs to the family of ABC transporters, which are ubiquitous, ATP-dependent transmembrane pumps or channels (Higgins, 1992; Schmitt and Tampé, 2002). The spectrum of transport substrates ranges from inorganic ions, nutrients such as amino acids, sugars, or peptides, hydrophobic drugs to large polypeptides, such as HlyA. Despite this enormous allocrite diversity, all ABC transporters share a common architecture, composed of two nucleotide-binding domains (NBD) and two transmembrane domains (TMD). Since the first discovery of an ABC transporter, the histidine permease in 1982 (Higgins et al, 1982), enormous progress has been made in characterizing their properties, recently extended by structural studies. Crystal structures of isolated NBDs in various states of the catalytic cycle of ATP hydrolysis are now available for ABC importers and exporters (Hung et al, 1998; Karpowich et al, 2001; Smith et al, 2002; Chen et al, 2003; Schmitt et al, 2003; Verdon et al, 2003a, 2003b; Lewis et al, 2004). Furthermore, three crystal structures of full-length ABC transporters have been published: the lipid flippase A (MsbA) from E. coli (Chang and Roth, 2001) and Vibrio cholera (Chang, 2003) and the vitamin B12 importer (BtuCD) from E. coli (Locher et al, 2002).
The motor domain of ABC transporters, the NBD, is an L-shaped protein with a two-domain architecture first observed for HisP (Hung et al, 1998). The catalytic domain, composed of the ABC subdomain, ABC-beta (Karpowich et al, 2001), and a RecA-like subdomain (Story and Steitz, 1992), contains the nucleotide-binding site. The helical domain, ABC-alpha, interacts with the TMD and is thought to shuttle signals back and forth between the NBD and TMD, coupling the action of the NBD to allocrite transport. An NBD contains several conserved motifs: the Walker A (Walker et al, 1982) and B motifs, common to all P-loop NTPases (Vetter and Wittinghofer, 1999), the Q-loop and Pro-loop (Schmitt et al, 2003), which link the catalytic and helical domains, the H-loop and the two hallmarks of ABC transporters, the C-loop or ABC signature motif (LSGGQ) (Schmitt and Tampé, 2002), and the equally distinctive feature, the D-loop (atSALDye in HlyB). Despite this obvious conservation, all NBDs, which have been characterized by X-ray crystallography, contain a structurally diverse region (SDR) within their helical domains, which is unique to each individual NBD (Schmitt et al, 2003). Only recently, structural (Smith et al, 2002; Chen et al, 2003) and biochemical data (Chen et al, 2001; Loo et al, 2002; Moody et al, 2002; Janas et al, 2003) have demonstrated that NBDs can dimerize stably in the ATP-bound state and that the dimer interface in the ATP-bound state includes a close association of the Walker A motif of one monomer and the C-loop of the other monomer.
Despite major advances in the understanding of ABC proteins, the exact mechanism of ATP hydrolysis remains an enigma (Schmitt and Tampé, 2002; Jones and George, 2004). Particularly, the exact function of the conserved amino-acid residues in the vicinity of the ATP-binding pocket, the reaction path of ATP hydrolysis, and the molecular communication between NBD monomers during ATP binding and hydrolysis are poorly understood. While the currently dominant view is that a glutamate residue C-terminal to the Walker B motif acts as the general or catalytic base (Moody et al, 2002), some data contradict such a role in catalysis (Urbatsch et al, 2000; Sauna et al, 2002; Verdon et al, 2003a; Tombline et al, 2004). On the other hand, mutation of a conserved histidine (H662 in HlyB) in both the histidine and maltose importers invariably appears to result in deficient transport and ATPase activity (Shyamala et al, 1991; Davidson and Sharma, 1997; Nikaido and Ames, 1999). This suggested an important (Hung et al, 1998) but so far poorly defined functional role for this amino acid.
Here, we report the crystal structure of the HlyB-NBD H662A with bound ATP/Mg2+ at 2.5 Å resolution. The protein adopted a composite dimer architecture in the crystal structure. Despite the fact that all residues suggested to be essential for hydrolysis are in a productive conformation in the HlyB-NBD, ATP is bound but remains uncleaved. We propose a model in which H662 acts as a ‘linchpin' holding together all parts of a complicated network, both intermolecular and across the monomer–monomer interface, arranged efficiently to provide a framework that allows ATP hydrolysis. We also report that ATPase activity was not reduced in the presence of D2O. As will be discussed, this and other data prompted us to consider whether substrate-assisted catalysis (SAC) rather than a general base mechanism might underlie ATP hydrolysis in ABC-ATPases.
Results and discussion
The H662A mutant of the HlyB-NBD, composed of the C-terminal residues 467–707 of HlyB, was overexpressed and purified as described for the wild-type NBD (Zaitseva et al, 2004). Crystals were obtained in the presence of ATP/Mg2+ (see Materials and methods) and diffracted to 2.5 Å. The structure was determined using a combination of a SAD data set from a seleno-methionine-substituted protein crystal and molecular replacement (see Materials and methods). The refined structure has an _R_F value of 21.9% and an _R_free of 26.4% without noncrystallographic symmetry (NCS) restraints. All amino acids except the N-terminal His tag were visible in the electron density. The final structure contains 964 amino acids together with four ATP, four Mg2+, and 172 water molecules. The latter were located in 1_F_o−_F_c difference maps at a conservative threshold of 3.5σ. Further details of the refinement and the geometry of the structure are given in Table I.
Table 1.
Data quality and refinement statistics
| Crystal parameters at 100 K | |
|---|---|
| Space group | P21 |
| Cell constants | |
| a, b, c (Å) | 44.926, 194.917, 63.706 |
| β (deg) | 110.68 |
| Data collection and processing | |
| Wavelength (Å) | 1.05 |
| Resolution (Å) | 95–2.5 (2.56–2.50) |
| Mean redundancy | 6.8 |
| Completeness (%) | 99.4 (99.8) |
| I/σ | 23.4 (2.8) |
| _R_sym (%) | 5.3 (40.1) |
| Refinement | |
| _R_F (%) | 21.9 (31.3) |
| _R_free (%) | 26.4 (37.1) |
| R.m.s.d. | |
| Bond (Å) | 0.006 |
| Angle (deg) | 1.037 |
| Average _B_-factor (Å2) | 51.1 |
| Ramachandran plot | |
| Most favored (%) | 91.1 |
| Allowed (%) | 8.9 |
| Model content | |
| Protein | 964 residues |
| Ligands | 4 ATP |
| 4 Mg2+ | |
| Water molecules | 172 |
| Crystal parameters and data collection statistics are from SCALEPACK (Otwinowski and Minor, 1997). Refinement statistics were derived from REFMAC5 (Murshudov et al, 1997) and Ramachandran analysis was performed using PROCHECK (Laskowski et al, 1993). |
Overall structure of the composite dimer of HlyB-NBD H662A
HlyB-NDB H662A, in complex with ATP/Mg2+, crystallized as a dimer of dimers in the asymmetric unit. This arrangement is likely not physiological, as the buried surface area between each dimer pair is only 480 Å2, and no biochemical evidence for such an architecture exists. The overall structure of the ATP/Mg2+-loaded composite dimer of the HlyB-NBD H662A is shown in Figure 1, with the H-loop (colored in green) normally containing H662 clearly visible within the dimer interface. ATP is coordinated by the Walker A motif of monomer 1 (_cis_-site) and the C-loop (LSGGQ) of monomer 2 (_trans_-site). This arrangement is similar to that reported for the two ABC importers, MJ0796 from Methanococcus jannaschii (Smith et al, 2002) and MalK from E. coli (Chen et al, 2003). A comparison with these structures reveals an r.m.s.d. of 1.87 Å for MJ0796 (378 Cα atoms) and 1.85 Å for MalK (404 Cα atoms), respectively. The similarity between the three structures suggests that this dimer arrangement is likely to be a universal feature of the NBDs of all ABC transporters in their ATP-bound form. The buried surface area between the two monomers of the individual dimers of HlyB-NBD H662A is 1440 Å2 with ATP providing another 450 Å2. Since the r.m.s.d. between the individual monomers is below 0.7 Å for 241 Cα atoms, the homodimer displays a highly symmetrical organization. In contrast to the structures of MJ0796 (Smith et al, 2002) and MalK (Chen et al, 2003), HlyB-NBD H662A crystallized in the presence of ATP/Mg2+.
Figure 1.
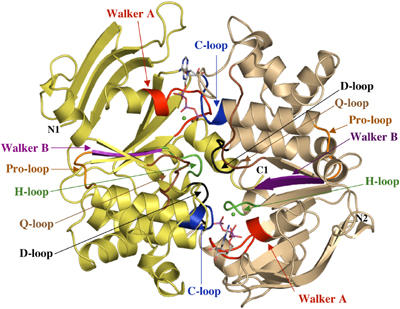
Crystal structure of the HlyB-NBD H662A dimer with bound ATP/Mg2+. ATP in stick representation and Mg2+ (green spheres) are sandwiched at the interface of the two HlyB-NBD monomers (shown in light tan and light yellow). N- and C-termini of the individual monomers are labeled. Conserved motifs are colored in red (Walker A motif; Walker et al, 1982), brown (Q-loop), blue (C-loop or ABC signature motif), magenta (Walker B), black (D-loop), and green (H-loop) and labeled accordingly. The figure was prepared using PyMol (www.pymol.org).
Notably, the ATPs are intact in the structure (Supplementary Figure S1), despite the fact that all residues considered to be essential for ATP hydrolysis, such as K508 of the Walker A motif, D630 (Walker B), the proposed catalytic carboxylate E631, and S607 and G609 of the C-loop in the trans monomer, are present in a productive conformation for hydrolysis. Indeed, the H662A mutant in solution displayed no detectable steady-state ATPase activity (Zaitseva et al, 2004; Benabdelhak et al, 2005). A 1_F_o−_F_c omit map, contoured at 4σ, confirmed that no cleavage of the β–γ phosphate bond occurred with this mutant (Supplementary Figure S1). Consequently, the conserved H662 must play a crucial role in prehydrolysis events or in the stabilization of the transition state, rather than simply acting as a ‘γ-phosphate sensor' as suggested previously (Geourjon et al, 2001).
Comparison of the ATP/Mg2+-loaded composite dimer and the nucleotide-free structure of HlyB-NBD
The most obvious difference between the nucleotide-free and ATP/Mg2+-loaded forms of the NBDs is their oligomeric state. While the former was crystallized as a monomer, the latter formed dimers upon crystallization. A comparison of the nucleotide-free structure of the HlyB-NBD (Schmitt et al, 2003) and the ATP/Mg2+-bound monomer reveals an r.m.s.d. of 2.2 Å over 241 Cα atoms. This is due to an 18.5° inward rigid-body rotation of the helical domain (r.m.s.d. of the catalytic domain, residues 467–549 and 625–707 of 0.9 Å, r.m.s.d. of the helical domain, residues 550–625 of 2.4 Å). This induced fit motion has already been described for MJ1267 and proposed to be a general feature of ABC-NBDs (Karpowich et al, 2001). Most striking is the conformational change of the C-terminal residues of the Walker A motif (residues 508–510). In the nucleotide-free state, these residues adopt a 310 helical conformation, while a canonical α-helical conformation is observed in the ATP/Mg2+-bound form. This further supports our earlier notion (Schmitt et al, 2003) that this local conformational difference might be used to control ATPase activity of the HlyB-NBD in vivo. Moreover, ATP/Mg2+ binding results in a significant rearrangement of the residues surrounding the ATP-binding pocket of the HlyB-NBD (see below).
Conservation of H662 in the NBD of ABC transporters—a residue apparently essential for catalysis
A sequence alignment of the NBDs for which crystal structures have been published (not shown) emphasizes the conservation and importance of H662, which is located in the H-loop preceding helix 7 (colored green in Figure 1). Mutation of the conserved histidine in HisP (H211) (Shyamala et al, 1991) and MalK (H192) (Davidson and Sharma, 1997) resulted in background steady-state ATPase activity (<2%), and completely abrogated transport (Davidson and Sharma, 1997; Nikaido and Ames, 1999). The reported high residual activity for a MalK mutant lacking the histidine (Walter et al, 1992) has not been confirmed (E Schneider, personal communication). In the case of the HlyB-NBD (Zaitseva et al, 2004), substitution of H662 for alanine reduced the steady-state ATPase activity to background levels (<0.1%).
Another residue so far considered essential for catalytic activity, the conserved glutamate immediately following the Walker B aspartate (E631 in HlyB), has been proposed to act as the catalytic base in ATP hydrolysis in ABC proteins (Moody et al, 2002). However, mutations of this glutamate, for example, to glutamine, variously affect ATP hydrolysis depending on the ABC protein. In MJ1267, MJ0796 (Moody et al, 2002), and BmrA (Orelle et al, 2003), no steady-state ATPase activity was detectable. In contrast, a very low ATPase activity was determined for the NBD of Mdl1p (Janas et al, 2003), at least a single turnover for human and mouse P-gp (Urbatsch et al, 2000; Sauna et al, 2002; Tombline et al, 2004), and around 20% residual ATPase activity for GlcV (Verdon et al, 2003b). These different outcomes question the previous proposals that the conserved glutamate acts as the general or catalytic base.
The importance of the conserved histidine is further supported by the structural alignment (data not shown) of HlyB-NBD H662 with other ATPases and GTPases containing the Walker A motif. Position 662 in HlyB coincides with those residues of the proteins suggested to be essential for catalysis: H885 (Rad50), Y311 (α-chain of F1-ATPase), E328 (β-chain of F1-ATPase) (Abrahams et al, 1994), Q194 (RecA) (Story and Steitz, 1992; Datta et al, 2003), Q61 (p21ras) (Pai et al, 1990), H85 (EF-Tu) (Bertchold et al, 1993), and H465 (T7 helicase) (Sawaya et al, 1999).
Architecture of the ATP-binding site
Intramolecular (in cis) interactions. Figure 2A shows the intra- and intermolecular interactions that make up the composite arrangement of the ATP-binding site of the HlyB-NBD. As observed in all nucleotide-bound crystal structures of ABC-NBDs, the adenine ring of ATP interacts with an aromatic amino acid (Y477) via π–π stacking. As in all other P-loop ATPases (Vetter and Wittinghofer, 1999), the Walker A motif (G502–T510) wraps around the triphosphate moiety, interacting with the ligand via main-chain and side-chain interactions. The conserved lysine (K508) interacts with the β- and γ-phosphates, thereby fixing both groups in a defined orientation. The β- and γ-phosphates also participate in Mg2+ coordination (green sphere). Further coordination to Mg2+ is provided by the side chain of S509 and three water molecules (blue spheres) completing the octahedral coordination sphere. One of these coordinating water molecules, 36, is hydrogen bonded to D630 of the Walker B motif. This whole area is further stabilized by interactions of D630 with S509, S509 with the α-phosphate group, and by a hydrogen bond between T510 and Y477 connecting the adenine binding region with the Walker A motif. Next to the three water molecules, which participate in Mg2+ coordination, two more water molecules, 90 and 109, were unambiguously identified in one of the binding sites. Water 109 connects the γ-phosphate of ATP with Q550 of the Q-loop. Furthermore, while a van der Waals attraction is observed between the backbone of D630 and Q550 in the nucleotide-free structure (4.1 Å), a hydrogen bond is present in the ATP/Mg2+-bound state (3.2 Å). Such an arrangement allows Q550 to connect ATP and Mg2+ to the helical domain by means of an induced fit mechanism via D630 (Karpowich et al, 2001). The second water molecule, 90, bridges the γ-phosphate group to E631 and via water 36 (part of the Mg2+ coordination sphere) connects E631 to the bound Mg2+ (Figure 2B). In the Rad50 structure (Hopfner et al, 2000), the corresponding E823 interacts with the bound Mg2+ via one water molecule.
Figure 2.
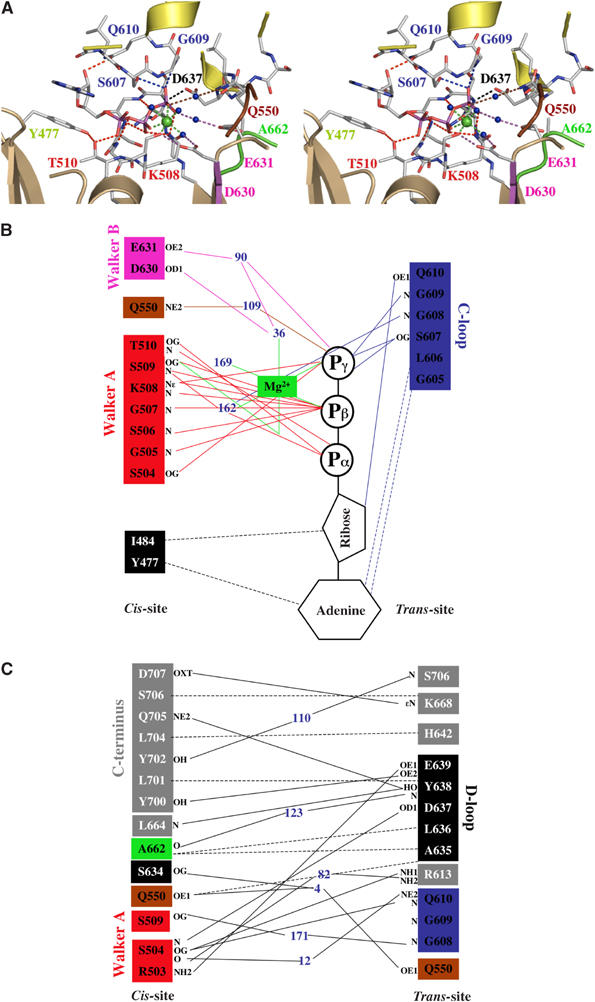
(A) Stereo view of one of the ATP-binding sites in the HlyB-NBD H662A dimer. ATP is shown in stick representation, Mg2+ as a green sphere, and water molecules as blue spheres. (B) Schematic diagram of the interactions between one of the two ATP/Mg2+ complexes and HlyB-NBD H662A. (C) Schematic diagram of the interactions across the monomer–monomer interface. Color coding is identical to Figure 1. Residues involved in monomer–monomer contact are highlighted by gray boxes, while residues involved in protein–ATP contacts are colored in boxes according to Figure 1. Blue numbers indicate water molecules. Hydrogen bonds and salt bridges are shown as solid lines, while van der Waals and hydrophobic interactions are shown as dashed lines. Letters represent the atoms involved in the interaction.
Intermolecular interactions. Further interactions with ATP arise from the C-loop in the trans monomer. This canonical arrangement of the composite interface of the dimer was described for Rad50 (Hopfner et al, 2000), MJ0796 (Smith et al, 2002), and MalK (Chen et al, 2003). ATP acts like a molecular glue with the γ-phosphate of ATP linking the Walker A and B motifs and the Q-loop of the cis monomer with the C-loop of the trans monomer (Figure 2B). Further symmetric interactions stabilize the monomer–monomer interface as illustrated in Figure 2C. These include the C-terminus of HlyB-NBD (amino acids 700–707), A662 (H-loop, H662 in wild-type HlyB), and the D-loop region (residues 634–637). Since the last 12 C-terminal amino acids of HlyB (helix 8 and 9) have no counterpart in MJ0796 and have a different orientation in MalK due to the presence of an additional C-terminal regulatory domain, the stabilizing effect of these interactions in the dimer have not been described before. It remains to be established whether the role of such additional residues is specific to HlyB or is more universally important.
Intermolecular interactions involving the conserved atSALDye motif. In addition to the above intermolecular interactions, D637 located in the ABC hallmark D-loop forms a hydrogen bond with the amide backbone of S504 of the Walker A in the trans monomer (Figure 2A and C). In fact, all residues of the D-loop participate in trans interactions (Figure 2C) linking together both subunits, including the interaction with the backbone of A662. We suggest that this conserved motif might be involved in monomer–monomer communication to transmit the functional state of one ATP-binding site to the other.
ATP–protein interactions. In contrast to the MalK dimer structure (lacking Mg2+), the bound Mg2+ in HlyB-NBD recruits additional water molecules. These water molecules act as connecting points to side chains of amino acids located within the ATP-binding site. This allows the formation of a complex network of interactions (Figure 2A and B). Overall, ATP makes 17 direct contacts with the homodimer of which 13 are hydrogen bonds (<3.4 Å) and four are van der Waals interactions (<4 Å). As shown in Figure 2B, 12 contacts are made with residues in the cis monomer and the remaining five with the trans monomer C-loop. These latter interactions of the trans monomer very likely complete the ATP-binding site, preventing uncontrolled dissociation of a bound nucleotide. This also implies that conformational changes associated with ATP hydrolysis (Karpowich et al, 2001), for example, the reverse rotation of the helical domain away from the catalytic domain, are a prerequisite for dissociation of ADP. After cleavage of ATP and dissociation of inorganic phosphate, the interactions between S607 and G609 of the trans monomer and the resulting ADP are abolished. Then, the nucleotide interacts only in trans via the hydrogen bond between Q610 and the ribose and via van der Waals contacts between G605/L606 and the adenine ring. In fact, the C-loop Q610 seems to perform a dual role (Figure 2B and C). In addition to the trans interaction with the ribose, this residue hydrogen bonds in trans to S504 of the Walker A motif. S504 in turn links the two monomers via interaction with the D-loop of the trans monomer.
A chemical reaction is the rate-limiting step of ATP hydrolysis in the HlyB-NBD
The structural issues discussed above placed H662 as a key player in catalysis, perhaps directly involved in the limiting step in ATP hydrolysis, rather than the previously proposed glutamate. This prompted us to examine further the possible mechanism of catalysis. The catalytic cycle of an NBD is composed of nucleotide association, NBD dimerization, ATP hydrolysis, and dissociation of the NBDs and nucleotide. Any of these steps: representing chemical or diffusion-controlled reactions: might be the rate-limiting step under steady-state conditions. To investigate the relative contribution of these steps, the reaction velocity of the ATPase activity of wild-type HlyB-NBD in solutions of different viscosity was first analyzed (Supplementary Figure S2). Since molecular diffusion coefficients depend inversely on the viscosity of the solution, diffusion-controlled reactions are slowed down in the presence of increasing concentrations of a viscogenic reagent such as sucrose (Martin and Hergenrother, 1999). As shown in Supplementary Figure S2, the turnover number of ATPase activity was not significantly affected by changes in viscosity. Therefore, diffusion-controlled reactions such as nucleotide association or dissociation or NBD dimerization seem unlikely to be rate-limiting factors (Cole et al, 1994). Rather, we suggest that a chemical reaction such as proton abstraction or ATP hydrolysis itself governs the reaction velocity under steady-state conditions.
Since a chemical reaction such as ATP hydrolysis seems to be rate limiting, one can assume two different and extreme scenarios: ‘general base catalysis' (Fersht, 1997) or substrate assisted catalysis (SAC) (Dall'Acqua and Carter, 2000) (Figure 3A). If ATP hydrolysis depends on the action of a general base, for example, E631, proton abstraction or polarization represents the rate-limiting step of hydrolysis. If SAC applies, no proton abstraction is involved in the rate-limiting step. One possibility to distinguish both extreme cases is the use of solvent isotope effects (Schowen and Schowen, 1982). For example, bovine F1-ATPase (Abrahams et al, 1994) or phospholipase C (Martin and Hergenrother, 1999), which are assumed to follow general base catalysis, showed an increase in 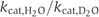 ratio of more than two (Dorgan and Schuster, 1981). On the other hand, the GTPase p21ras, which is presumed to use SAC, displayed only a slight decrease in
ratio of more than two (Dorgan and Schuster, 1981). On the other hand, the GTPase p21ras, which is presumed to use SAC, displayed only a slight decrease in 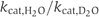 in the presence of D2O (Schweins et al, 1995; Schweins and Warshel, 1996).
in the presence of D2O (Schweins et al, 1995; Schweins and Warshel, 1996).
Figure 3.
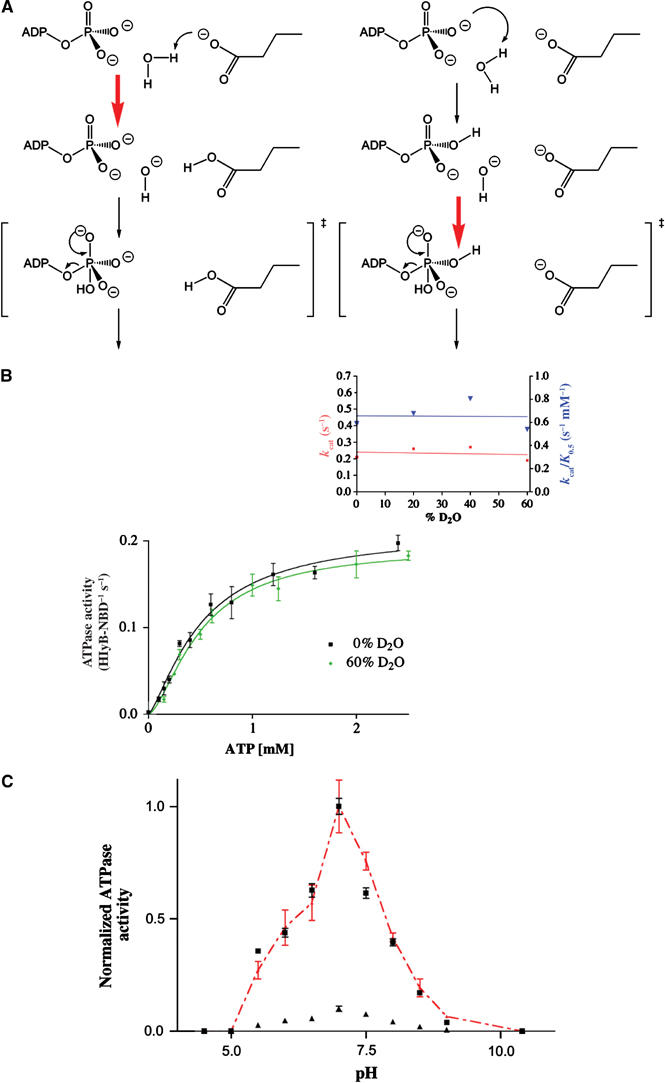
(A) Schematic drawing of the two extreme mechanisms of ATP hydrolysis. Left panel: general base catalysis; right panel: SAC. The red arrow indicates the rate-limiting step. (B) ATPase activity of wild-type HlyB-NBD in the presence of 0 and 60% D2O. The inset shows the determined _k_cat and _k_cat/_K_0.5 values for individual D2O concentrations used. (C) pH dependence of the wild-type HlyB-NBD (black squares) and the E631Q mutant (black triangles). To allow comparison of the wild-type and mutant protein, ATPase activity of the E631Q mutant was scaled to the activity of wild-type enzyme (red triangles). To guide the eye, the red, dashed line connects individual data points. Assays were performed in the presence of 1 mM ATP and 10 mM Mg2+.
As shown in Figure 3B, the ATPase activity remained essentially the same over the range 0–60% D2O with respect to _v_max. The Hill coefficient also was identical (data not shown). Similarly, no decrease in the _k_cat values or in the catalytic efficiency (_k_cat/_K_0.5) could be observed (Figure 3B, inset). Extrapolation of the _k_cat values to 100% D2O resulted in a 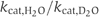 of 0.79 for HlyB-NBD, similar to the value of 0.71 reported for p21ras (Schweins and Warshel, 1996).
of 0.79 for HlyB-NBD, similar to the value of 0.71 reported for p21ras (Schweins and Warshel, 1996).
The experiment in D2O implied that proton abstraction might not be the rate-limiting step in this system. Unfortunately however, this result is inconclusive, leaving open the possibility of a base-dependent catalytic step that is not rate limiting. We chose therefore to assess further the relative roles of the H662 and E631 residues and analyzed the pH dependence of ATPase activity for the wild type and the E631Q mutant. If E631 is directly involved in catalysis, a change in pH dependence would be expected in the E631Q mutant since Q is a poorer base than E. As shown in Figure 3C, the pH dependence of the wild type and that of the E631Q mutant (retaining around 10% residual activity) are virtually identical, providing further evidence that E631 does not participate directly in catalysis. Again this result does not exclude the possibility that a base mechanism may still operate upstream of the rate-limiting step in the catalytic cycle. Nevertheless, all these results, combined with the evidence that H662 plays a crucial role in catalysis, compelled us to consider alternative mechanisms. Thus, we propose the hypothesis of SAC where the substrate participating in catalysis might be either ATP itself, as it has been suggested for some GTPases (Schweins et al, 1995), or conceivably the cofactor, Mg2+. Further studies should be directed toward an unambiguous determination of the precise nature of the chemical reaction, which represents the rate-limiting step of ATP hydrolysis for this and other ABC-ATPases.
Comparing the ATP-bound crystal structures of E/Q and H/A mutants of NBDs
As stated above, mutation of the conserved histidine in ABC transporter NBDs abolished both ATPase activity and substrate transport to background levels in all systems reported so far (Shyamala et al, 1991; Davidson and Sharma, 1997; Nikaido and Ames, 1999). This prompted us to carry out a structural comparison of the ATP-bound dimers of MJ0796 E171Q (Smith et al, 2002) and the HlyB-NBD H662A, in order to determine if this would reveal more on the molecular roles of the histidine and glutamate residues.
The two dimer structures, HlyB H662A and MJ0796 E171Q, were aligned using the Walker A and B motifs as anchor points (Figure 4). The r.m.s.d. of the 12 Cα atoms used in the superimposition was below 0.5 Å. As shown in Figure 4, the structural alignment results in a perfect overlay of the C-loop regions of both proteins. However, Q550 (HlyB) and Q91 (MJ0796), the conserved glutamines of the Q-loop, and Y477 (HlyB) and Y11 (MJ0796), which participate in adenine fixation, did not align well (not shown). In the case of the two tyrosines, the sequence flexibility in the loop, which connects strand 1 harboring the tyrosine at its C-terminus with strand 2, might explain the misalignment. Similarly, the displacement of the conserved glutamine might be due to the inherent flexibility of the helical domain (Yuan et al, 2003) and/or the presence of the SDR within the helical domain (Schmitt et al, 2003).
Figure 4.
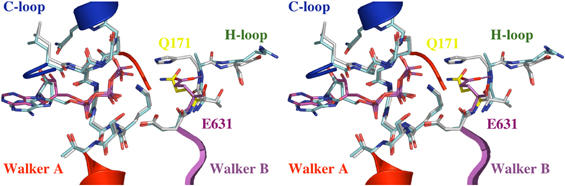
Stereo view of the superimposition of HlyB-NBD H662A in complex with ATP/Mg2+ and the ATP sandwich dimer of MJ0796 (Smith et al, 2002). The backbone of the Walker A and B motifs, C-loop, and H-loop of HlyB-NBD H662A are shown in cyan and the corresponding backbone of MJ0796 E171Q in gray. ATPs are highlighted in cyan (HlyB-NBD H662A) and magenta (MJ0796 E171Q). The interaction between the side chain of E631 (magenta) and the backbone amide of A662 is highlighted. As shown, the flip-out of Q171 (yellow) destroys this interaction in the MJ0796 E171Q structure.
Most striking in the comparative alignment, however, is the nearly 90° displacement of the side chain of Q171 in the MJ0796 E171Q structure compared to the side chain of E631 (HlyB-NBD). This flip-out (Figure 4) cannot be so easily explained by sequence variability or structural diversity since it occurs in a region of high conservation. In the H662A crystal structure, the side chain of E631 forms a hydrogen bond with the backbone amide of the substituted A662 (2.9 Å) in the same monomer. In the case of the MalK/ATP structure, this hydrogen bond is indeed present between the glutamate and the histidine (distance of 3.0 Å). However, in the case of the MJ0796 E171Q/ATP structure, because of the 90° displacement, the distance between the side chain of Q171 and the backbone amide of H204 is 4.4 Å. This suggests that the E/Q mutation in MJ0796 disrupts the hydrogen bond between the backbone of H204 and the glutamate.
A model for ATP hydrolysis of ABC transporters
In cis interaction of H662 and E631. To test the possibility that H662 and E631 normally form a vital interaction, we reintroduced H662 ‘in silico' into the HlyB-NBD structure and energy minimized the model. The hypothetical structure obtained after this procedure is shown in Figure 5. The most important conclusion from this model is that E631 in HlyB does form intramolecular hydrogen bonds with the backbone and the side chain of H662 simultaneously. Furthermore, while one of the two nitrogens of the imidazole side chain of the reintroduced H662 interacts with the side chain of E631, the other nitrogen interacts with the γ-phosphate of ATP and water 90. This organization reinforces the idea of an important role for H662 since no other side chain is available to fulfill such a dual task of simultaneously coordinating the γ-phosphate of ATP and the side chain of E631.
Figure 5.
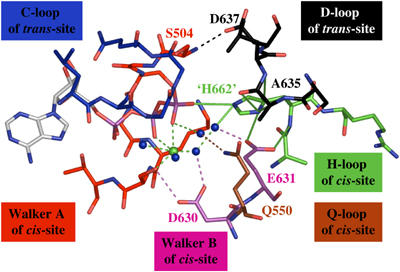
Simulated model of the prehydrolysis state of the HlyB-NBD after restoring H662. The figure demonstrates the key role of H662 through interactions with the γ-phosphate, E631, and the D-loop. Color coding is identical to Figure 1. For further details, see text.
Based on the model proposed in Figure 5, one can envision two extreme scenarios for the function of E631 and H662: in the first, E631 serves as a platform to position H662 in the proper orientation to catalyze ATP hydrolysis while H662 acting as a ‘linchpin', or vice versa. However, as argued above, E631 and H662 form a ‘catalytic dyad', which can be subdivided into a chaperone or platform function (E631) and a linchpin (H662). Consequently, we suggest that a glutamine instead of a glutamate is not able to form the proposed bidentate interaction with the backbone and side chain of H662 (Figure 4). Therefore, the histidine side chain might be no longer fixed in place and may adopt multiple, mainly nonproductive, conformations. Nevertheless, in different NBDs containing an E/Q exchange, this plasticity may be more or less tolerated.
In trans interaction of H662 and the highly conserved D-loop. In addition to the postulated bidentate interaction between H662 and E631 in our model, H662 also serves to connect the two monomers via an imidazole side chain hydrogen bonded with the backbone of S634 (3.0 Å) and van der Waals interactions with L636 and A635 (Figure 2C). These three residues are all part of the D-loop. Thus, we suggest that H662 not only participates in ATP hydrolysis, but could also serve as a key player in mediating the communication between the two monomers in the ATP-loaded composite dimer. The same interaction pattern between the histidine and the D-loop is also present in the MalK (Chen et al, 2003) and MJ0796 dimers (Smith et al, 2002). It appears that Mg2+, which is missing in the MalK/ATP structure, is not essential for this kind of monomer–monomer interaction. Rather, as discussed above, Mg2+ is important to connect the aspartate (D630 in HlyB, D170 in MJ0796, and D158 in MalK) to the Mg2+/ATP site and to orient all residues active in catalysis in a productive formation. The coordination of H662 with the D-loop at the monomer interface may finally provide a satisfactory explanation for the conservation of this second diagnostic motif of ABC-NBDs (atSALDye in HlyB): a mechanism for communication between monomers.
A putative function of the ‘linchpin' H662. What is the precise role of the ‘linchpin' H662? In subtilisin from Bacillus amyloliquefaciens, a histidine (H64) is thought to stabilize the transition state of the enzyme reaction (Carter et al, 1991). Addition of imidazole converts the inactive subtilisin H64A mutant into a moderately active enzyme (Carter et al, 1991). Similarly, imidazole also restores the inactive HlyB-NBD H662A mutant (Supplementary Figure S3). This observation suggests that H662 may stabilize the transition state of the reaction first by interacting with the γ-phosphate moiety of ATP and water 90 and second via the D-loop in the ATP-binding site of the opposing monomer. Water 90, which forms hydrogen bonds to H662, E631, and via water 109 to Q550, could serve as the catalytic water since a slight movement would be sufficient to place it in a proper orientation for in-line attack of the scissile bond.
Materials and methods
Protein purification, crystallization, and data collection
HlyB-NBD H662A was purified as described for the wild-type protein (Zaitseva et al, 2004). Crystals in the ATP/Mg2+-bound form were obtained by mixing 5 mg/ml HlyB-NBD H662A in 2 mM CAPS pH 10.4, 30% glycerol, 2 mM ATP, and 10 mM MgCl2 with 100 mM sodium malonate pH 5.6, 0.25 M sodium acetate, 5% isopropanol, and 10% PEG-MME 5500 at 4°C in a 1:1 ratio. Data from a single crystal were collected at beamline BW-6, DESY, Hamburg with a MAR CCD. Data were processed using DENZO and SCALEPACK (Otwinowski and Minor, 1997). Data statistics are summarized in Table I.
Structure determination and refinement
Native Patterson analysis revealed the presence of a dimer of dimers or a tetramer in the asymmetric unit. Initial attempts to solve the structure of the ATP/Mg2+-bound HlyB-NBD H662A by molecular replacement using the nucleotide-free structure of the HlyB-NBD (Schmitt et al, 2003) failed. In order to solve the phase problem, crystals were grown from seleno-methionine-substituted protein under conditions similar to the one used for the wild-type protein. From these crystals, a SAD data set was obtained at beamline BW-6, DESY Hamburg. In contrast to native H662A crystals, seleno-methionine-substituted crystals were perfect pseudo-merohedrally twinned (α∼0.5). Nevertheless, it was possible to obtain a molecular replacement solution from this crystal using the catalytic and helical domains of the nucleotide-free structure of the HlyB-NBD (Schmitt et al, 2003) simultaneously as search models in MOLREP (Vagin and Teplyakov, 2000). The solution obtained was subsequently used as a template for molecular replacement of the ‘native' data set using AmoRe (Navaza, 1994). The initial solution was refined using REFMAC5 (Murshudov et al, 1997), followed by repetitive rounds of manual rebuilding into 2_F_o−_F_c and 1_F_o−_F_c maps using O (Jones et al, 1991). In the initial state of refinement, strict NCS was applied to all four monomers, which was released in the last eight cycles of rebuilding and refinement. The final model consists of a dimer of dimers (964 residues), four ATP, four Mg2+, and 172 water molecules, which were determined using ARP/wARP (Lamzin and Wilson, 1993) at a threshold of 3.5σ. Refinement statistics and model geometry are given in Table I.
ATPase assay
ATPase assays of the wild-type HlyB-NBD in the presence of various amounts of D2O were performed by the malachite green assay as described in detail (Zaitseva et al, 2004) at a protein concentration of 0.2 mg/ml. To account for the deuterium-induced pH shift, buffer in 100% D2O (100 mM HEPES, 20% glycerol) was adjusted to pH 7.4 (Schowen and Schowen, 1982) prior to mixing with buffer in 100% H2O (100 mM HEPES, 20% glycerol) to obtain the indicated concentrations of D2O. Data were analyzed applying the Hill equation. Data points present the average of three independent experiments. Further details of generation and characterization of the E631Q mutant, pH dependence, and the effect of protein concentration on ATPase reaction will be given elsewhere (manuscript in preparation). ATPase assays of the HlyB-NBD H662A mutant with increasing concentrations of imidazole (pH 7.0) were performed in the presence of 1 mM ATP and 10 mM Mg2+ as described above. Experiments in solutions of different viscosity were performed in the above-mentioned buffer supplemented with 0, 5, 17.5, 25, 30, and 35% sucrose corresponding to a relative viscosity of 1, 1.1, 1.5, 2.2, 2.8, and 3.7 as measured with an Ostwald viscometer. All data shown represent the average of at least two independent experiments with the standard deviation reported as errors.
Structural alignment and energy minimization of the H662 model
Structural alignments of HlyB-NBD H662A in complex with ATP/Mg2+ and the ATP-bound form of MJ0796 E171Q (Smith et al, 2002) or MalK from E. coli (Chen et al, 2003) were performed with LSQMAN (Kleywegt, 1996) using the Walker A and B motifs as anchor points. LSQMAN was also used to structurally align HlyB-NBD H662A ATP/Mg2+ with RecA from E. coli (Story and Steitz, 1992), from Mycobacterium tuberculosis (Datta et al, 2003), EF-Tu (Bertchold et al, 1993), the α- and β-chain of F1-ATPase (Abrahams et al, 1994), ras21 (Pai et al, 1990), and T7 helicase-replicase (Sawaya et al, 1999) using the Walker A and B motifs as anchor points.
A662 of the refined structure of HlyB-NBD H662A was replaced ‘in silico' by histidine to simulate the wild-type protein. To reduce model bias, energy minimization using the GROMOS96 force field (van Gunsteren and Berendsen, 1990) of the initial model was performed with variations of the initial side-chain rotamer (±10°) of the reintroduced H662 (χ1=–65°). All other main rotamers (χ1=65° and 180°) produced steric clashes with other parts of the protein. Although small quantitative differences in the individual models were observed, qualitatively (χ1 of the final minimized model 62±3°), the described bidentate interaction was preserved.
Protein Data Bank accession code
Coordinates have been deposited in the Protein Data Bank under accession number 1XEF.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We thank Chris van der Does, Rupert Abele, Julian Chen, Alfred Wittinghofer, and Jochen Reinstein for many helpful discussions and Uli Ermler and Hartmut Michel for in-house facilities. We are indebted to Gleb Bourenkov, BW6 DESY Hamburg for his excellent support and advice during data acquisition and structure determination. LS thanks R Tampé for constant encouragement and support. This work was supported by CNRS and University of Paris-Sud (IBH) and the Deutsche Forschungsgemeinschaft (grants Schm1279/2-3 and SFB 628 to LS).
References
- Abrahams JP, Leslie AG, Lutter R, Walker JE (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria [see comments]. Nature 370: 621–628 [DOI] [PubMed] [Google Scholar]
- Benabdelhak H, Schmitt L, Horn C, Jumel K, Blight MA, Holland IB (2005) Positive cooperative activity and dimerization of the isolated ABC-ATPase domain of HlyB from E. coli. Biochem J 368: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertchold H, Reshetnikova L, Reiser COA, Schirmer NK, Sprinzl M, Hilgenfeld R (1993) Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature 365: 126–132 [DOI] [PubMed] [Google Scholar]
- Carter P, Abrahmsen L, Wells JA (1991) Probing the mechanism and improving the rate of substrate-assisted catalysis in subtilisin BPN′. Biochemistry 30: 6142–6148 [DOI] [PubMed] [Google Scholar]
- Chang G (2003) Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation. J Mol Biol 330: 419–430 [DOI] [PubMed] [Google Scholar]
- Chang G, Roth CB (2001) Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 293: 1793–1800 [DOI] [PubMed] [Google Scholar]
- Chen J, Lu G, Lin J, Davidson AL, Quiocho FA (2003) A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol Cell 12: 651–661 [DOI] [PubMed] [Google Scholar]
- Chen J, Sharma S, Quiocho FA, Davidson AL (2001) Trapping the transition state of an ATP-binding cassette transporter: evidence for a concerted mechanism of maltose transport. Proc Natl Acad Sci USA 98: 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole PA, Burn P, Takacs B, Walsh CT (1994) Evaluation of the catalytic mechanism of recombinant human Csk (C-terminal Src kinase) using nucleotide analogs and viscosity effects. J Biol Chem 269: 30880–30887 [PubMed] [Google Scholar]
- Dall'Acqua W, Carter P (2000) Substrate-assisted catalysis: molecular basis and biological significance. Protein Sci 9: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Ganesh N, Chandra NR, Muniyappa K, Vijayan M (2003) Structural studies on MtRecA–nucleotide complexes: insights into DNA and nucleotide binding and the structural signature of NTP recognition. Proteins 50: 474–485 [DOI] [PubMed] [Google Scholar]
- Davidson AL, Sharma S (1997) Mutation of a single MalK subunit severely impairs maltose transport activity in Escherichia coli. J Bacteriol 179: 5458–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorgan LJ, Schuster SM (1981) The effect of nitration and D2O on the kinetics of beef heart mitochondrial adenosine triphosphatase. J Biol Chem 256: 3910–3916 [PubMed] [Google Scholar]
- Fersht A (1997) Enzyme, Structure and Mechanism. New York: Freemann [Google Scholar]
- Geourjon C, Orelle C, Steinfels E, Blanchet C, Deleage G, Di Pietro A, Jault JM (2001) A common mechanism for ATP hydrolysis in ABC transporter and helicase superfamilies. Trends Biochem Sci 26: 539–544 [DOI] [PubMed] [Google Scholar]
- Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8: 67–113 [DOI] [PubMed] [Google Scholar]
- Higgins CF, Haag PD, Nikaido K, Ardeshir F, Garcia G, Ames GF (1982) Complete nucleotide sequence and identification of membrane components of the histidine transport operon of S. typhimurium. Nature 298: 723–727 [DOI] [PubMed] [Google Scholar]
- Holland IB, Benabdelhak H, Young J, De Lima Pimenta A, Schmitt L, Blight MA (2003) Bacterial ABC transporters involved in protein translocation. In ABC Proteins: From Bacteria to Man, Holland IB, Cole SP, Kuchler K, Higgins C (eds) pp 209–241. London: Academic Press [Google Scholar]
- Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101: 789–800 [DOI] [PubMed] [Google Scholar]
- Hung LW, Wang IXY, Nikaido K, Liu PQ, Ames GFL, Kim SH (1998) Crystal structure of the ATP-binding subunit of an ABC transporter. Nature 396: 703–707 [DOI] [PubMed] [Google Scholar]
- Janas E, Hofacker M, Chen M, Gompf S, van der Does C, Tampe R (2003) The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J Biol Chem 278: 26862–26869 [DOI] [PubMed] [Google Scholar]
- Jarchau T, Chakraborty T, Garcia F, Goebel W (1994) Selection for transport competence of C-terminal polypeptides derived from Escherichia coli hemolysin: the shortest peptide capable of autonomous HlyB/HlyD-dependent secretion comprises the C-terminal 62 amino acids of HlyA. Mol Gen Genet 245: 53–60 [DOI] [PubMed] [Google Scholar]
- Jones PM, George AM (2004) The ABC transporter structure and mechanism: perspectives on recent research. Cell Mol Life Sci 61: 682–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Karpowich N, Martsinkevich O, Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, Hunt JF (2001) Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure 9: 571–586 [DOI] [PubMed] [Google Scholar]
- Kenny B, Chervaux C, Holland IB (1994) Evidence that residues −15 to −46 of the haemolysin secretion signal are involved in early steps in secretion, leading to recognition of the translocator. Mol Microbiol 11: 99–109 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ (1996) Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D 52: 842–857 [DOI] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C (2000) Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405: 914–919 [DOI] [PubMed] [Google Scholar]
- Lamzin VS, Wilson KS (1993) Automated refinement of protein models. Acta Crystallogr D 49: 129–147 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, Emtage S (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23: 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher KP, Lee AT, Rees DC (2002) The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science 296: 1091–1098 [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM (2002) The ‘LSGGQ' motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing walker A sequence. J Biol Chem 277: 41303–41306 [DOI] [PubMed] [Google Scholar]
- Martin SF, Hergenrother PJ (1999) Catalytic cycle of the phosphatidylcholine-preferring phospholipase C from Bacillus cereus. Solvent viscosity, deuterium isotope effects, and proton inventory studies. Biochemistry 38: 4403–4408 [DOI] [PubMed] [Google Scholar]
- Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ (2002) Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J Biol Chem 277: 21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr D 50: 157–163 [Google Scholar]
- Nikaido K, Ames GF (1999) One intact ATP-binding subunit is sufficient to support ATP hydrolysis and translocation in an ABC transporter, the histidine permease. J Biol Chem 274: 26727–26735 [DOI] [PubMed] [Google Scholar]
- Orelle C, Dalmas O, Gros P, Di Pietro A, Jault JM (2003) The conserved glutamate residue adjacent to the Walker-B motif is the catalytic base for ATP hydrolysis in the ATP-binding cassette transporter BmrA. J Biol Chem 278: 47002–47008 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology, Carter CW, Sweet RM (eds) Vol. 276. London: Academic Press [DOI] [PubMed] [Google Scholar]
- Pai EF, Krengel U, Petsko GA, Goody RS, Kabsch W, Wittinghofer A (1990) Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 Å resolution: implications for the mechanism of GTP hydrolysis. EMBO J 9: 2351–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna ZE, Muller M, Peng XH, Ambudkar SV (2002) Importance of the conserved Walker B glutamate residues, 556 and 1201, for the completion of the catalytic cycle of ATP hydrolysis by human P-glycoprotein (ABCB1). Biochemistry 41: 13989–14000 [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Guo S, Tabor S, Richardson CC, Ellenberger T (1999) Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell 99: 167–177 [DOI] [PubMed] [Google Scholar]
- Schmitt L, Tampé R (2002) Structure and mechanism of ABC-transporters. Curr Opin Struct Biol 12: 754–760 [DOI] [PubMed] [Google Scholar]
- Schmitt L, Benabdelhak H, Blight MA, Holland IB, Stubbs MT (2003) Crystal structure of the ABC-domain of the ABC-transporter HlyB: identification of a variable region within the hecial domain of ABC-domains. J Mol Biol 330: 333–342 [DOI] [PubMed] [Google Scholar]
- Schowen KB, Schowen RL (1982) Solvent isotope effects on enzyme systems. Methods Enzymol 87: 551–606 [PubMed] [Google Scholar]
- Schweins T, Geyer M, Scheffzek K, Warshel A, Kalbitzer HR, Wittinghofer A (1995) Substrate-assisted catalysis as a mechanism for GTP hydrolysis of p21ras and other GTP-binding proteins. Nat Struct Biol 2: 36–44 [DOI] [PubMed] [Google Scholar]
- Schweins T, Warshel A (1996) Mechanistic analysis of the observed linear free energy relationships in p21ras and related systems. Biochemistry 35: 14232–14243 [DOI] [PubMed] [Google Scholar]
- Shyamala V, Baichwal V, Beall E, Ames GF (1991) Structure–function analysis of the histidine permease and comparison with cystic fibrosis mutations. J Biol Chem 266: 18714–18719 [PubMed] [Google Scholar]
- Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell 10: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story RM, Steitz TA (1992) Structure of the recA protein–ADP complex. Nature 355: 374–376 [DOI] [PubMed] [Google Scholar]
- Tombline G, Bartholomew LA, Tyndall GA, Gimi K, Urbatsch IL, Senior AE (2004) Properties of P-glycoprotein with mutations in the ‘catalytic carboxylate' glutamate residues. J Biol Chem 279: 46518–46526 [DOI] [PubMed] [Google Scholar]
- Urbatsch IL, Julien M, Carrier I, Rousseau ME, Cayrol R, Gros P (2000) Mutational analysis of conserved carboxylate residues in the nucleotide binding sites of P-glycoprotein. Biochemistry 39: 14138–14149 [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr D 56 ((Part 12): 1622–1624 [DOI] [PubMed] [Google Scholar]
- van Gunsteren WF, Berendsen HJC (1990) Computer simulation of molecular dynamics: methodology, applications and perspectives in chemistry. Ang Chem Int Ed Eng 29: 992–1023 [Google Scholar]
- Verdon G, Albers SV, Dijkstra BW, Driessen AJ, Thunnissen AM (2003a) Crystal structures of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: nucleotide-free and nucleotide-bound conformations. J Mol Biol 330: 343–358 [DOI] [PubMed] [Google Scholar]
- Verdon G, Albers SV, van Oosterwijk N, Dijkstra BW, Driessen AJ, Thunnissen AM (2003b) Formation of the productive ATP-Mg2+-bound dimer of GlcV, an ABC-ATPase from Sulfolobus solfataricus. J Mol Biol 334: 255–267 [DOI] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A (1999) Nucleoside triphosphate-binding proteins: different scaffolds to achieve phosphoryl transfer. Q Rev Biophys 32: 1–56 [DOI] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, Gray NJ (1982) Distantly related sequneces in the a- and b-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1: 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter C, Wilken S, Schneider E (1992) Characterization of site-directed mutations in conserved domains of MalK, a bacterial member of the ATP-binding cassette (ABC) family [corrected]. FEBS Lett 303: 41–44 [DOI] [PubMed] [Google Scholar]
- Welch RA (1991) Pore-forming cytolysins of gram-negative bacteria. Mol Microbiol 5: 521–528 [DOI] [PubMed] [Google Scholar]
- Yuan YR, Martsinkevich O, Hunt JF (2003) Structural characterization of an MJ1267 ATP-binding cassette crystal with a complex pattern of twinning caused by promiscuous fiber packing. Acta Crystallogr D 59: 225–238 [DOI] [PubMed] [Google Scholar]
- Zaitseva J, Holland IB, Schmitt L (2004) The role of CAPS in expanding the crystallisation space of the nucleotide binding domain of the ABC-transporter from E. coli. Acta Crystallogr D 60: 1076–1084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3