Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination (original) (raw)
Abstract
Activating transcription factor 3 (ATF3) is rapidly induced by diverse environmental insults including genotoxic stress. We report herein that its interaction with p53, enhanced by genotoxic stress, stabilizes the tumor suppressor thereby augmenting functions of the latter. Overexpression of ATF3 (but not a mutated ATF3 protein (Δ102–139) devoid of its p53-binding region) prevents p53 from MDM2-mediated degradation and leads to increased transcription from p53-regulated promoters. ATF3, but not the Δ102–139 protein, binds the p53 carboxy-terminus and diminishes its ubiquitination and nuclear export. Genotoxic-stressed ATF3-null mouse embryonic fibroblasts, or cells in which ATF3 was reduced by small interference RNA, show inefficient p53 induction and impaired apoptosis compared with wild-type cells. ATF3-null cells (but not wild-type cells), which poorly accumulate p53, are transformed by oncogenic Ras. Thus, ATF3 is a novel stress-activated regulator of p53 protein stability/function providing the cell with a means of responding to a wide range of environmental insult, thus maintaining DNA integrity and protecting against cell transformation.
Keywords: ATF3, p53, stability, stress, ubiquitination
Introduction
The importance of p53 induction in response to stress in determining cell fate (cell cycle arrest or apoptosis) is well established (Levine, 1997). p53 protein levels are elevated by stress via an increased stability. Consequently, p53-responsive genes are _trans_-activated and cells either arrest (to allow DNA repair) or undergo apoptosis to eliminate the damaged cells. In contrast, cells disrupted for p53 are unable to repair DNA damage, leading to uncontrolled cell proliferation and malignancy. Indeed, p53 gene mutations contribute to tumorigenesis in almost half of human cancers (Levine, 1997).
In normal, unstressed cells, the p53 protein is short-lived (_t_1/2 ∼ 20 min), reflecting a rapid turnover through ubiquitin-mediated proteolysis (Vousden, 2002). The main physiological antagonist of p53 is the MDM2 protein (Jones et al, 1995; Montes et al, 1995; Haupt et al, 1997; Kubbutat et al, 1997), which binds to the amino-terminus of the tumor suppressor and catalyzes the addition of ubiquitin moieties to lysine residues at positions 370, 372, 373, 381, 382 and 386 at the p53 carboxy-terminus (Nakamura et al, 2000; Rodriguez et al, 2000). p53 ubiquitination exposes a nuclear export signal promoting cytoplasmic relocation and degradation by proteasomes (Lohrum et al, 2001). Conversely, interruption in ubiquitination either by interfering with MDM2 binding to p53 (e.g. pRb; Michael and Oren, 2003) or by impairment of its E3 ubiquitin ligase (as for p14/19ARF; Honda and Yasuda, 1999) increases p53 protein levels.
Activating transcription factor 3 (ATF3), a 181-amino-acid protein, is a member of the ATF/CREB family of transcription factors that, like p53, is maintained at a low level in quiescent cells (Hai et al, 1999). However, ATF3 is rapidly induced by a wide-range of stresses including post-seizure brain, nerve axotomy and hepatotoxicity (Hai et al, 1999). In tissue culture, genotoxic stress (ultraviolet/ionizing radiation, DNA-damaging agents) is a strong inducer of ATF3 (Hai et al, 1999; Hai and Hartman, 2001). Interestingly, these same stress conditions also induce p53 (An et al, 1998; Nagata et al, 1999; Cheng et al, 2003; Schafer et al, 2003; Morrison et al, 2004). Although ATF3 has been described as a transcription factor (Hai et al, 1999), few target genes have been reported to date. Since ATF3 expression (a) is induced by a broad spectrum of environmental insults and (b) coincides with p53 activation in many stress responses, we hypothesized that this protein regulates p53 stability and function. Here we report that, in response to stress, ATF3 interacts with the p53 protein thereby preventing ubiquitination and degradation of the latter and consequently augmenting p53 function. Conversely, ATF3 deficiency, evident in some cancers, which yields inefficient p53 accumulation, is permissive for Ras-induced cell transformation of mouse embryonic fibroblasts (MEFs).
Results
ATF3 increases p53 stability
Since ATF3, like p53, is rapidly induced by cellular stresses, we hypothesized that the tumor suppressor is regulated by ATF3. Considering that p53 levels are mainly controlled post-translationally, we determined the effect of ATF3 on the stability of the former protein. p53, with or without ATF3, was expressed in the p53-null H1299 cells and p53 protein levels measured. ATF3 expression dramatically increased the amount of p53 (Figure 1A), an effect unlikely due to transcription, since the amount of GFP protein also expressed from a CMV promoter was not enhanced (Figure 1B). Cycloheximide chase experiments revealed that increased p53 protein stability achieved with ATF3 expression reflected reduced turnover of the former (Figure 1C). We next determined if endogenous p53 protein level was also increased by ATF3 overexpression by coexpressing ATF3 and GFP in HCT116 cells that bear wild-type p53. Endogenous p53 protein and its downstream target, p21, were elevated in the FACS-sorted GFP-positive cells made to express ATF3 (Figure 1D). Again, the elevated p53 protein level was not due to increased mRNA as evidenced by the unchanged p53/GAPDH ratio (Figure 1E). Together, these data suggest that ATF3 regulates p53 protein stability.
Figure 1.
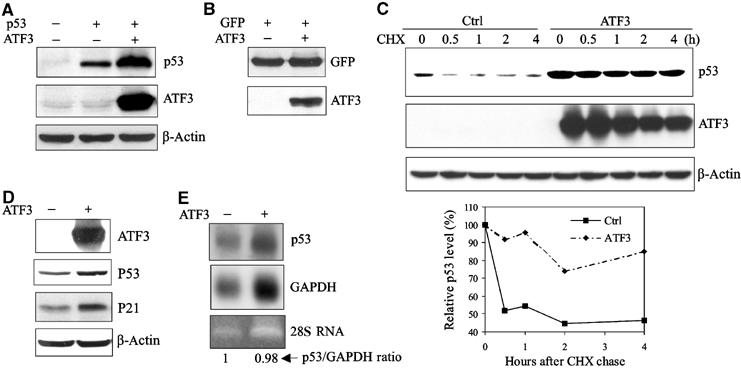
ATF3 increases p53 protein stability. (A) H1299 cells were transfected with, as indicated, 0.05 μg p53, 0.8 μg ATF3 or its vector pCG, and 0.1 μg RSV-luc. Cell lysates normalized according to luciferase activity were subjected to Western blotting. (B) H1299 cells were transfected, as indicated, with 0.05 μg pEGFP-N1, 0.8 μg ATF3 or pCG (vector), and 0.1 μg RSV-luc. Cell lysates normalized as described in (A) were subjected to Western blotting. (C) H1299 cells were transfected with a p53 expression construct and, where indicated, an expression plasmid bearing ATF3 and then treated with cycloheximide. Cells were harvested at the indicated time points, and cell lysates with an equal protein amount were subjected to Western blotting. Relative p53 levels were quantified by densitometry (D, E) HCT116 cells were transfected with pEGFP-N1 and, where indicated, with ATF3 or its vector. GFP-positive cells were collected by FACS, and subjected to Western blotting (D) or Northern blotting (E).
ATF3 directly binds p53, an interaction enhanced by genotoxic stress
Since p53 protein stability can be regulated by associated proteins (Vousden, 2002), we considered the possibility that its enhanced stability requires an interaction with ATF3. To answer this question, p53 and ATF3 were expressed in H1299 cells and co-immunoprecipitation assays performed. Indeed, p53 protein was detected in the immuno-complex captured by the ATF3 antibody (Figure 2A, lane 4). To corroborate these data, we purified a recombinant ATF3 protein (Supplementary Figure S1C, lane 2), incubated it with immobilized GST-p53 fusion protein (Figure 2C, lane 2) and performed GST pulldown assays (Figure 2B). Again, ATF3 bound p53 (Figure 2B, lane 3), suggesting a direct interaction. Note that ATF3 did not directly bind MDM2 in this assay (Figure 2B, lane 4) arguing against the possibility that the latter mediates the interaction of ATF3 and p53.
Figure 2.
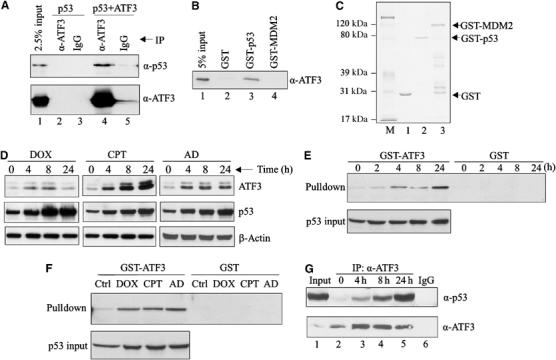
Genotoxic stress increases the direct interaction of the ATF3 and p53 proteins. (A) H1299 cells were transfected with p53 and, where indicated, with pCG/ATF3. Cell lysates (1 mg protein) were incubated with 1 μg of anti-ATF3 antibody or IgG at 4°C overnight, immunocomplexes captured with protein A-agarose and subjected to Western blotting for p53 and ATF3. (B) The indicated GST fusion proteins from bacteria culture were immobilized on glutathione-agarose and incubated with a purified recombinant ATF3 protein 4 h at 4°C. The agarose was extensively washed and eluates subjected to Western blotting for ATF3. (C) The indicated GST fusion proteins were resolved by SDS–PAGE and stained with Coomassie blue. (D) HCT116 cells were treated with 0.4 μg/ml DOX, 2 μM CPT or 5 nM AD as indicated. After the indicated times, cells were lysed and subjected to Western blotting for ATF3 and p53. (E, F) HCT116 cell lysates, adjusted to equal p53 amounts (p53 input), from unstressed and stressed cells (treated as in (D); for (E), the cells were treated with DOX) were incubated with glutathione-agarose beads bound with GST-ATF3 fusion protein or GST protein. After extensive washes, bound material was eluted and immunoblotted for p53. (G) HCT116 cells were treated with 0.4 μg/ml DOX and harvested at the indicated time points. Cell lysates (2 mg protein) were incubated with 0.5 μg ATF3 antibody overnight. Immunoprecipitates were captured and subjected to Western blotting for p53 and ATF3. Lysate from 24-h-treated cells was also immunoprecipitated with 0.5 μg normal IgG as a control (lane 6).
Next, since both ATF3 and p53 are induced by a wide range of stress, we determined if genotoxic stress affects the interaction between these two proteins. We first assessed if ATF3 and p53 were coinduced by various genotoxic agents by treating HCT116 cells with doxorubicin (DOX), camptothecin (CPT) and actinomycin D (AD), and measuring both proteins. As expected, these three agents elevated ATF3 in parallel with p53 protein levels (Figure 2D).
Interestingly, when the cells were DOX-treated, p53 protein remained elevated at 24 h despite a decline in ATF3 protein amount (Figure 2D). Therefore, we considered the possibility that, like Pin1 (Zacchi et al, 2002; Zheng et al, 2002), the ATF3–p53 interaction is enhanced during the late stage of the stress response. Pulldown assays were performed capturing p53 protein from HCT116 cell lysates prepared at varying DOX treatment times with an immobilized GST-ATF3 fusion protein (Supplementary Figure S1A). The amount of lysate used was adjusted to equal p53 input amounts as determined by Western blotting (Figure 2E, lower panel). Interestingly, while relatively little p53 was captured by the immobilized ATF3 using cell lysates from untreated cells, DOX dramatically increased the amount of pulled down p53 (Figure 2E), indicating an enhanced interaction between the two proteins. This effect was time dependent with an 18-fold augmentation evident after 24 h (Figure 2E). Similarly, CPT and AD also increased the interaction between ATF3 and p53 (Figure 2F). To explore this possibility further, lysate from DOX-treated HCT 116 cells was immunoprecipitated with an anti-ATF3 antibody and the material Western blotted for both p53 and ATF3. More ATF3–p53 protein complex was detected in lysates from DOX-treated cells than from unstressed cells (Figure 2G). These findings are reminiscent of the nuclear corepressor mSin3a that forms a persistent complex with p53 (Zilfou et al, 2001).
The ATF3 region spanning 102–139 is required for p53 binding and stabilization of the tumor suppressor
We next mapped the ATF3 region required for interaction with p53 using truncated ATF3-GST fusion proteins (Supplementary Figure S1A) in pulldown experiments with 35S-labeled p53 (Figure 3A). The full-length ATF3 protein, and peptides spanning ATF3 amino acids 1–121 and 1–141 all bound p53 (Figure 3A, lanes 6 and 7). In contrast, ATF3 peptides spanning amino acids 1–100 and 142–181 failed to bind the tumor suppressor (Figure 3A, lanes 5 and 8). Thus, the region spanning ATF3 amino acids 100–140 is required for p53 binding. This region includes the leucine zipper motif necessary for its interaction with other proteins (Hsu et al, 1992; Chen et al, 1996; Kang et al, 2003) and its deletion (Δ102–139) abrogated p53 binding (Figure 3A, lane 10). Further, expression of this ATF3 mutant in H1299 cells failed to stabilize p53 (Figure 3C) but retained its ability to bind the full-length ATF3 protein in GST pulldown assays (Figure 3B), arguing against the possibility that the former is misfolded. We therefore conclude that stabilization of p53 by ATF3 requires the interaction of these two proteins.
Figure 3.
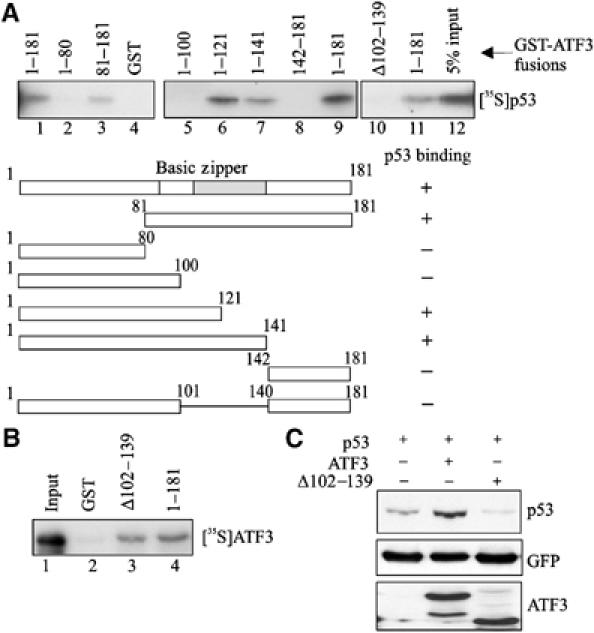
Interaction of the ATF3 and p53 proteins is required for stabilization of the tumor suppressor. (A) GST-ATF3 fusion proteins were adsorbed to glutathione-agarose, and incubated with _in vitro_-translated [35S]methionine-labeled p53 protein. After extensive washes, the bound p53 was eluted, subjected to SDS–PAGE and visualized by fluorography. (B) GST or GST fused to the mutant (Δ102–139) or full-length ATF3 protein (1–181) was adsorbed to glutathione-agarose, incubated with _in vitro_-translated [35S]methionine-labeled ATF3 protein (full length) and subjected to pulldown assays as in (A). (C) Plasmids encoding p53, the full-length ATF3 protein or mutant ATF3 protein (Δ102–139) were transfected where indicated with 0.05 μg pEGFP-N1 into H1299 cells and Western blotting performed as described in Figure 1A with the exception that the HA-tagged ATF3 proteins were detected with an anti-HA antibody.
ATF3 protects p53 from MDM2-mediated degradation by blocking ubiquitination of the tumor suppressor
How might ATF3 binding to p53 protect the latter from degradation? To answer this question, the p53 region bound by ATF3 was identified in pulldown assays with truncated GST-p53 fusion proteins (Supplementary Figure S1B). While ATF3 did not bind the p53 amino-terminus region (Figure 4A, lanes 3 and 4), a peptide spanning amino acids 362–393 of the tumor suppressor efficiently recognized ATF3 (Figure 4A, lane 8). Interestingly, this p53 region spans lysine residues 370, 372, 373, 381, 382 and 386, which are ubiquitin acceptors (Michael and Oren, 2003). Indeed, the stability of the tumor suppressor is mainly controlled by MDM2, which ubiquitinates p53 at these residues, thereby targeting it for degradation (Michael and Oren, 2003). Thus, we tested the possibility that ATF3 modulates MDM2-mediated p53 ubiquitination by utilizing an _in vitro_-reconstituted ubiquitination reaction system with E1, E2 enzymes and a GST-MDM2 recombinant protein (Supplementary Figure S1C, lane 1). In the presence of all the required ubiquitination reaction components (URCs), MDM2 catalyzed the addition of ubiquitin moieties to the _in vitro_-translated p53 protein (Figure 4B, lane 4) whereas omission of any of the URCs prevented p53 ubiquitination (Figure 4B, lanes 1–3). Importantly, addition of recombinant ATF3 protein (Supplementary Figure S1C, lane 2) reduced p53 ubiquitination in a dose-dependent manner (Figure 4B, lanes 5–7) with the highest ATF3 amounts (but not an equivalent amount of BSA; Figure 4B, lane 8) completely blocking this reaction (Figure 4B, lanes 6 and 7). This protective effect was also evident with higher input amounts of MDM2 (Figure 4C, lanes 7 and 8) that gave rise to higher molecular weight complexes that may reflect polyubiquitination.
Figure 4.
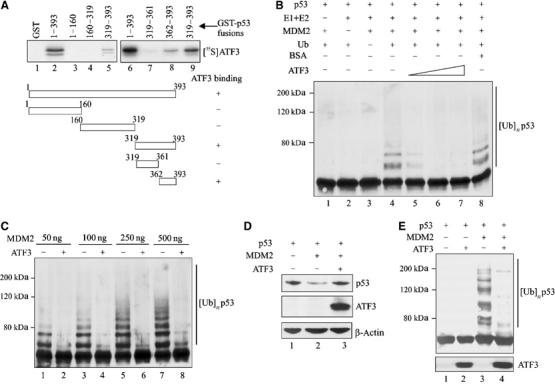
ATF3 prevents MDM2-mediated p53 degradation by blocking its ubiquitination. (A) Truncated GST-p53 proteins were immobilized onto glutathione-agarose and incubated with 35S-labeled ATF3 protein. After extensive washes, the bound ATF3 was eluted, subjected to SDS–PAGE and visualized by fluorography. (B) A 0.5 μl portion of p53 protein prepared with rabbit reticulocyte lysate was preincubated for 1 h with different amounts of purified ATF3 protein (200, 250 and 400 ng) or 400 ng BSA and then mixed with 50 ng MDM2 and other URCs at 37°C for 1 h. The reactions were then subjected to Western blotting for p53. (C) A 0.5 μl portion of p53 protein was preincubated with 250 ng ATF3 protein and subjected to ubiquitination reactions with the indicated amounts of MDM2. (D) H1299 cells were transfected, where indicated, with 0.05 μg p53, 0.4 μg MDM2, 1.5 μg ATF3 or pCG, and 0.1 μg RSV-luc plasmids, and harvested for Western blotting. (E) H1299 cells were transfected with 0.4 μg p53, 0.9 μg MDM2, 0.9 μg HA-ub, and 3.8 μg ATF3 or pCG, as indicated, and treated with 25 μM MG125 and 100 μM MG101 for 3 h before harvest. Cell lysates were adjusted to equal p53 level and subjected to Western blotting with DO-1.
We then determined if ATF3 also protected against MDM2-directed p53 ubiquitination in vivo. Expectedly (Haupt et al, 1997; Kubbutat et al, 1997), MDM2 dramatically reduced exogenous p53 levels in H1299 cells (Figure 4D, lane 2), a response effectively countered by coexpression of ATF3 (Figure 4D, lane 3). To determine if ATF3 reduced p53 ubiquitination in vivo, similar cotransfection experiments including a ubiquitin-expressing plasmid were performed. Coexpression of MDM2 with p53 dramatically increased p53 ubiquitination (Figure 4E, lane 3) whereas ATF3 coexpression antagonized this effect (Figure 4E, lane 4). Together, these in vitro and in vivo data suggest that ATF3 protects p53 from MDM2-mediated ubiquitination and degradation.
ATF3 does not inhibit the intrinsic E3 ubiquitin ligase activity of MDM2 or prevent binding of the latter to p53
ATF3 might conceivably (a) disrupt the binding of MDM2 to p53 or (b) interfere with the E3 ubiquitin ligase activity of MDM2. In pulldown assays, ATF3 did not reduce the amount of p53 bound to the immobilized MDM2 protein (Figure 5A), arguing against the first possibility. The inability of ATF3 to interfere with the p53–MDM2 interaction was unlikely due to its titration by reticulocyte proteins in the assay, since higher ATF3 inputs generated more p53-bound ATF3 (Figure 5A, lower panel, lanes 4 and 5). Alternatively, ATF3 may inhibit the intrinsic ubiquitin ligase activity of MDM2. MDM2 undergoes self-ubiquitination and this auto-ubiquitination reaction was used to determine if ATF3 directly affects the enzymatic activity. Ubiquitinated MDM2 protein was readily detected with an anti-polyubiquitin antibody FK1 (Figure 5B, lane 5) whereas no conjugates were evident upon omission of the URCs (E1, E2 or Ub; lanes 1–4). However, incubation of MDM2 with ATF3 at levels comparable to the previous experiments had no effect on MDM2 self-ubiquitination (Figure 5B, lane 7), suggesting that ATF3 does not function to inhibit E3 ubiquitin ligase. Altogether, these findings are consistent with the notion that ATF3 blocks p53 ubiquitination by binding to a region spanning amino acids 362–393 of the tumor suppressor thereby preventing ubiquitination of the latter. If this is true, the ATF3 mutant, unable to bind p53 (see Figure 3A), should show low activity in preventing p53 ubiquitination. Unlike the full-length protein, the ATF3 protein deleted of amino acids 102–139 (Supplementary Figure S1D), and unable to bind p53, was less effective in blocking p53 ubiquitination in vitro (Figure 5C) and failed to prevent MDM2-mediated degradation (Figure 5D). Therefore, binding of ATF3 to the carboxy-terminal region of p53 regulates post-translational modification of the latter, yielding a stabilized p53 protein.
Figure 5.
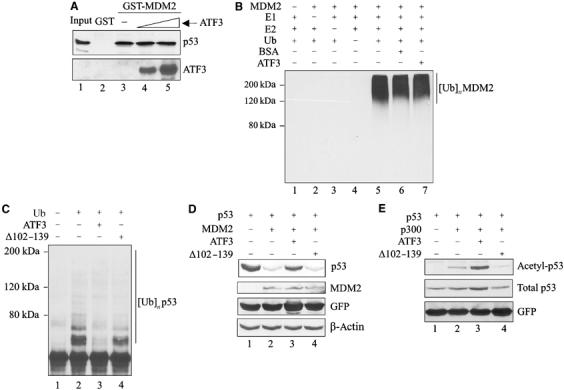
ATF3 does not interfere with either the binding of MDM2 to p53 or the ubiquitinating activity of MDM2. (A) p53 protein prepared from rabbit reticulocyte lysates (2 μl) was incubated with (1 and 1.6 μg) purified ATF3 protein at 37°C and then incubated with immobilized GST-MDM2. After extensive washes, the bound p53 was eluted and subjected to Western blotting for p53 and then reprobed with an anti-ATF3 antibody. (B) Purified ATF3 protein (250 ng) was mixed with 250 ng MDM2, URCs and BSA (250 ng) for ubiquitination reactions as indicated. The reaction mixtures were subjected to Western blotting for ubiquitinated MDM2 using anti-polyubiquitin antibody (FK-1). (C) p53 protein was preincubated with 250 ng of the full-length or the mutant ATF3 protein and then subjected to ubiquitination reactions as in Figure 4C. (D) Transfections of H1299 cells were as in Figure 4E with the exception that a plasmid encoding ATF3 deleted of the p53-binding domain (Δ102–139) and pEGFP-N1 were included. (E) H1299 cells were transfected with the following expression vectors: 0.1 μg p53, 0.1 μg RSV-luc and 0.05 μg pEGFP-N1 and, where indicated, 2 μg p300, 1.75 μg pCG, the mutant or full-length ATF3. The cells were lysed in a buffer containing 5 μM TSA and subjected to Western blotting with antibodies against Lys382 acetylated p53, total p53 (DO-1) or GFP.
Since p53 acetylation and ubiquitination may target the same residues (Ito et al, 2001), we also determined if ATF3 regulates acetylation of p53. As expected, the acetylase/coactivator p300 catalyzed the acetylation of p53 lysine 382 residue (Figure 5E, lane 2), while coexpressing ATF3, if anything, augmented acetylation (Figure 5E, lane 3). The mutant ATF3, unable to bind p53, did not increase p53 acetylation (Figure 5E, lane 4). These findings further indicate that ATF3 targets the C-terminus of the tumor suppressor.
ATF3 prevents MDM2-mediated nuclear export
P53 is a transcription factor and its nuclear localization is a likely prerequisite for its activity. We therefore considered the possibility that ATF3 regulates localization of the tumor suppressor. Nuclear export of p53 is controlled by two nuclear export signals and dependent on ubiquitination at its carboxy-terminus (Lohrum et al, 2001; Li et al, 2003). Since ATF3 prevents p53 ubiquitination, we speculated that ATF3 also blocks p53 nuclear export. To address this possibility, we transfected H1299 cells with expression vectors bearing p53, MDM2 and ATF3, and used immunofluorescence to localize the p53 and ATF3 proteins. Expectedly (Lohrum et al, 2001), MDM2 promoted p53 export to the cytoplasm (Figure 6A), with ∼40% of the transfected cells showing cytoplasmic staining with an anti-p53 antibody (Figure 6). In contrast, ATF3 expression reduced the percentage of cells positive for cytoplasmic p53 staining to ∼18% (Figure 6B). Consistent with its failure to block ubiquitination, the ATF3 mutant deficient in p53 binding (Δ102–139) lost the ability to counter MDM2-mediated p53 nuclear export (Figure 6). This lack of effect is unlikely caused by cytoplasmic redistribution of the mutant protein, since its targeting to the nucleus by N-terminal fusion of an SV40 nuclear localization sequence (NLS Δ102–139) did not prevent MDM2-mediated p53 nuclear export (Figure 6) and failed to stabilize p53 (Supplementary Figure S2A). Thus, ATF3 promotes nuclear retention of p53, presumably by blocking MDM2-directed ubiquitination of the tumor suppressor.
Figure 6.
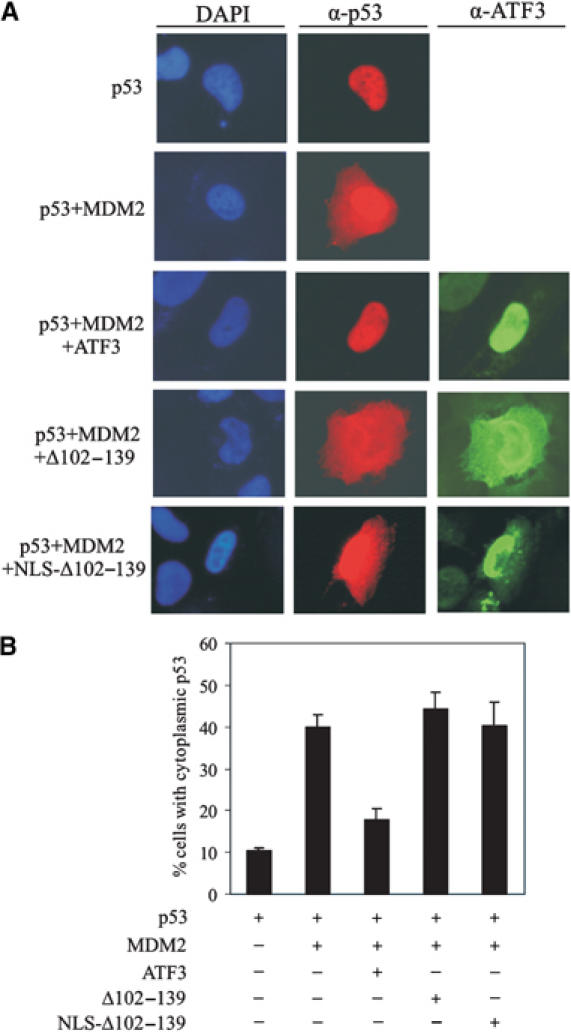
ATF3 prevents MDM2-mediated p53 nuclear export. H1299 cells were cotransfected, where indicated, with 0.1 μg p53, 0.2 μg MDM2 and 0.7 μg ATF3 expression vectors, fixed and stained using anti-p53 and anti-ATF3 antibodies for visualization (A) and counting (B). At least 300 transfected cells were scored for p53 subcellular distribution.
ATF3 augments trans-activation of p53 target promoters
If ATF3 stabilizes p53, leading to an increased nuclear level of the latter, augmented _trans_-activation of downstream target promoters would be expected. Indeed, coexpression of ATF3, but not the mutant Δ102–129 (unable to bind p53) or the NLS-tagged mutant, increased transcription from a synthetic promoter comprised of tandem repeats of a p53 response element (Figure 7A and Supplementary Figure S2B) in H1299 cells. ATF3 alone had no discernible effect on this promoter. Similarly, ATF3 augmented p53's ability to _trans_-activate several natural p53-dependent promoters (MDM2, p21, PIG3 and PUMA) (Figure 7B and C). Moreover, elevated p53 levels were evident in cells made to express the full-length but not the mutated (Δ102–139) ATF3 protein (Figure 7A). Therefore, p53 stabilization by ATF3 augments transcription from p53-dependent promoters.
Figure 7.
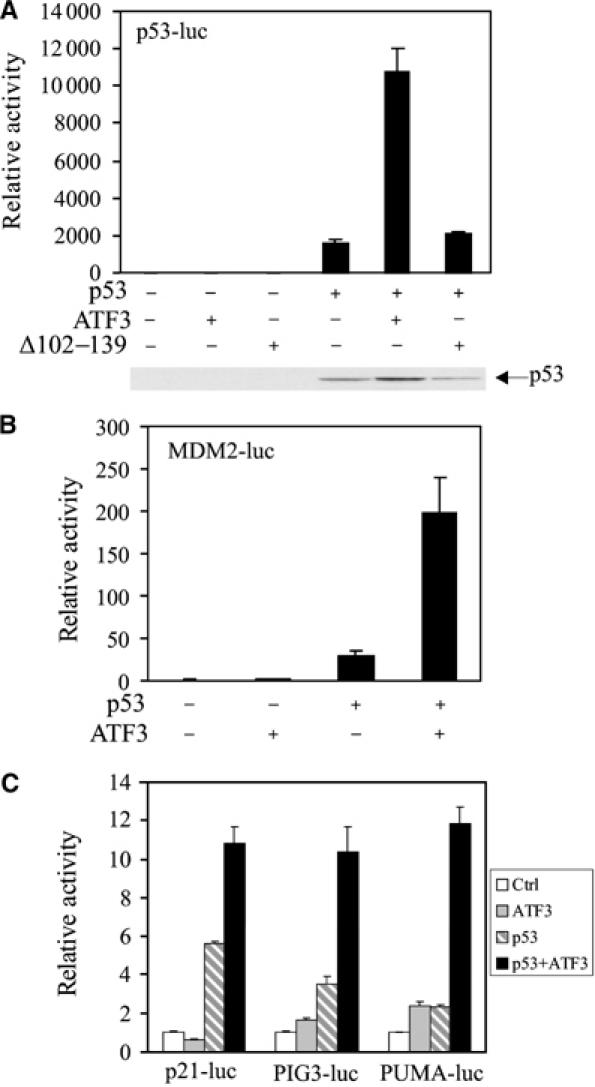
ATF3 augments _trans_-activation of p53 responsive promoters. H1299 cells were transfected, where indicated, with vectors bearing p53 (0.02 μg), ATF3 (0.4 μg) or Δ102–139 (0.4 μg), pRL-TK (0.001 μg), and a reporter regulated by tandem repeated p53 response elements (0.01 μg) (A) or 0.1 μg of the following promoters: MDM2 (B), p21, PIG3 or PUMA (C). Cells were lysed 30 h later and subjected to luciferase assays using the Dual-luciferase Assay System. p53 protein level was determined by Western blotting and shown for (A).
Loss of ATF3 impairs p53 stabilization in the genotoxic response
Since ATF3 stabilizes p53 and binds the tumor suppressor avidly in genotoxic-stressed cells, we determined whether DNA damage-induced p53 activation is impaired in ATF3-null MEFs (Hartman et al, 2004). As expected, treatment of cells with CPT induced ATF3 in parallel with p53 in ATF3+/+ (WT) cells (Figure 8A), whereas this effect was compromised in ATF3−/− cells (Figure 8A). Similar results were evident with DOX and AD treatments as well as ultraviolet and γ irradiation (see Supplementary Figure S3A). These effects were unlikely related to adaptive selection (toward p53 inactivation) occurring with in vitro culturing of the MEFs, since acute reduction of ATF3 levels in the p53-wild-type A549 cells using small interference RNA (siRNA) diminished the CPT-dependent induction of p53 protein and its downstream target p21 (Supplementary Figure S4A).
Figure 8.
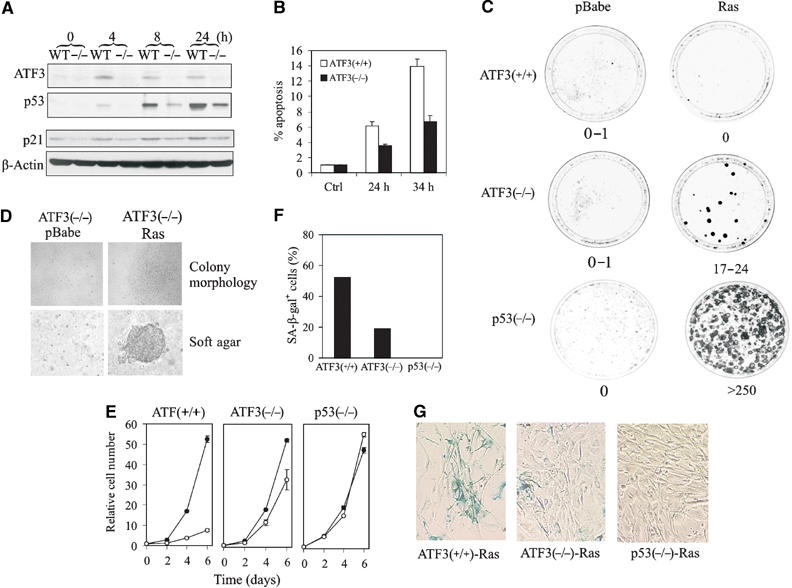
Loss of ATF3 impairs the p53-dependent cellular response to DNA damage and allows cellular transformation by an oncogenic Ras. (A) ATF3-null (−/−) or wild-type (WT) MEFs were treated with 2 μM CPT. Cells were lysed and subjected to Western blotting for ATF3, p53 or p21. (B) MEFs were infected with a retrovirus bearing E1A 12S, treated with 2 μM CPT and harvested and fixed at the indicated times. Apoptotic cells were defined as sub-G0/G1 cell as determined by FACS analysis. (C) MEFs were infected with a retrovirus bearing H-rasV12 or empty vector (pBabe). After selecting in puromycin for 4 days, the cells were plated in triplicate for colony formation assays. (D) Colony morphology of Ras-transformed ATF3−/− cells in monolayer and in soft agar. (E) Infected MEFs were plated in 12-well plates, harvested at the indicated times and cell numbers determined by crystal violet staining. Open circle indicates Ras and closed circle pBabe. (F, G) Ras-infected MEFs were stained with X-gal (pH 6.0) overnight. X-gal staining-positive cells were counted (F) under a microscope.
Loss of ATF3 impairs p53 proapoptotic and tumor suppressor functions
If ATF3 stabilizes p53, the function of the tumor suppressor should be compromised in cells null for the former gene. We first determined whether the p53-mediated apoptotic response was deficient in ATF3−/− MEFs. E1A sensitizes MEFs to apoptosis through a p53-dependent mechanism (Lowe et al, 1994) and ATF3-deficient and wild-type MEFs infected with a retrovirus bearing E1A 12S were exposed to CPT or DOX to examine their sensitivities to these agents. Consistent with their ability to elevate p53 protein levels (Figure 8A and Supplementary Figure S3A), CPT and DOX induced apoptosis in the ATF3 wild-type cells whereas the percentage of ATF3−/− apoptotic cells was reduced by ∼50% compared with the wild-type cells (Figure 8B and Supplementary Figure S3B). These data would indicate that ATF3 loss compromises the proapoptotic effect of p53. Although AD failed to induce apoptosis, more ATF3−/− cells (compared with wild-type cells) were evident in S phase upon treatment, again indicating impaired p53 function (Supplementary Figure S3C).
To rule out the possibility that ATF3-null cells had accumulated genetic defects in the p53 pathway thereby compromising apoptotic function, ATF3 levels in A549 cells were acutely reduced by siRNA and the effect on apoptosis was measured. Diminished ATF3 levels yielded a lower percentage of apoptotic nuclei in response to CPT treatment (Supplementary Figure S4B), arguing against the possibility that in vitro culturing of the ATF3-null MEFs leads to mutational inactivation of the p53 pathway thereby compromising the response to apoptotic inducers.
We next determined if p53 tumor suppressor function is compromised in ATF3-deficient cells. Expression of an oncogenic H-rasV12 alone is unable to transform MEFs due to the protective effect of p53, as evidenced by transformation of p53-null cells (Figure 8C; Serrano et al, 1997). We therefore determined if ATF3−/− MEFs would now demonstrate sensitivity to this oncogene. We infected ATF3-null, p53-null (as controls) and wild-type MEFs with a retrovirus expressing oncogenic Ras, and after puromycin selection, colony forming assays were performed. In triplicate experiments, the spontaneous transformation rate in the wild-type MEFs was 0–1 colonies per dish and oncogenic Ras expression failed to augment the transformation rate above background (0–1 colonies per dish) (Figure 8C). In contrast, Ras expression in the ATF3-deficient fibroblasts yielded 17–24 colonies per dish (Figure 8C) with cells exhibiting typical transformed phenotype including lack of contact inhibition and growth in soft agar (Figure 8D). On the other hand, the ATF3-deficient cells infected with the retrovirus bearing the empty vector (pBabe) neither produced transformed foci in monolayer culture or established colonies in soft agar (Figure 8D). These data are consistent with the notion that inefficient p53 stabilization, evident in ATF3-null cells, allows for cell transformation by Ras.
Compared with the control virus (pBabe), Ras expression arrested growth of wild-type MEFs (Figure 8E, left panel), presumably by inducing premature senescence (Serrano et al, 1997) as evident by cellular β-galactosidase activity (Figure 8G). Expectedly, this effect was completely abolished by loss of p53 (Figure 8E, right panel), and partially countered by ATF3 deficiency (Figure 8E, middle panel). Thus, while p53 loss abrogated Ras induction of β-galactosidase-positive cells, loss of ATF3 reduced by ∼60% the number of positive cells (Figure 8F and G). Thus, ATF3 loss partially abolishes p53 function, with the remaining p53 in ATF3-null cells accounting for incomplete abolition of senescence and inefficient transformation. Nevertheless, ATF3 deficiency increases the sensitivity of fibroblasts to oncogene-directed transformation, indicating a possible role of ATF3 in tumorigenesis.
Discussion
We have identified a novel regulator (ATF3) of p53 stability and function. The significance of this finding stems from the fact that ATF3 is rapidly induced by a wide range of stresses including genotoxic stress (Hai and Hartman, 2001). Indeed, to our knowledge, ATF3 is the first regulator of p53 stability that shows such a broad responsiveness to diverse stress inputs. This novel sensing mechanism thus allows the cell to be responsive to a wide range of environmental insults leading to p53 induction and hence allowing for cell repair or elimination, events necessary to protect against genomic instability and cell transformation.
Like mSin3a (Zilfou et al, 2001), ATF3 protects p53 by a direct interaction between these proteins. The ATF3–p53 interaction is achieved in two ways. First, ATF3 level, low in unstressed cells, is rapidly induced by cellular stresses (Hai et al, 1999) thus increasing the chance encounter between these proteins. Second, our study shows that genotoxic stress strongly enhances the interaction between ATF3 and p53. The latter mechanism provides for p53 stabilization in the face of declining ATF3 expression allowing for continued p53 function. This persistent interaction is also evident with mSin3a, which, like ATF3, protects p53 from degradation. The propyl isomerase Pin1 also stabilizes p53 via an increased affinity for the tumor suppressor (Zacchi et al, 2002; Zheng et al, 2002) although the former achieves this effect via a conformational change in p53.
ATF3 regulates p53 stability by interfering with ubiquitination of the latter. This block is achieved by ATF3 binding to the p53 carboxy-terminus region harboring most of the lysine residues targeted for ubiquitination (Michael and Oren, 2003). Thus, the binding of ATF3 to this region may prevent these sites from being ubiquitinated. Indeed, our data clearly showed a direct interaction of the ATF3 and p53 proteins, and deletion of the p53 interaction domain in ATF3 yielded a protein that failed to (a) stabilize the tumor suppressor and (b) prevent MDM2-directed ubiquitination. The in vitro pulldown assays would also argue against the possibility that ATF3 increases the expression of an intermediary protein that enhances p53 stability. This activity of ATF3 in antagonizing p53 ubiquitination is a novel function distinct from its well-recognized role as a transcription factor. Nevertheless, the dual functions of ATF3 are not without precedent for a transcription factor since Yin Yang 1 (YY1) increases p53 ubiquitination leading to an accelerated turnover (Sui et al, 2004).
We found little evidence that ATF3 modulates the MDM2 binding to p53, a mechanism evident for the coactivator protein TAF(II)31 (Buschmann et al, 2001) and YY1 (Sui et al, 2004). This is predictable considering that ATF3 (a) and MDM2 bind separate p53 regions, and (b) does not directly bind MDM2. In fact, ATF3 closely resembles the protection of p53 by p14/19ARF, c-Abl and HIF-1α (Honda and Yasuda, 1999; Sionov et al, 2001; Chen et al, 2003) where increased p53 stability is achieved via reduced ubiquitination. Nevertheless, it is worth noting that the mechanism by which ATF3 decreases p53 ubiquitination differs from these other regulators. HIF-1α directly binds MDM2 thereby antagonizing ubiquitination of the tumor suppressor (Chen et al, 2003) while p14/19ARF blocks the intrinsic ubiquitinating activity of MDM2 (Honda and Yasuda, 1999). However, ATF3 neither prevents MDM2 binding to p53 nor affects its intrinsic ubiquitinating activity. Rather, ATF3 binds the p53 carboxy-terminus thereby inhibiting the ubiquitination of target acceptor lysines. However, this effect may not be due to steric hindrance, since lysine 382 in this region could be acetylated in the presence of ATF3. More likely, the binding of ATF3 to this region renders a conformation change, thus favoring one modification over the other.
ATF3 also blocked the MDM2-mediated p53 nuclear export. The p53 protein contains two nuclear export signals one of which resides (amino acids 339–352) in the C-terminal oligomerization domain (Stommel et al, 1999) and binding of ATF3 proximal to the C-terminus nuclear export signal might block p53 nuclear export. However, it is known that the ability of MDM2 to efficiently export p53 from the nucleus depends on its function as a ubiquitin ligase targeting the lysine acceptors in the p53 carboxy-terminus (Geyer et al, 2000; Lohrum et al, 2001; Li et al, 2003). Thus, more likely, the role of ATF3 in blocking this ubiquitination accounts for the diminished p53 nuclear export. Interestingly, decreased p53 ubiquitination in the nucleus may have another consequence in light of recent findings (Joseph et al, 2003; Li et al, 2003) of p53 turnover by nuclear proteasomes. Thus, reduced p53 ubiquitination achieved with ATF3 may diminish turnover of the tumor suppressor by nuclear proteasomes. Another possibility worth considering relates to the relocalization of p53 to the mitochondria, a prerequisite for transcriptional-independent apoptosis (Schuler and Green, 2005). Although the ubiquitinated p53 (i.e. which escaped ATF3) presumably relocalizes to the cytoplasm, it is not presently known whether this p53 pool will enter the mitochondria to render transcription-independent apoptosis.
Interestingly, consistent with its role in regulating p53 stability, loss of ATF3 impaired p53 activation in response to genotoxic stress. These findings could have implications in cancer. Indeed, the potential significance of ATF3 deficiency as contributing to carcinogenesis resides in two separate observations: (a) sensitivity of ATF3-null cells to Ras transformation and (b) expression profiling studies showing reduced ATF3 expression in renal and lung cancers (Garber et al, 2001; Higgins et al, 2003). However, spontaneous tumor formation has not been observed in the ATF3-null mice, presumably due to residual p53 protecting against tumorigenesis. In line with this contention, hematopoietic stem cells reduced in p53 amounts by RNAi give rise, in some instances, to benign lymphoid hyperplasias in mice (Hemann et al, 2003) despite compromised apoptotic function in these cells. Certainly, the extensive number of independent regulators of p53 is well known and arguably may partially substitute for ATF3 deficiency. In this regard, whether deletion of the ATF3 gene, compromising p53 function, partially rescues the lethality of _mdm2_−/− embryos is debatable and worthy of future investigations. Nevertheless, in the context of cancer, it is probable that reduced ATF3 levels combined with defects in other regulators of p53 act in concert to diminish p53 levels, thereby contributing to tumorigenesis.
In conclusion, we have identified the stress-inducible ATF3 as a novel regulator of p53 stability. ATF3 interacts with p53 to block ubiquitination of the latter. Our data argue for a model in which, in response to stress, ATF3 expression is elevated increasing its interaction with p53 and protecting the latter from the prodegradation effect of MDM2. What sets ATF3 apart from other regulators of p53 stability is the fact that it is induced by a broad spectrum of cellular stresses. We propose that this novel ‘sensing mechanism' provides the cell with a means of responding to diverse environmental insult, thus maintaining cell homeostasis and protecting against cell transformation.
Materials and methods
Plasmids
We obtained plasmids for p53, HDM2, PUMA-luc and p21-luc (B Vogelstein), MDM2 (C Blattner), p300 (T Yao), PIG3-luc (X Lu), pWZL-E1A12S and pBabe-H-RasV12 (SW Lowe) and HA-ubiquitin (Y Gotoh). We constructed plasmids for GST-ATF3, GST-p53 or GST-MDM2 fusion proteins by PCR amplifying the corresponding ATF3/p53/MDM2 fragments and cloning these fragments into pGEX-3X (Amersham Biosciences). We also constructed ATF3Δ102–139 using the QuickChange Site-directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. This truncated cDNA and full-length ATF3 cDNA were also PCR amplified and cloned into pTrcHis2 (Invitrogen) to express His-tagged recombinant proteins. An NLS (PKKKRKV) or an HA tag was also added to the N-terminus of the truncated ATF3 cDNA by PCR.
Cell culture and transfections
H1299 and HCT116 cells were cultured in McCoys 5A medium/10% FCS, and MEFs from ATF3-null animals (congenic in C57BL6 background) (Hartman et al, 2004) were grown in DMEM/10% serum. Transfections were performed with Fugene 6.0 reagent (Roche) or lipofectamine 2000 (Invitrogen). For controlling transfection efficiency, an RSV-luc plasmid was included. Except where indicated, cells were harvested 30 h after transfections for Western blotting. Transfections to determine promoter activity were performed with lipofectamine 2000. Smartpool siRNAs specifically targeting ATF3 (Dharmacon) were transfected with DharmaFect 4 (Dharmacon) according to the manufacturer's recommendations.
Pulldown assays
GST pulldown assays to map the p53-binding or ATF3-binding domain were as described previously (Horikoshi et al, 1995). To determine the interaction between p53 and ATF3 in stressed cells, pulldown assays were performed following the protocol described (Shen et al, 1998) with modifications. After equalizing p53 input concentrations (by pre-running a Western blotting to determine p53 levels), cell lysates were incubated with glutathione-agarose beads bound with GST-ATF3 fusion protein for 4 h at 4°C. Beads were washed and the bound protein eluted and subjected to Western blotting for p53 protein.
Western blotting, immunoprecipitation and cycloheximide chase assays
Western blotting and immunoprecipitation were performed as previously described (Yan et al, 2003). Antibodies for p53 (DO-1, Pab1801, FL393) and ATF3 (C-19) were from Santa Cruz Biotechnology. A mixture of DO-1 and Pab1801 was used for immunoblotting human p53 protein, while FL393 was used to detect mouse p53 protein in MEFs. Antibodies for p21, acetylated p53 (Lys382), GFP and β-actin were from BD Pharmingen, Cell Signaling, Clontech and Sigma Chemicals, respectively. For cycloheximide chase assays, we treated cells with 50 μg/ml cycloheximide 30 h after transfections, harvested the cells at indicated time points and subjected cell lysates to Western blotting.
Cytoimmunostaining
Cells were fixed in 4% formaldehyde for 15 min at 4°C, permeabilized with 0.2% Triton X-100 (5 min) at room temperature and blocked with 5% normal horse serum/1% normal goat serum. Cells were sequentially incubated with p53 DO-1 antibody (1:150), Alexa Fluor 594-conjuated anti-mouse IgG (Molecular Probes), ATF3 antibody (1:50), Alexa Fluor 485-conjugated anti-rabbit IgG and then counterstained with DAPI.
Protein purification and in vitro ubiquitination assays
GST-MDM2 and ATF3 proteins were expressed in Escherichia coli BL21, and purified with glutathione-agarose (Sigma) or Ni+-NTA agarose (Invitrogen), respectively. For in vitro ubiquitination assays, we added 0.5 μl of _in vitro_-translated p53 protein, preincubated with or without ATF3 or Δ102–139 at 37°C for 1 h in a 30 μl reaction containing 40 mM Tri–HCl, pH 7.5, 5 mM MgCl2, 2 mM DTT, 2 mM ATP and, where indicated, 25 ng E1, 100 ng E2, 5 μg ubiquitin (E1, E2 and ubiquitin were obtained from Boston Biochem) and varying amounts of GST-MDM2. Ubiquitination reactions were performed at 37°C (1 h), terminated by boiling in SDS-loading buffer (5 min) and subjected to Western blotting for ubiquitinated p53 (with DO-1) or MDM2 (with FK-1 from BioMol).
Retrovirus infections and function assays
We transfected Phoenix packaging cells (a gift from P Nolan) with pWZL-E1A12S, pBabe-H-RasV12 or its vector using lipofectamine 2000. After 48 h, supernatants were harvested and used to infect MEFs in the presence of 4 μg/ml polybrene (Sigma) twice at 8 h intervals (Liu et al, 2004). MEFs were then selected with 75 μg/ml hygromycin B (E1A infection) or 2 μg/ml puromycin (Ras infection) for 4 days. For apoptosis assays, resistant cells were expanded and treated with CPT. Cells in the sub-G0/G1 fraction, as determined by FACS, were defined as apoptotic. For cell transformation assays, 104 virus-infected cells were mixed with 3 × 105 uninfected cells (Serrano et al, 1997). Colony formation was determined after 2 weeks. Infected cells (2 × 104) were also mixed with 0.3% agar, plated on a 0.5% agar layer and cultured for 2 weeks to determine anchorage-independent growth. Growth curves and staining for senescence-associated β-galactosidase activity were performed as described (Serrano et al, 1997).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
This work was supported by grants NIH DE10845 and CA58311 to DB. We thank Drs B Vogelstein, KH Vousden, C Blattner, X Lu, W Gu, Z-M Yuan, T Yao, Y Gotoh, P Nolan, M Murphy, T Shenk and SW Lowe for the materials. We are grateful to Dr Gigi Lozano for critical intellectual input. We also thank Dr Geng Liu for technical advice.
References
- An WG, Kanekal M, Aaronswon SA, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM (1998) Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature 392: 405–408 [DOI] [PubMed] [Google Scholar]
- Buschmann T, Lin Y, Aithmitti N, Fuchs SY, Lu H, Resnick-Silverman L, Manfredi JJ, Ronai Z, Wu X (2001) Stabilization and activation of p53 by the coactivator protein TAFII31. J Biol Chem 276: 13852–13857 [DOI] [PubMed] [Google Scholar]
- Chen BP, Wolfgang CD, Hai T (1996) Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153/Chop10. Mol Cell Biol 16: 1157–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Li M, Luo J, Gu W (2003) Direct interactions between HIF-1α and Mdm2 modulate p53 function. J Biol Chem 278: 13595–13598 [DOI] [PubMed] [Google Scholar]
- Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV (2003) Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med 9: 338–342 [DOI] [PubMed] [Google Scholar]
- Garber ME, Troyanskaya OG, Schluens K, Peterson S, Thaesler Z, Pacyna-Gengelbach M, van de Rijun M, Rosen GD, Peron CM, Whyte RI, Altman RB, Brown PO, Botsein D, Peterson I (2001) Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA 98: 13784–13789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer RK, Yu ZK, Maki CG (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol 2: 569–573 [DOI] [PubMed] [Google Scholar]
- Hai T, Hartman MG (2001) The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and hemeostasis. Gene 273: 1–11 [DOI] [PubMed] [Google Scholar]
- Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U (1999) ATF3 and stress responses. Gene Expression 7: 321–325 [PMC free article] [PubMed] [Google Scholar]
- Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T (2004) Role for activating transcription factor 3 in stress-induced β-cell apoptosis. Mol Cell Biol 24: 5721–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299 [DOI] [PubMed] [Google Scholar]
- Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ, Cordon-Cardo C, Hannon GJ, Lowe SW (2003) An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet 33: 396–400 [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Shinghal R, Gill H, Reese JH, Terris M, Cohen RJ, Fero M, Pollack JR, van de Rijun M, Brooks JD (2003) Gene expression patterns in renal cell carcinoma assessed by complementary DNA microarrys. Am J Pathol 162: 925–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Yasuda H (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J 18: 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi N, Usheva A, Chen J, Levine AJ, Weinmann R, Shenk T (1995) Two domains of p53 interact with the TATA-binding protein, and the adenovirus 13S E1A protein disrupts the association, relieving p53-mediated transcriptional repression. Mol Cell Biol 15: 227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JC, Bravo R, Taub R (1992) Interactions among LRF-1, JunB, c-Jun, and c-Fos define a regulatory program in the G1 phase of liver regeneration. Mol Cell Biol 12: 4654–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Lai C, Zhao X, Saito S, Hamilton M, Appella E, Yao T (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 20: 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A (1995) Rescue of embryonic lethality in mdm2-deficient mice by absence of p53. Nature 378: 206–208 [DOI] [PubMed] [Google Scholar]
- Joseph TW, Zaika A, Moll UM (2003) Nuclear and cytoplasmic degradation of endogenous p53 and HDM2 occurs during down-regulation of the p53 response after multiple types of DNA damage. FASEB J 17: 1622–1630 [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen C, Massague J (2003) A self-enabling TGFβ response coupled to stress signaling: smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 11: 915–926 [DOI] [PubMed] [Google Scholar]
- Kubbutat MHG, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W (2003) Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302: 1972–1975 [DOI] [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 36: 63–68 [DOI] [PubMed] [Google Scholar]
- Lohrum MAE, Woods DB, Ludwig RL, Balint E, Vousden KH (2001) C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol 21: 8521–8532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Jacks T, Housman DE, Ruley HE (1994) Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci USA 91: 2026–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael D, Oren M (2003) The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 13: 49–58 [DOI] [PubMed] [Google Scholar]
- Montes OL, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378: 203–206 [DOI] [PubMed] [Google Scholar]
- Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA (2004) p53-dependent cell death signaling in neurons. Neurochem Res 28: 15–27 [DOI] [PubMed] [Google Scholar]
- Nagata M, Takenaka H, Shibagaki R, Kishimoto S (1999) Apoptosis and p53 protein expression increase in the process of burn wound healing in guinea-pig skin. Br J Dermatol 140: 829–838 [DOI] [PubMed] [Google Scholar]
- Nakamura S, Roth JA, Mukhopadhyay T (2000) Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol Cell Biol 20: 9391–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Desterro JMP, Lain S, Lane DP, Hay RT (2000) Multiple C-terminal lysine residues target p53 for ubiquitin–proteasome-mediated degradation. Mol Cell Biol 20: 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer T, Scheuer C, Roemer K, Menger MD, Vollmar B (2003) Inhibition of p53 protects liver tissue against endotoxin-induced apoptotic and necrotic cell death. FASEB J 17: 660–667 [DOI] [PubMed] [Google Scholar]
- Schuler M, Green DR (2005) Transcription, apoptosis and p53: catch-22. Trends Genet 21: 182–187 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Shen M, Stukenberg PT, Kirschner MW, Lu KP (1998) The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phsophorylation. Genes DeV 12: 706–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sionov RV, Coen S, Goldberg Z, Berger M, Bercovich B, Ben-Neriah Y, Ciechanover A, Haupt Y (2001) C-Abl regulates p53 levels under normal and stress conditions by preventing its nuclear export and ubiquitination. Mol Cell Biol 21: 5869–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J 18: 1660–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui G, Affar EB, Shi Y, Brignone C, Wall NR, Yin P, Donohoe M, Luke MP, Calvo D, Grossman SR, Shi Y (2004) Yin Yang 1 is a negative regulator of p53. Cell 117: 859–872 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2002) Activation of the p53 tumor suppressor protein. Biochim Biophys Acta 1602: 47–59 [DOI] [PubMed] [Google Scholar]
- Yan C, Wang H, Toh Y, Boyd DD (2003) Repression of 92-kDa type IV collagenase expression by MTA1 is mediated through direct interactions with the promoter via a mechanism, which is both dependent on and independent of histone deacetylation. J Biol Chem 278: 2309–2316 [DOI] [PubMed] [Google Scholar]
- Zacchi P, Gostissa M, Uchida T, Salvagno C, Avollo F, Vollnia S, Fonal Z, Blandlno G, Schneider C, Sal GD (2002) The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419: 853–857 [DOI] [PubMed] [Google Scholar]
- Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZJ (2002) The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419: 849–853 [DOI] [PubMed] [Google Scholar]
- Zilfou JT, Hoffman WH, Sank M, George DL, Murphy M (2001) The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. Mol Cell Biol 21: 3974–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4