Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes (original) (raw)
Abstract
We have conducted a comprehensive search for conserved elements in vertebrate genomes, using genome-wide multiple alignments of five vertebrate species (human, mouse, rat, chicken, and Fugu rubripes). Parallel searches have been performed with multiple alignments of four insect species (three species of Drosophila and Anopheles gambiae), two species of Caenorhabditis, and seven species of Saccharomyces. Conserved elements were identified with a computer program called phastCons, which is based on a two-state phylogenetic hidden Markov model (phylo-HMM). PhastCons works by fitting a phylo-HMM to the data by maximum likelihood, subject to constraints designed to calibrate the model across species groups, and then predicting conserved elements based on this model. The predicted elements cover roughly 3%–8% of the human genome (depending on the details of the calibration procedure) and substantially higher fractions of the more compact Drosophila melanogaster (37%–53%), Caenorhabditis elegans (18%–37%), and Saccharaomyces cerevisiae (47%–68%) genomes. From yeasts to vertebrates, in order of increasing genome size and general biological complexity, increasing fractions of conserved bases are found to lie outside of the exons of known protein-coding genes. In all groups, the most highly conserved elements (HCEs), by log-odds score, are hundreds or thousands of bases long. These elements share certain properties with ultraconserved elements, but they tend to be longer and less perfectly conserved, and they overlap genes of somewhat different functional categories. In vertebrates, HCEs are associated with the 3′ UTRs of regulatory genes, stable gene deserts, and megabase-sized regions rich in moderately conserved noncoding sequences. Noncoding HCEs also show strong statistical evidence of an enrichment for RNA secondary structure.
Despite tremendous progress in vertebrate genomics, it is still not clear how much of the human and other vertebrate genomes are directly functional, in the sense of encoding proteins or RNAs helping to regulate transcription and translation, enabling replication, altering chromatin structure, or performing other important cellular tasks. It is even less clear exactly which regions are functional. More is known about the functional roles of sequences in the genomes of model eukaryotes such as Drosophila melanogaster, Caenorhabditis elegans, and Saccharomyces cerevisiae, but much remains to be learned in these genomes as well. Especially in larger genomes, where functional elements are believed to account for only a small fraction of all bases, effective general-purpose methods for identifying sequences likely to be functional are of critical importance.
One of the best strategies known for finding functional sequences is to look for sequences that are conserved across species (e.g., Hardison et al. 1997; Loots et al. 2000; Boffelli et al. 2003; Kellis et al. 2003; Margulies et al. 2003; Woolfe et al. 2005). While orthologous sequences from related species might appear “conserved” (i.e., unusually similar) because of reduced mutation rates (Wolfe et al. 1989; Clark 2001; Ellegren et al. 2003; Hardison et al. 2003), the primary reason for cross-species sequence conservation is believed to be negative (purifying) selection. Thus, orthologous sequences that are significantly more similar than would be expected if they were evolving under some reasonable model of neutral evolution are likely to have critical functional roles. Thanks to a recent explosion in the number of sequenced genomes, and to the development of tools that allow whole genomes to be aligned (Brudno et al. 2003; Blanchette et al. 2004; Bray and Pachter 2004), cross-species conservation is emerging as a primary research tool in genomics. It is now possible to conduct large-scale searches for conserved sequences and to use the results of such searches to help stimulate new hypotheses and drive experimentation (Nobrega et al. 2003, 2004; Frazer et al. 2004; Woolfe et al. 2005).
Comparative studies suggest that mammalian genomes contain large numbers of functional elements that have yet to be identified and characterized. Analyses of human and rodent genomes (Mouse Genome Sequencing Consortium 2002; Chiaromonte et al. 2003; Roskin et al. 2003; Cooper et al. 2004; Rat Genome Sequencing Project Consortium 2004) indicate that about 5% or more of bases in mammalian genomes are under purifying selection, while protein-coding genes are believed to account for only about 1.5% of bases, leaving at least 3.5% that are thought to be functional, but not to code for proteins. (A high rate of turnover of constrained bases might put this fraction considerably higher; see Smith et al. 2004.) This conserved noncoding sequence—the “dark matter” of the genome—has been the subject of intense recent interest (e.g., Frazer et al. 2001, 2004; Shabalina et al. 2001; Dermitzakis et al. 2002; Bejerano et al. 2004a,b; Nobrega et al. 2004; Woolfe et al. 2005) but remains, for the most part, poorly understood. While there is less “dark matter” in the genomes of insects, worms, and yeasts, these genomes also contain many conserved sequences whose functions are not yet known (Bergman et al. 2002; Kellis et al. 2003; Stein et al. 2003).
Most groups have used pairwise alignments and simple, percent identity-based methods for identifying conserved elements. For example, Dermitzakis et al. (2002) and Nobrega et al. (2003) have defined conserved elements as intervals of at least 100 bp with >70% identity. Tools such as VISTA (Mayor et al. 2000), PipMaker (Schwartz et al. 2000), and zPicture (Ovcharenko et al. 2004) can be used to construct alignments, visualize annotations and percent identity levels, and/or define conserved elements according to length and identity thresholds. As more genomes have become available, however, it has become essential to make use of multiple (_n_-way) rather than just pairwise (2-way) alignments, and to consider the phylogeny of the species that are represented. A few methods for detecting conserved elements in multiple alignments have been described, some using a phylogeny (e.g., Stojanovic et al. 1999; Boffelli et al. 2003; Margulies et al. 2003; Chapman et al. 2004; Cooper et al. 2004; Ovcharenko et al. 2005a). Of the methods described so far, however, only the “phylogenetic shadowing” method (Boffelli et al. 2003) (to our knowledge) makes use of the branch lengths of the phylogeny, allows for multiple substitutions per site on single branches of the tree, and considers the “pattern” of substitution (e.g., the tendency for transitions to occur more frequently than transversions). In addition, most methods (including phylogenetic shadowing) use a sliding window of fixed size, which can be a limitation. For example, if the window size is small, it may be difficult to discriminate effectively between conserved and nonconserved regions, but if it is large, small conserved elements may be missed, even if highly conserved.
In this study, we describe a new program, called phastCons, that is designed to identify conserved elements in multiply aligned sequences. PhastCons is based on a phylogenetic hidden Markov model (phylo-HMM), a type of statistical model that considers both the process by which nucleotide substitutions occur at each site in a genome and how this process changes from one site to the next (Yang 1995; Felsenstein and Churchill 1996; Siepel and Haussler 2004). Phylo-HMMs provide a principled, mathematically rigorous framework in which to address problems of “segmentation” using comparative sequence data—i.e., problems in which aligned sequences are to be parsed into segments of different classes (e.g., “conserved” and “nonconserved” or “coding” and “noncoding”). For several reasons, they are attractive tools for the problem of identifying conserved elements; they can be used with a general phylogeny and the best available continuous-time Markov models of nucleotide substitution, they do not require a sliding window of fixed size, they allow nearly all parameters to be estimated from the data by maximum likelihood, and they permit all necessary computations to be carried out efficiently on large-scale data sets.
Using phastCons, we have conducted comprehensive searches for conserved elements in four separate genome-wide multiple alignments, consisting of five vertebrate genomes, four insect genomes, two Caenorhabditis genomes, and seven Saccharomyces genomes. This study contains a detailed discussion of our results. Some highlights are as follows:
- Roughly 3%–8% of the human genome consists of sequences conserved in vertebrates and/or other eutherian mammals. Much higher fractions of the more compact D. melanogaster (37%–53%), C. elegans (18%–37%), and S. cerevisiae (47%–68%) genomes are conserved across closely related species. From yeasts to vertebrates, in order of increasing genome size and general biological complexity, increasing fractions of conserved bases are found to lie outside of known or suspected exons of protein-coding genes, apparently reflecting the importance of regulatory and other noncoding sequences in complex eukaryotes.
- In all species groups, the most highly conserved elements (HCEs), by log-odds score, are hundreds or thousands of bases long and show extreme levels of conservation, but not the perfect identity seen in ultraconserved elements. Less than half (42%) of the vertebrate HCEs overlap exons of known protein-coding genes, in contrast to insects, worms, and yeasts, where nearly all (>93%) HCEs overlap such exons.
- Some of the most extreme conservation in vertebrates is seen in 3′ UTRs, particularly of genes that regulate other genes, possibly reflecting widespread post-transcriptional regulation. This trend is less pronounced in insects and was not observed in worms. (Data for yeasts was not available.)
- HCEs in vertebrate 3′ UTRs, and to a lesser extent, HCEs in 5′ UTRs, show strong statistical evidence of an enrichment for local RNA secondary structure, consistent with the hypothesis of a role in post-transcriptional regulation. HCEs in introns and intergenic regions also appear to be enriched for local RNA secondary structure, indicating that many may encode functional RNAs.
- In vertebrates, intergenic HCEs are strongly enriched (nearly fivefold) in stable gene deserts, suggesting that many of them may act as distal _cis_-regulatory elements for precisely regulated genes (Ovcharenko et al. 2005b).
Results
Predicted conserved elements
Four separate genome-wide multiple alignments were prepared for the four species groups, with the human, D. melanogaster, C. elegans, and S. cerevisiae genomes serving as reference genomes (see Methods and Table S2 in the Supplemental material). Using the phastCons program, a two-state phylogenetic hidden Markov model (phylo-HMM) (see Fig. 1) was then fitted separately to each alignment by maximum likelihood, subject to certain constraints (see Methods). The estimated parameters included branch lengths for all branches of the phylogeny and a parameter ρ representing the average rate of substitution in conserved regions as a fraction of the average rate in nonconserved regions (Fig. 2). The tree topologies were assumed to be known (see Supplemental material).
Figure 1.

State-transition diagram for the phylo-HMM used by phastCons, which consists of a state for conserved regions (c) and a state for nonconserved regions (n). Each state is associated with a phylogenetic model (ψ_c_ and ψ_n_); these models are identical except for a scaling parameter ρ (0 ≤ ρ ≤ 1), which is applied to the branch lengths of ψ_c_ and represents the average rate of substitution in conserved regions as a fraction of the average rate in nonconserved regions (see Methods). Two parameters, μ and ν (0 ≤ μ, ν ≤ 1), define all state-transition probabilities, as illustrated. The probability of visiting each state first (indicated by arcs from the node labeled “begin”) is simply set equal to the probability of that state at equilibrium (stationarity). The model can be thought of as a probabilistic machine that “generates” a multiple alignment, consisting of alternating sequences of conserved (dark gray) and nonconserved (light gray) alignment columns (see example at bottom).
Figure 2.

The assumed tree topologies for the vertebrate, insect, worm, and yeast data sets (top to bottom) and the branch lengths estimated for the conserved (left) and nonconserved (right) states of the phylo-HMM. The conserved and nonconserved phylogenies are identical, except for the scaling constant ρ, which was estimated at 0.33, 0.24, 0.36, and 0.32 (top to bottom). Horizontal lines indicate branch lengths and are drawn to scale, both within and between species groups. The estimated trees were unrooted; arbitrary roots were chosen for display purposes. Note that some distortions in the branch lengths occur due to alignment-related ascertainment biases (see text and Supplemental material).
The estimated “nonconserved” branch lengths for vertebrates were fairly consistent with recent results based on (apparently) neutrally evolving DNA in mammals (Cooper et al. 2004), but were not accurate representations of the neutral substitution process in all respects. In particular, the branches to the more distant species (chicken and Fugu) were significantly underestimated, because the genomes of these species are, in general, alignable to the human, mouse, and rat genomes only in regions that are under at least partial constraint. Similar effects were observed with the insect, worm, and yeast phylogenies. Nevertheless, inaccuracies in the estimates of some (particularly longer) nonconserved branch lengths do not appear to have strongly influenced our results (see Supplemental material). Moreover, our method has certain advantages over more traditional methods for estimating neutral substitution rates, such as by using fourfold degenerate (4d) sites in coding regions—e.g., it does not depend on 4d sites being free from selection or being suitable proxies for neutrally evolving sites in general; and as an “unsupervised” learning method (see Methods), it is not dependent on possibly incomplete and/or erroneous annotations.
As an approximate way of calibrating our methods across species groups, we constrained the model parameters such that the coverage of known coding regions by predicted conserved elements (i.e., the fraction of coding bases falling in conserved elements) was equivalent in all groups. We chose a target coverage of 65% (±1%), as estimated from human/mouse comparisons (Chiaromonte et al. 2003). This number was adjusted for alignment coverage in coding regions, yielding 56% for the worm data set and 68% for the insects and yeasts. The degree of “smoothing” of the phylo-HMM was also constrained by forcing the expected amount of phylogenetic information (in an information theoretic sense) required to predict a conserved element to be equal for all data sets (see Methods). Our results are, in general, not highly sensitive to the precise level of target coverage used in this calibration procedure (see Supplemental material).
Based on the estimated parameters, conserved elements were then identified in each set of multiple alignments, using the phastCons program (see Methods). About 1.31 million conserved elements were predicted for the vertebrate data set, about 472,000 for the insects, about 98,000 for the worms, and about 68,000 for the yeasts. Each predicted element was assigned a log-odds score indicating how much more likely it was under the conserved state of the phylo-HMM than under the nonconserved state (see Supplemental material). A synteny filter, designed to eliminate predictions that were based on alignments of nonorthologous sequence (especially transposons or processed pseudogenes), reduced the numbers of predictions for vertebrates and insects to about 1.18 million and 467,000, respectively; alignments of nonorthologous sequence were less prevalent in the worm and yeast data sets, so the filter was omitted in these cases. The remaining predicted elements cover 4.3% of the human genome, 44.5% of D. melanogaster, 26.4% of C. elegans, and 55.6% of S. cerevisiae. These numbers are somewhat sensitive to the methods used for parameter estimation. Various different methods produced coverage estimates of 2.8%–8.1% for the vertebrates, 36.9%–53.1% for the insects, 18.4%–36.6% for the worms, and 46.5%–67.6% for the yeasts (see Supplemental material). Note that the vertebrate coverage is similar to recent estimates of 5%–8% for the share of the human genome that is under purifying selection (Chiaromonte et al. 2003; Roskin et al. 2003; Cooper et al. 2004), despite the use of quite different methods and data sets.
(In the discussion that follows, specific estimates of quantities of interest will be given, rather than ranges of estimates. The reader should bear in mind that, while these estimates are generally not highly sensitive to the method used for parameter estimation, they do change somewhat from one method to another. Further details are given in the Supplemental material.)
The 1.18 million vertebrate elements, in addition to covering 66% of the bases in known coding regions (approximately the target level), cover 23% of the bases in known 5′ UTRs and 18% of the bases in known 3′ UTRs—15.5-fold, 5.3-fold, and 4.3-fold enrichments, respectively, compared with the expected coverage if the predicted conserved elements were distributed randomly across 4.3% of the genome (Fig. 3). Almost nine of 10 (88%) known protein-coding exons are overlapped by predicted elements, as well as almost two of three known UTR exons (63% of 5′-UTR exons and 64% of 3′-UTR exons; when an exon contains both UTR and coding sequence, the UTR portion is considered to be a separate “UTR exon”). Regions not in known genes, but matching publicly available mRNA or spliced EST sequences (“other mRNA” in Fig. 3) show 9.2% coverage by conserved elements (a 2.1-fold enrichment), and regions not in known genes or other mRNAs, but transcribed according to data from the Affymetrix/NCI Human Transcriptome project (“other trans”; see Methods), which presumably include a mixture of undocumented coding regions, UTRs, noncoding RNAs, and other [poly(A)+] transcripts, show 7.5% coverage by conserved elements (a 1.8-fold enrichment). Introns of known genes and unannotated (putative intergenic) regions contain significant fractions of conserved bases (3.6% coverage for introns and 2.7% coverage for unannotated regions), but smaller fractions than would be expected by chance. The predicted elements also include 42% of the bases in a set of 561 putative RNA genes (see Methods), and 56% of these genes are overlapped by predicted elements, indicating that our methods are reasonably sensitive for detecting functional noncoding as well as protein-coding sequences. (If only RNA genes that align syntenically across species are considered, the base-level coverage increases to 65%, about the same as in protein-coding genes). The predicted elements include <1% of the bases in mammalian ancestral repeats (ARs) (see Methods), which are believed, for the most part, to be neutrally evolving, suggesting that the false-positive rate for predictions is quite low. (Simulation experiments indicate a false-positive rate of <0.3% in all species groups; see Supplemental material.)
Figure 3.

Fractions of bases of various annotation types covered by predicted conserved elements (left) and fractions of bases in conserved elements belonging to various annotation types (right). Annotation types include coding regions of known genes (CDS), 5′ and 3′ UTRs of known genes, other regions aligned to mRNAs or spliced ESTs from GenBank (other mRNA), other transcribed regions according to data from Phase 2 of the Affymetrix/NCI Human Transcriptome project (other trans; see Methods), introns of known genes, and other regions (unannotated). All annotations were for the reference genome of each species group and all fractions were computed with respect to these genomes (see Methods). Dashed lines in column graphs indicate expected coverage if conserved elements were distributed uniformly. Transcriptome data was available for the vertebrates only, and UTRs and other mRNAs were omitted for yeast because of sparse data. Note that these graphs change somewhat (but not dramatically) under alternative calibration methods (see Supplemental material).
In the more compact insect, worm, and yeast genomes, less dramatic differences are observed across annotation classes in the coverage by conserved elements (Fig. 3). In all three cases, coding regions show substantially higher coverage than would be expected if conserved elements were distributed randomly, as do UTRs and other mRNAs in worms (but not in insects). Introns and unannotated regions show lower than expected coverage by conserved elements in all three species groups, but still appear to contain substantial numbers of conserved bases. The fractions of introns and intergenic regions in conserved elements are similar, with introns showing slightly higher fractions in all groups but yeast (where they are few in number). In worms, our estimates of the fractions of coding regions, introns, and intergenic regions that are conserved are fairly similar to estimates based on an early comparative study of C. elegans and C. briggsae (Shabalina and Kondrashov 1999), while in insects, our estimates of these fractions for intronic and intergenic regions are roughly 1-1/2–2′ higher than estimates based on D. melanogaster/D. virilis comparisons (Bergman and Kreitman 2001). Note that the results for worm may be influenced by the difficulty of aligning noncoding regions in C. elegans and C. briggsae and by the limited phylogenetic information in pairwise alignments (see Discussion and Supplemental material).
Conversely, looking at how the predicted conserved elements are composed, we find that only about 28% of the bases predicted to be conserved in vertebrates fall in known or likely exons, including UTRs (Fig. 3). In vertebrates, 18.0% of conserved bases fall in known coding regions (CDSs), 1.1% and 3.6% fall in known 5′ and 3′ UTRs, respectively, and another 5.2% fall in other mRNAs. Another 2.4% fall in other transcribed regions, leaving about 70% unannotated. (The percentage in RNA genes and other known noncoding functional elements is negligible.) These numbers are in good agreement both with bulk statistical estimates, based on genome-wide human/mouse and human/mouse/rat alignments, of the share of the human genome that is under selection (Chiaromonte et al. 2003; Roskin et al. 2003; Cooper et al. 2004), and with an analysis of conserved elements in the region of the CFTR gene (Margulies et al. 2003; Thomas et al. 2003). Broadly speaking, if ∼5% of the human genome is conserved, and if ∼1.5% codes for proteins (and these are mostly conserved), then noncoding regions must account for about 0.035/0.05 = 70% of conserved elements. Margulies et al. (2003), using two different methods, found that 72% of bases in predicted conserved elements in the CFTR region were not in exons. Cooper et al. (2004) reported similar results based on whole-genome human/mouse/rat alignments.
Interestingly, a non-negligible fraction (3.7%) of the predicted conserved elements are found in ARs. Simulation experiments (see Supplemental material) and inspection of individual cases suggest that most of these conserved ARs are not likely to be false-positive predictions. While most bases in ARs have evolved neutrally (ARs are underrepresented fivefold in conserved elements), some have apparently taken on critical functions that may help to differentiate mammals from ancestral vertebrates (Britten 1997; Jordan et al. 2003; van de Lagemaat et al. 2003). Many conserved ARs show relatively weak conservation, but some are more strongly conserved. For example, one highly conserved element of more than 700 bp, in a gene desert between the zinc finger genes ZNF537 and ZNF507, contains a 351-bp L1MCa repeat. Three conserved elements in introns of the RNA-processing gene SRRM2, ranging from 478 to 975 bp in length, contain L2 or L3b repeats.
Moving from vertebrates to insects and then to worms and to yeasts, in decreasing order of genome size and general biological complexity, a progressively larger fraction of conserved elements can be seen to fall in coding regions and UTRs, and a progressively smaller fraction in introns and unannotated regions (Fig. 3). In particular, the fraction of bases in predicted conserved elements that fall in known or likely protein-coding exons increases from 28% in vertebrates to 34% in insects, 59% in worms, and 86% in yeasts, so that while most conserved bases in vertebrates and insects apparently do not code for proteins, most in worms and yeasts do. This trend can be seen as an expected consequence of increasing gene density (the more gene-dense genomes have smaller fractions of noncoding bases), but it nevertheless underscores the importance of noncoding regions in the genomes of complex eukaryotes, whose complexity apparently derives not so much from increased numbers of protein-coding genes as from more elaborate mechanisms for gene regulation. Note that the fraction of conserved elements in introns and intergenic regions may be underestimated for the two-species worm data set (see Discussion and Supplemental material).
The lengths of the predicted elements for all four species groups are approximately geometrically distributed, averaging about 100–120 bp for the vertebrate, insect, and yeast groups and about 270 bp for the less phylogenetically informative worm group. In all groups, elements range in length from 5 bp to thousands of basepairs. A more detailed analysis in vertebrates revealed noticeable differences in the length distributions of the elements associated with different types of annotations; elements in ARs are shortest, on average, those in introns and intergenic regions are slightly longer, those in UTRs are longer still, and those in CDSs are longest (Supplemental Fig. S3). Accordingly, the composition of conserved elements is strongly dependent on the length-dependent element scores (Supplemental Fig. S3). In particular, the fractions of elements in coding regions, UTRs, and other mRNAs tend to increase with score, while the fraction in introns tend to decrease. The fraction in 3′ UTRs is particularly large among the highest scoring elements, suggesting some special role for highly conserved 3′ UTRs in vertebrates (see below). The percentage of bases in ARs also decreases sharply with element score. Additional details are given in the Supplemental material.
Base-by-base conservation scores
In addition to predictions of discrete conserved elements, phastCons produces a continuous-valued “conservation score” for each base of the reference genome. These scores are plotted along the genome and displayed as part of a conservation track in the version of the UCSC Genome Browser (Karolchik et al. 2003) dedicated to the reference genome. Beneath the plot of conservation scores, the conservation track also has an alignment display, which shows either a graphical summary of the pairwise alignments between each genome and the reference genome, or (at appropriate zoom levels) the actual bases of the multiple alignment. Conservation tracks have been produced for all four data sets discussed in this study and are displayed in the UCSC Genome Browsers for the human, D. melanogaster, C. elegans, and S. cerevisiae genomes (Fig. 4).
Figure 4.

Screen shots of the conservation tracks in the (A) human and (B) S. cerevisiae UCSC Genome Browsers. Each conservation track has two parts, a plot of conservation scores, and beneath it, a display showing where each of the other genomes aligns to the reference genome. (Darker shading indicates higher BLASTZ scores; white indicates no alignment.) A separate track labeled “PhastCons Conserved Elements” shows predicted conserved elements and log-odds scores. In A, exons 7–11 of the RNA-edited human gene GRIA2 are shown. Peaks in the conservation plot generally correspond to exons and valleys to noncoding regions, but a 158-bp conserved noncoding element can be seen near the 3′ end of exon 11. This conserved element includes the editing complementary sequence (ECS) of the RNA editing site in exon 11. The displays seen when zooming in to the base level at a typical exon (left) and in the region of the RNA editing site (right; see arrow) are shown as insets. On the left, several synonymous substitutions are visible (highlighted bases) and the elevated conservation abruptly ends after the splice site, while on the right, there are fewer synonymous substitutions and the elevated conservation extends into the intron. In the base-level display, the vertical orange bars and numbers above them indicate “hidden” indels and their lengths—i.e., deletions in the human genome or insertions in other genomes. In B, the S. cerevisiae GAL1 gene and 5′-flanking region are shown. Strong cross-species conservation can be seen in the regulatory region upstream of the promoter, as well as in the protein-coding portion of the gene. The conserved element shown at bottom overlaps three _GAL4_-binding sites (highlighted in base-level view). A fourth _GAL4_-binding site also is reflected by a small bump in the conservation scores (left arrow), as is the promoter itself (right arrow).
Like the predicted elements, the base-by-base conservation scores are derived from the two-state phylo-HMM. The conservation score at each base in the reference genome is defined as the posterior probability that the corresponding alignment column was generated by the conserved state (rather than the nonconserved state) of the phylo-HMM, given the model parameters and the multiple alignment. (Thus, the scores range between 0 and 1.) The conservation scores can be interpreted as probabilities that each base is in a conserved element, given the assumptions of the model and the maximum-likelihood parameter estimates. The scores are also influenced by the values of two user-defined tuning parameters (see Methods). The same parameter estimates and user-defined parameters are used for both the conservation scores and the predicted elements.
The conservation tracks are useful devices for visualizing cross-species conservation along a genome, and are complementary to tracks in the browser describing known protein-coding and RNA genes, known regulatory regions, aligned mRNA and EST sequences, gene predictions, and so on. With appropriate parameter settings, many functional elements stand out clearly as “mesas” of cross-species conservation against a “plain” of neutral or nearly neutral evolution (Fig. 4). Sometimes the conservation track lends support to independent annotations such as gene predictions; in other cases, it highlights conserved sequences that are not supported by any existing annotations and helps to stimulate further investigation into possible functions of these sequences. Using the UCSC Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables), it is possible to define regions of the genome having scores that exceed (or fall below) some threshold, and to conduct searches that intersect the conservation scores with other annotations (e.g., “find all intervals with conservation scores above 0.9 that do not overlap known genes”). The conservation track has been popular with users of the UCSC Genome Browser and the phastCons conservation scores are already in use in several other research projects (e.g., ENCODE Project Consortium 2004; Elnitski et al. 2005; Ovachrenko et al. 2005b; King et al. 2005).
Highly conserved elements
The genome-wide sets of conserved elements predicted for each of the four species groups were ranked by log-odds score, and the top-scoring elements were extracted for further analysis. The top 5000 elements were selected from the vertebrate and insect sets, and the top 1000 were selected from the smaller worm and yeast sets. (The numbers of elements selected were essentially arbitrary—they were chosen to be small enough that only truly extreme cases of cross-species conservation would be included, but large enough to allow meaningful statistics to be obtained. The results discussed below are not highly sensitive to these numbers.)
These highly conserved elements (HCEs) are like ultraconserved elements (UCEs) (Bejerano et al. 2004b) in that they show extreme evolutionary conservation (defined by some arbitrary threshold), but HCEs tend to be longer than UCEs and tend to have less extreme sequence conservation, due to the length dependency of the log-odds scores. In addition, the set of vertebrate HCEs is about 10-fold larger than the set of UCEs and is based on a different set of species (including chicken and Fugu). HCEs are different from (and in some ways complementary to) UCEs; nevertheless, the vertebrate HCEs do include about 80% of the human/rodent UCEs identified by Bejerano et al. (2004b). (The 20% that are not included tend to be short, mean length 231 bp).
The vertebrate HCEs cover 0.14% of the human genome. They are considerably longer on average than elements in the full set (lengths ranged from 318 to 4922 bp, with mean 781.4 bp) and they have a larger fraction of bases in CDS and UTR regions (Supplemental Fig. S3). At the base level, coding regions are enriched 22-fold for HCEs, while 3′ UTRs and 5′ UTRs are enriched 11-fold and eightfold, respectively. Nevertheless, only 42% of HCEs overlap known exons (36% overlap CDS exons, 9% overlap 5′ UTR exons, and 16% overlap 3′ UTR exons), with 19% falling completely in known introns, and another 32% completely in unannotated regions. The fraction of HCEs overlapping known exons is somewhat higher than the 23% observed for UCEs (Bejerano et al. 2004b), presumably because of the length dependency of the log-odds scores and the tolerance for a small number of substitutions.
The HCEs identified for the other three sets of genomes cover a higher percentage of each reference genome (2.5% in insect, 1.9% in worm, and 8.0% in yeast) and are much more likely to overlap coding regions (93% of HCEs in insect, 98% in worm, and 99% in yeast overlapped CDSs). As with the vertebrates, the HCEs for the other three species groups are quite long, with lengths ranging from 197 to 5783 bp (mean 627.9 bp) for the insects, 622 to 12646 bp (mean 1889.6 bp) for the worms, and 323 to 4005 bp (mean 973.5 bp) for the yeasts. The fractions of HCEs in insects overlapping UTRs are similar to those in vertebrates (6.1% and 15.5% overlap 5′ and 3′ UTRs, respectively), but in worm, these fractions are considerably lower (1.9% and 3.7%). (Sparse data on UTRs in yeasts did not allow for a comparison with this group.) In insect, worm, and yeast, only about 1%–5% of highly conserved elements fall completely in introns or intergenic regions. In general, highly conserved elements appear to become more strongly associated with genes as genome sizes become smaller and gene densities increase, consistent with the trend discussed above for the larger set of conserved elements (Fig. 3).
HCEs in the 3′ UTRs of vertebrate genes
As noted above, 3′ UTRs account for an unexpectedly large fraction of bases in vertebrate HCEs, and this trend becomes more pronounced as higher scoring subsets of all conserved elements are considered—3′ UTRs account for 9.6% of bases in the top 5000 elements, 12.5% in the top 1000, and 14.3% in the top 100, compared with 5.6% in all conserved elements (Supplemental Fig. S3). In contrast, 5′ UTRs are only slightly overrepresented in HCEs (1.5% of bases, compared with 1.1% in all conserved elements), and they are almost absent in the top 100 elements. Some of the most extreme conservation in vertebrate genomes is seen in the 3′ UTRs of DNA- and RNA-binding genes such as NOVA1, ELAVL4, ZFHX1B, BCL11A, and SYNCRIP, which, in turn, are regulators of other genes (Supplemental Table S3; Fig. 5), suggesting that regulation in 3′ UTRs plays a key role in critical regulatory networks. These findings are consistent with earlier reports of widespread conservation in 3′ UTRs (Duret et al. 1993; Lipman 1997), some of which have noted an enrichment for genes for DNA-binding proteins (Duret et al. 1993). It is likely that many conserved 3′-UTR sequences are involved in posttranscriptional regulatory mechanisms, e.g., by influencing subcellular localization, transcript stability, or translatability (Duret et al. 1993; Grzybowska et al. 2001; Mignone et al. 2002).
Figure 5.

Extreme conservation at the 3′ end of the ELAVL4 (HuD) gene, an RNA-binding gene associated with paraneoplastic encephalomyelitis sensory neuropathy and homologous to Drosophila genes with established roles in neurogenesis and sex determination (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM). The 3117-bp conserved element that overlaps the 3′ UTR of this gene (arrow) is the fifth highest scoring conserved element in the human genome (log odds score 2475). Several conserved elements in introns are also visible.
Post-transcriptional regulation by microRNA (miRNA) binding in 3′ UTRs is of particular interest, as it is believed that miRNAs may regulate the translation of a large fraction of eukaryotic genes (e.g., John et al. 2004; Krek et al. 2005; Lewis et al. 2005; Xie et al. 2005). Most genes known and predicted to be targeted by miRNAs—in D. melanogaster and C. elegans as well as human and mouse—show only moderate conservation in their 3′ UTRs, with short conserved elements alternating with nonconserved regions. There are exceptions, however, such as HOXB8, which is targeted by miR-196 in mouse and has a 1135-bp HCE in its 3′ UTR. MiR-196 is unusual among known animal miRNAs for its near-perfect complementarity to its HOXB8 target site and for inducing cleavage of the HOXB8 mRNA rather than inhibiting translation (Yekta et al. 2004). Human genes with predicted miRNA targets appear to be somewhat enriched for 3′-UTR HCEs; examples include the genes for the eukaryotic translation initiation factor EIF4E, the methyl CpG-binding protein MECP, the DNA-binding proteins (and oncoproteins) MYC and MYCN, the homeobox protein NBPHOX, and the ubiquitin protein ligase UBE3A, which is mutated in Angelman syndrome (John et al. 2004). We also found several examples of D. melanogaster genes with predicted miRNA target sites and HCEs in their 3′ UTRs, such as the Hox cluster genes Abd-B, Antp, Scr, and Ubx (Enright et al. 2003). Correlations between HCEs and predicted miRNA target sites must be treated cautiously, because they may be artifacts of considering conservation in target-site prediction. Still, miRNA binding may provide at least a partial explanation for extreme conservation in some 3′ UTRs, e.g., because of multiple, possibly overlapping, target sites and/or requirements for near-perfect complementarity.
Three groups of known RNA-binding proteins and the mRNAs they bind provide further circumstantial evidence for a connection between HCEs in 3′ UTRs and post-transcriptional regulation, and moreover (if predictions of target sites are accurate), for a connection with miRNAs. John et al. (2004) found a substantial enrichment for predicted miRNA targets among the genes for the fragile X mental retardation protein FMR1, the ELAV-like proteins, and the polyadenylation-binding proteins (CPEBs), and among the genes whose mRNAs are known to be bound by these proteins, suggesting that miRNAs play a critical role in the regulatory networks in which these genes participate. Interestingly, the same genes are also highly enriched for HCEs in 3′ UTRs (Fig. 5). The FMR1 gene and its mRNA cargoes BASP1, CACNA1D, CIC, DDX5, HNRPA28, and HTR1B, all contain both predicted miRNA target sites and HCEs in their 3′ UTRs, as do the genes ELAVL1, ELAVL2, and ELAVL4, the genes GAP-43, FOS, and MYC, whose mRNAs are bound by ELAV-like proteins, and all four known human CPEB genes. The PURA and PURB genes, which interact with FMR1 at the protein level, also have HCEs in their 3′ UTRs.
Another possible reason for highly conserved sequences in 3′ UTRs might be gene regulation via antisense transcription. (Here, we mean _cis-_acting rather than _trans_-acting antisense transcription—i.e., transcription of both DNA strands at the same locus.) For example, if long perfect RNA duplexes were essential for regulation, then sequence conservation might result from selection against allelic divergence (Lipman 1997). This possibility is of particular interest in light of the recent identification of a large number of apparent sense/antisense transcriptional units in eukaryotic genomes, many of which overlap in their 3′ UTRs (Shendure and Church 2002; Yelin et al. 2003) and in light of accumulating evidence for the importance of antisense transcription in various kinds of transcriptional and post-transcriptional regulation (Lavorgna et al. 2004; Dahary et al. 2005). However, we have not observed a strong correlation between antisense transcription and extreme conservation (HCEs) in 3′ UTRs, or for that matter, extreme conservation in 5′ UTRs or coding regions. Only a few of the 40 known sense/antisense pairs reviewed by Shendure and Church (2002) contain HCEs coinciding with regions of sense/antisense overlap. (A striking example is the nuclear receptor NR1D1, whose 3′ UTR overlaps the 3′-most exon of the thyroid hormone receptor THRA, as well as a 1651-bp HCE and an ultraconserved element). Most sense/antisense pairs show only moderate conservation.
Secondary structure in noncoding HCEs
Because several known mechanisms for post-transcriptional regulation involve stem-loop (and other) structures in UTRs (Ross 1996; Mignone et al. 2002), strong conservation in UTRs may occur partly as a result of structural constraints. We tested HCEs in UTRs for statistical evidence of secondary structure using a model analogous to a phylo-HMM, but with a stochastic context-free grammar (SCFG) in place of a hidden Markov model. SCFGs are richer computational models than HMMs, which can accommodate the long distance base pairing that occurs in RNA structures (Durbin et al. 1998). Compared with an ordinary SCFG, a “phylo-SCFG” gains additional power for detecting secondary structure by picking up on the tendency for compensatory substitutions in stem-pairing sites (Knudsen and Hein 1999; Pedersen et al. 2004) (see also Rivas and Eddy 2001). We evaluated the HCEs in UTRs using a “folding potential score” (FPS), a log-odds score derived from two phylo-SCFGs—one allowing for both stem-pairing and nonpairing sites and one allowing only for nonpairing sites (see Methods). The FPS reflects possible (local) stem-pairings within each sequence in a multiple alignment and compensatory substitutions along the branches of the phylogeny, but is designed to avoid biases related to base composition, overall conservation level, and sequence length (see Methods and Supplemental material).
Compared with a random sample of 3′ UTRs without HCEs, the HCEs in 3′ UTRs have considerably higher FPSs on average, indicating a significant enrichment for local secondary structure (Fig. 6A). The HCEs in 5′ UTRs, in contrast, do not have significantly higher FPSs than those of non-HCE 5′ UTRs (P = 0.26; data not shown). However, this finding appeared to be partly a consequence of spurious stem pairings in CpG islands. (CG dinucleotides are sometimes erroneously predicted to pair with one another.) When elements overlapping CpG islands are excluded, the 5′-UTR HCEs do show a modest, but statistically significant enrichment for secondary structure (P = 0.05). The 3′-UTR HCEs also have significantly higher FPSs than do the 5′-UTR HCEs (Fig. 6B). These results provide bulk statistical support for widespread secondary structure in highly conserved 3′ UTRs, and suggest that secondary structure is present, although probably less wide-spread, in highly conserved 5′ UTRs. It is worth noting that the non-HCE 3′ UTRs had significantly higher FPSs than the non-HCE 5′ UTRs, suggesting that there is also widespread secondary structure in 3′ UTRs outside of highly conserved elements.
Figure 6.

Histograms of folding potential scores (FPSs) for (A) highly conserved elements (HCEs) in 3′ UTRs vs. a random sample of 3′ UTRs without HCEs, (B) HCEs in 3′ UTRs vs. HCEs in 5′ UTRs, and (C) HCEs in introns vs. HCEs in coding regions (vertebrate data in all cases). Scores are based on a phylogenetic stochastic context-free grammar, and represent the potential for local secondary structure in a sliding window of 150 bp (see Methods). In all three cases, the difference between the distributions is highly statistically significant (P = 8.8 e-66, P = 1.1 e-8, and P = 4.4 e-215, respectively; Wilcoxon rank sum test).
Secondary structure in intronic and intergenic conserved elements is also of interest, because it may indicate the presence of novel noncoding RNAs. We tested the intronic and intergenic HCEs and found strong evidence there as well for local secondary structure. FPSs in intronic HCEs are, on average, about the same as those in 3′-UTR HCEs, while FPSs in intergenic HCEs are, on average, intermediate between those in 3′- and 5′-UTR HCEs. We also computed FPSs for HCEs in coding regions, which are not expected to have extensive secondary structure. The FPSs of both intronic and intergenic HCEs, as well as those of 3′- and 5′-UTR HCEs, are significantly higher than those of coding HCEs (Fig. 6C), suggesting that many intronic and intergenic HCEs may function at the RNA level.
A similar analysis was performed with the insect HCEs. Here, the 3′-UTR HCEs show a statistically significant enrichment for secondary structure (P = 0.02), but the 5′-UTR, intronic, and intergenic HCEs (for which the sample sizes are quite small) do not. As with the vertebrates, the 3′ UTRs without HCEs have significantly higher FPSs than do the 5′ UTRs without HCEs (P = 1.2 e-29). Several of the intergenic HCEs overlap known functional RNA structures annotated in FlyBase. We did not analyze the noncoding HCEs for secondary structure in worm and yeast because data for these species groups was too sparse to allow meaningful statistics to be obtained.
Clearly, much more can be done on the topic of secondary structure in conserved elements in UTRs, introns, and intergenic regions—specific structures can be predicted and analyzed, structures can be correlated with particular categories of genes, and so on. A manuscript devoted to this topic is in preparation (J.S. Pedersen, G. Bejerano, and D. Haussler, in prep.).
Functional enrichment of genes associated with HCEs
Using the Gene Ontology (GO) database (Ashburner et al. 2000), we examined the molecular functions and biological processes of known genes overlapped by HCEs in coding regions, 5′ UTRs, 3′ UTRs, and introns. In vertebrates, these genes are enriched for many of the same GO categories that are associated with mammalian/vertebrate ultraconserved elements (Bejerano et al. 2004b; International Chicken Genome Sequencing Consortium 2004), regions with high fractions of conserved noncoding bases (International Chicken Genome Sequencing Consortium 2004), stable gene deserts (Ovcharenko et al. 2005b), and human/Fugu conserved noncoding elements (Woolfe et al. 2005)—e.g., “DNA binding”, “transcription regulator activity,” and “development” (Table 1). These “trans-dev” (Woolfe et al. 2005) categories are significantly enriched in genes overlapped by HCEs in UTRs and introns as well as in coding regions. Other categories are more strongly associated with high conservation in some parts of genes than in other parts of genes. For example, genes overlapped by HCEs in coding regions are more strongly enriched for “ion channel activity,” “glutamate receptor activity,” and “synaptic transmission,” than are genes overlapped in other regions (Table 1), suggesting a possible connection with RNA editing. There are several known cases of RNA-edited ion channel genes involved in neurotransmission, and both editing sites and complementary sequences are known to be highly conserved (Aruscavage and Bass 2000; Hoopengardner et al. 2003). Indeed, the RNA-editing site in GRIA2, shown in Figure 4, corresponds to an HCE, as do the editing sites in the related genes GRIA3 and GRIA4. A recently identified RNA-editing site in KCNA1 (Hoopengardner et al. 2003) also corresponds to an HCE, one of the top 100 by log-odds score. Other categories, such as “ubiquitin cycle,” “RNA binding,” “mRNA metabolism,” and “mRNA processing” are particularly strongly enriched in genes overlapped by HCEs in 3′ UTRs, suggesting that many of these HCEs may have functional roles in post-transcriptional regulation. The known human genes overlapped by HCEs include many well-studied disease genes (Supplemental Table S3).
Table 1.
Selected gene ontology (GO) categories of vertebrate genes overlapped by highly conserved elements
| CDS | 5′ UTR | 3′ UTR | Intron | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Term | Description | Na | exp.b | obs.c | Pd | exp. | obs. | P | exp. | obs. | P | exp. | obs. | P |
| GO:0003677 | DNA binding | 1914 | 164.5 | 378 | 1.3e-62 | 59.4 | 158 | 1.5e-33 | 84.4 | 221 | 1.0e-45 | 28.6 | 80 | 5.1e-19 |
| GO:0030528 | transcription regulator activity | 1125 | 96.7 | 251 | 1.7e-49 | 34.9 | 119 | 2.4e-34 | 49.6 | 140 | 8.5e-31 | 16.8 | 54 | 6.2e-15 |
| GO:0007275 | development | 1746 | 150.1 | 266 | 1.2e-22 | 54.2 | 115 | 1.0e-15 | 77.0 | 122 | 1.1e-07 | 26.0 | 47 | 3.8e-05 |
| GO:0005216 | ion channel activity | 334 | 28.7 | 79 | 3.8e-17 | 10.3 | 24 | 1.2e-04 | 14.7 | 16 | 4.0e-01 | 4.9 | 2 | 1.2e-01 |
| GO:0006333 | chromatin assembly/disassembly | 153 | 13.1 | 47 | 3.1e-15 | 4.7 | 11 | 8.3e-03 | 6.7 | 17 | 4.2e-04 | 2.2 | 2 | 6.0e-01 |
| GO:0007399 | neurogenesis | 384 | 33.0 | 82 | 5.2e-15 | 11.9 | 38 | 2.7e-10 | 16.9 | 36 | 1.7e-05 | 5.7 | 15 | 6.7e-04 |
| GO:0009887 | organogenesis | 880 | 75.6 | 144 | 1.0e-14 | 27.3 | 67 | 6.2e-12 | 38.8 | 64 | 5.2e-05 | 13.1 | 27 | 3.0e-04 |
| GO:0009653 | morphogenesis | 1099 | 94.4 | 169 | 1.3e-14 | 34.1 | 76 | 2.2e-11 | 48.5 | 77 | 3.1e-05 | 16.4 | 34 | 3.8e-05 |
| GO:0008066 | glutamate receptor activity | 38 | 3.2 | 19 | 3.6e-11 | 1.1 | 6 | 1.0e-03 | 1.6 | 5 | 2.5e-02 | – | – | – |
| GO:0008134 | transcription factor binding | 251 | 21.5 | 54 | 1.9e-10 | 7.7 | 21 | 3.8e-05 | 11.0 | 35 | 1.5e-09 | 3.7 | 10 | 4.5e-03 |
| GO:0005515 | protein binding | 2179 | 187.3 | 252 | 1.4e-07 | 67.7 | 98 | 6.9e-05 | 96.1 | 141 | 8.9e-07 | 32.5 | 41 | 6.7e-02 |
| GO:0007018 | microtubule-based movement | 55 | 4.7 | 18 | 3.9e-07 | – | – | – | 2.4 | 8 | 2.6e-03 | 0.8 | 2 | 2.0e-01 |
| GO:0003723 | RNA binding | 601 | 51.6 | 88 | 4.2e-07 | 18.6 | 26 | 5.6e-02 | 26.5 | 66 | 5.5e-12 | 8.9 | 7 | 3.2e-01 |
| GO:0007268 | synaptic transmission | 240 | 20.6 | 44 | 1.1e-06 | 7.4 | 12 | 7.2e-02 | 10.5 | 10 | 5.1e-01 | – | – | – |
| GO:0030154 | cell differentiation | 200 | 17.1 | 37 | 6.4e-06 | 6.2 | 17 | 1.7e-04 | 8.8 | 15 | 3.2e-02 | 2.9 | 7 | 3.1e-02 |
| GO:0007267 | cell-cell signaling | 532 | 45.7 | 77 | 3.5e-06 | 16.5 | 23 | 6.9e-02 | 23.4 | 24 | 4.9e-01 | 7.9 | 2 | 1.3e-02 |
| GO:0016071 | mRNA metabolism | 188 | 16.1 | 35 | 9.8e-06 | 5.8 | 10 | 6.9e-02 | 8.2 | 29 | 3.7e-09 | 2.8 | 3 | 5.4e-01 |
| GO:0006397 | mRNA processing | 170 | 14.6 | 30 | 1.2e-04 | 5.2 | 8 | 1.6e-01 | 7.5 | 24 | 4.5e-07 | 2.5 | 3 | 4.7e-01 |
| GO:0006512 | ubiquitin cycle | 542 | 46.6 | 69 | 5.9e-04 | 16.8 | 22 | 1.2e-01 | 23.9 | 45 | 3.4e-05 | 8.1 | 3 | 3.6e-02 |
In insects, worms, and yeasts, genes overlapped by HCEs in coding regions are enriched for some of the same GO categories as in vertebrates, but there are also substantial differences across species groups (Supplemental Table S4). The insects show the greatest similarity to the vertebrates, with enrichment for several trans-dev categories, as well as for categories such as “protein binding,” “cell–cell signaling,” “synaptic transmission,” and “voltage-gated ion channel activity.” The apparent connection with RNA editing occurs also in insects; the RNA-edited potassium channel genes shaker, ether-a-go-go, and slowpoke (Hoopengardner et al. 2003) each overlap four or more HCEs (see related observations by Glazov et al. 2005). Some new categories also appear in insects, such as “metamorphosis” and “oogenesis.” The worm and yeast sets are generally quite different from the vertebrate and insect sets, although overlap does occur in several categories, including “RNA binding,”“ion transport,” and “chromatin assembly or disassembly.” Categories unique to worm and yeast are among the most strongly enriched for each species group: “structural constituent of cuticle” in worm and “structural constituent of ribosome” in yeast. The enrichment for cuticle (primarily collagen) genes is intriguing, as these genes seem to require the same kind of precise regulation required by many development genes—the nematode cuticle is synthesized multiple times in different forms during the nematode life cycle, in a complex process involving the differential expression of more than 150 individual collagen genes (Johnstone 2000).
As in vertebrates, the insect genes overlapped by HCEs in 3′ and 5′ UTRs are enriched for several trans-dev categories. Insect genes overlapped in 3′ UTRs, however, are not enriched for the “ubiquitin cycle,” “RNA binding,” “mRNA metabolism,” and “mRNA processing” categories, which are strongly enriched in their vertebrate counterparts, and are enriched for new categories such as “structural constituent of ribosome,” “cell–cell signaling,” and “synaptic transmission.” We did see an association in insects, as in vertebrates, between 3′-UTR HCEs and certain known post-transcriptional regulatory networks. For example, the insect orthologs of the vertebrate _FMR1, ELAV_-like, and CPEB genes all have HCEs overlapping their 3′ UTRs. Due to sparse data, a comparison across all species groups was not possible with the genes overlapped by HCEs in UTRs and introns.
The general conclusions of this section remain unchanged if the number of conserved elements considered is altered by a factor of two—e.g., if the top-scoring 500 or 2000 worm elements are analyzed instead of the top-scoring 1000.
Vertebrate HCEs and segments rich in conserved noncoding sequence
Based on human/chicken comparisons, the “conserved noncoding fraction” (CNF) of the human genome (fraction of nonrepetitive noncoding sequence that aligns to chicken) in regions on the order of a megabase in size has been observed to vary considerably across the genome. Segments of particularly high CNF tend to be gene-poor and G+C-poor. In addition, these high-CNF segments contain 60% of human/chicken ultraconserved elements, while themselves occupying only 2.3% of the human genome, and the genes overlapping them are significantly enriched for particular (mostly trans-dev) GO categories (International Chicken Genome Sequencing Consortium 2004).
We defined an alternative set of (vertebrate) high-CNF segments (“phastCons high-CNF segments,” as opposed to “human/chicken high-CNF segments”) as maximal intervals of at least 250 kb having CNF_pc_ of at least 10%, where CNF_pc_ is the fraction of noncoding bases that fall in the complete set of conserved elements predicted by phastCons (genome-wide average: 3.4%; repetitive regions are included here). There are 101 phastCons high-CNF segments covering 2.1% of the human genome and averaging 601 kb in length and 13.8% CNF_pc_. Unlike human/chicken high-CNF segments, these segments are not significantly depleted for genes, but like human/chicken high-CNF segments, they show a significant enrichment for trans-dev genes. Certain phastCons high-CNF segments with below-average human/chicken noncoding conservation appear to contain significant mammal-specific conservation (see Supplemental material).
Even if redefined such that the HCEs are excluded when computing the CNF_pc_, the phastCons high-CNF segments overlap 13% of all HCEs and 18% of intronic/intergenic HCEs—enrichments of eightfold and 13-fold, respectively. Thus, there appears to be a strong correlation between moderate conservation in megabase-sized regions and extreme conservation in smaller regions of hundreds or thousands of bases. These independently defined phastCons high-CNF segments also include 23% of human/rodent ultraconserved elements, a 15-fold enrichment.
HCEs and gene deserts
We also examined the question of whether vertebrate HCEs are associated with unusually large intergenic regions in the human genome (“gene deserts”), using 545 such regions recently analyzed by Ovcharenko et al. (2005b). These gene deserts have a minimum length of 640 kb and cover 25% of the human genome; they tend to have low G+C content, high single nucleotide polymorphism (SNP) rates, and decreased fractions of bases conserved between human, chicken, and mouse. On the basis of human/chicken conservation, Ovcharenko et al. (2005b) divided them into “stable” deserts (higher conservation) and “variable” deserts (lower conservation), and found several differences between these two classes that are not direct consequences of conservation level—e.g., flanking genes of stable deserts were primarily enriched for trans-dev GO categories, while different categories were associated with variable deserts. Human/chicken synteny breaks are almost completely absent in stable deserts, suggesting that these regions may harbor _cis_-regulatory elements whose order, orientation, and position with respect to flanking genes are maintained by purifying selection (consistent with recent experimental results—Nobrega et al. 2003; Kimura-Yoshida et al. 2004; Uchikawa et al. 2004) (but, see also Nobrega et al. 2004).
Stable gene deserts account for only 12% of bases in intergenic regions, yet 53% of the 1578 intergenic HCEs fall within or overlap stable deserts, 4.5 times the expected number. In contrast, variable gene deserts account for 30% of bases in intergenic regions and only 2.2% of intergenic HCEs fall within or overlap variable deserts. Conversely, 75% of stable deserts include or are overlapped by at least one HCE, while this is true for only 15% of variable deserts. Thus, HCEs are substantially enriched in stable gene deserts and depleted in variable gene deserts, and most stable deserts have HCEs, while most variable deserts do not. These results lend additional support to the claim that stable and variable gene deserts are fundamentally different, and further suggest that many intergenic HCEs may be distal _cis_-regulatory elements, particularly of trans-dev genes. See related findings by Woolfe et al. (2005).
The largest human/chicken high-CNF segment, a 3.5-Mb region of human chromosome 2, spans the ARHGAP15, GTDC1, and ZFHX1B genes and about two-thirds of a 3.3-Mb gene desert on one side of ZFHX1B (International Chicken Genome Sequencing Consortium 2004; Hillier et al. 2005). Both ZFHX1B, which encodes a zinc finger/homeodomain transcription factor mutated in Hirschsprung disease syndrome (Supplemental Table S3), and ARHGAP15 have numerous HCEs overlapping coding regions, introns, and UTRs (23 in ZFHX1B, ranging from 351 to 2476 bp, and 22 in ARHGAP15, ranging from 500 to 1316 bp). The 2.1-Mb portion of this high-CNF segment falling in the gene desert contains 38 HCEs covering 1.3% of its bases—more than nine times the genome-wide average. This region vividly illustrates the associations among high-CNF segments, gene deserts, trans-dev genes, and highly conserved elements. The gene deserts flanking the developmental genes DACH1, OTX2, and SOX2, all of which have been shown experimentally to harbor distal enhancers (Nobrega et al. 2003; Kimura-Yoshida et al. 2004; Uchikawa et al. 2004), are also rich in HCEs.
Discussion
We have conducted genome-wide searches for conserved elements in four groups of eukaryotic species, using a new method for identifying conserved elements that considers the phylogeny of each species group, makes use of continuous-time Markov models of nucleotide substitution, and allows key parameters to be estimated by maximum likelihood. To our knowledge, this is the first genome-wide survey and comparison of conserved elements in different groups of eukaryotic species (excluding comparisons primarily of proteomes; e.g., Rubin et al. 2000). Our results generally support previous estimates of the fraction of the human genome under selection and the fraction of conserved human bases that fall in noncoding regions, and they allow for an approximate quantitative comparison of these fractions across species groups. In addition, we have identified highly conserved elements (HCEs), similar in some ways to ultraconserved elements, but on average, longer and less perfectly conserved, in all four species groups. Several interesting properties of these HCEs have been noted, including an association in vertebrates and insects with 3′ UTRs, particularly of genes for DNA- and RNA-binding proteins, an enrichment in vertebrates and insects for statistical evidence of RNA secondary structure, and associations in vertebrates with high-CNF segments and stable gene deserts.
As with ultraconserved elements, the reasons for the extreme conservation observed in most vertebrate HCEs remain unknown, but statistical enrichments and individual cases suggest that at least some of these sequences function as _cis_-regulatory binding sites, as RNA genes, in mRNA secondary structures important for RNA editing or post-transcriptional regulation, or as microRNA targets. Similar evidence was found for insect HCEs. The lengths of the conserved sequences, however, remain puzzling. What could explain such sustained conservation, spanning hundreds or thousands of bases? This kind of conservation is not seen ordinarily with sequences of any known functional class. One possible explanation is that HCEs result from cases of multiple, overlapping constraints—e.g., overlapping binding sites, binding sites overlapping with RNA structural or protein-coding constraints, or overlapping protein-coding and RNA structural constraints (as in RNA editing sites within coding regions). A related possibility is that these sequences are “hubs” of regulatory networks, which because of their interactions with many other RNAs or proteins (each interaction possibly involving a slightly different subset of bases), have become evolutionarily “frozen.” The presence of 3′-UTR HCEs in the FMR1, CPEB, and _ELAV_-like genes, as well as in related genes, seems to support this hypothesis. Still, it is possible that some HCEs have single, as-yet-undiscovered functions, which are capable of producing such extreme conservation individually. We also cannot rule out the possibility that their conservation has a mutational, rather than a selectional explanation—i.e., that somehow these sequences have been shielded from mutations and/or subjected to hyperefficient repair (Bejerano et al. 2004b).
Space has not allowed for a detailed discussion of another phenomenon known to be associated with unusual levels of cross-species conservation; that of alternative splicing (e.g., Sorek and Ast 2003; Bejerano et al. 2004b; Rahman et al. 2004; Sugnet et al. 2004). Alternative splicing might (as noted by a reviewer of this work) provide some useful clues about how unusual noncoding conservation arises. For example, sequences flanking (and within) alternatively spliced exons—which are believed to mediate splicing by weakly binding various interacting proteins (e.g., Black 2003)—may tend to be conserved because they have been fine tuned by evolution to promote splicing in certain tissue types or development stages, but not in others. In addition, the same sequence may bind more than one factor, or may have roles both in protein binding and in determining secondary structure, and thus, may provide another example of conservation due to multiple, overlapping constraints. It is worth noting that an association between HCEs and alternative splicing might explain some of the functional enrichments we have observed, since alternative splicing is known to affect some classes of genes (including development genes and ion channel genes) more than others. However, we have not found an enrichment for HCEs in a set of about 5000 alternatively retained cassette exons (Sugnet et al. 2004), as compared with a background set of exons. On the other hand, the flanking intronic regions of these exons do show a 1.5 to twofold enrichment for conserved elements (including non-HCEs). These elements tend to be short, e.g., compared with those in 3′ UTRs, and may fail to be identified as HCEs simply for this reason. The relationship between alternative splicing and cross-species conservation is explored further in a forthcoming study by C. Sugnet, K. Srinivasan, T.A. Clark, G. O'Brien, M. Cline, A. Williams, D. Kulp, J. Blume, D. Haussler, and M. Ares, (in prep.).
Clearly, our comparison of conserved elements across species groups is dependent on the procedure used to calibrate the model. Our approach of holding fixed the coverage of coding regions by predicted conserved elements assumes that coding regions evolve in fundamentally similar ways across species groups (more similar than noncoding regions), and that the fraction of sites in coding regions that are conserved is not highly sensitive to the phylogeny. This approach has some obvious deficiencies. First, there undoubtedly are differences between groups in how coding regions evolve, potentially making a fixed threshold effectively more or less stringent in certain groups than in others. Some possible reasons for such differences include differences in effective population size, in the strength and type of codon bias, in the fraction of coding sites subject to noncoding constraints (e.g., related to splicing or RNA editing), and in neighbor dependencies in substitution rates. Second, the sensitivity and specificity of methods for detecting conserved elements inevitably depend on the number of species considered, their phylogeny, and the amount of missing data (Margulies et al. 2003), all of which differ across species groups (see Supplemental material). Third, what is actually conserved across species (as distinct from what is predicted to be conserved) is a function of the evolutionary divergence of the species being considered and the rate at which turnover of functional elements occurs over evolutionary time (Smith et al. 2004)—factors which also may differ across species groups.
It is difficult to imagine a calibration procedure that would address all of these problems. Indeed, there is probably no perfect way to perform a quantitative comparison of conserved elements across groups having diverse numbers of species, phylogenies, substitution patterns, and genome sizes, and the results of any such comparison should be interpreted cautiously. Nevertheless, alternative calibration methods—based on full maximum-likelihood parameter estimation, estimation of neutral rates from fourfold degenerate sites, and alternative coverage targets in coding regions—have led to generally similar results (see Supplemental material), and certain basic conclusions appear to be fairly robust. In particular, the fractions of bases in each reference genome that are conserved across related species are smallest for vertebrates (3%–8%), intermediate for worms and insects (18%–37% and 37%–53%, respectively), and largest for yeasts (47%–68%). In addition, the fractions of conserved bases that fall in protein-coding regions are lowest for vertebrates (11%–24%), slightly higher for insects (26%–27%), substantially higher for worms (49%–60%), and highest for yeasts (84%–87%). Finally, while the HCEs for each species group change slightly under different calibration methods, the general properties of these elements are quite insensitive to the calibration method.
Probably the weakest part of our analysis concerns the worm data set. The large degree of divergence between C. elegans and C. briggsae led to low-alignment coverage, and may have created a bias toward alignment of conserved elements in coding rather than noncoding regions (because conserved noncoding regions tend to be shorter on average; hence, harder to align.) In addition, having only two species considerably reduced the amount of phylogenetic information per site, forcing the tuning parameter ω (expected length) to be increased, and in turn, causing short, conserved elements to tend to be missed, and larger numbers of nonconserved bases to be contained within predicted conserved elements (see Methods). Together, these factors have probably resulted in an overestimate of the fraction of conserved bases that fall in coding regions (estimated at 49%–60%), and may have resulted in an underestimate of the total fraction conserved (estimated at 18%–37%). With additional data, the estimates of these fractions will probably move toward those for the insects, although it seems likely that they will fall short of matching the insect estimates. It will soon be possible to carry out an improved analysis of conserved elements in both worms and insects with the sequencing of five additional nematodes and nine additional species of Drosophila. Several more vertebrate genomes will also soon become available. We have carried out a preliminary analysis of a larger set of insect genomes, including draft assemblies of D. ananassae, D. virilis, and D. mojavensis, and found that the total coverage by conserved elements and the fraction of conserved elements in noncoding regions both decreased somewhat, but did not change dramatically; the general properties of conserved elements and HCEs were essentially unchanged.
The phylo-HMM used by phastCons is a fairly rich probabilistic model, but it is clearly not realistic in several respects. The assumptions that all sites evolve at one of two evolutionary rates (conserved and nonconserved), that these rates are uniform across the genome, that sites evolve independently conditional on whether they are in conserved or nonconserved regions, and that the phylogenetic models for conserved and nonconserved regions have the same branch-length proportions, base compositions, and substitution patterns, all represent oversimplifications of the complex process of sequence evolution in eukaryotic genomes (e.g., Hardison et al. 2003; Hwang and Green 2004; Siepel and Haussler 2004). In addition, treating alignment gaps as missing data ignores an important source of phylogenetic information. We have experimented with versions of phastCons that address various of these deficiencies, e.g., by introducing states for additional evolutionary rates (Yang 1995; Felsenstein and Churchill 1996), allowing the phylogenetic models to have different branch-length proportions or substitution patterns, or using substitution models that consider context dependencies in substitution rates (Siepel and Haussler 2004). In general, the more parameter-rich and complex versions increase the computational burden of parameter estimation and prediction without producing an appreciable improvement in the quality of the program's output. More complex parameterizations also increase the danger of converging on biologically uninteresting local maxima of the likelihood function. Thus, we have settled on the relatively simple model described here for its efficiency, interpretability, and apparent effectiveness at discriminating between conserved and nonconserved sequences. Still, some additional complexity in the model may turn out to be warranted. Possible extensions include better handling of indels (alignment gaps), allowing for lineage-specific conserved elements, and detecting elements with other types of evolutionary signatures, such as those under positive selection.
Finally, it is important to note that our entire analysis is conditional on whole-genome alignments produced by the MULTIZ program. This program shows good accuracy in simulation experiments (Blanchette et al. 2004), and our synteny filters help to ensure that only orthologous conserved elements are considered, but whole-genome alignment is a difficult problem, and programs that address it are still in their infancy. Some conserved elements are undoubtedly missed because of alignment failures, and even with synteny filtering, other predicted elements are probably spurious in that they are based on alignments of nonorthologous sequence. Furthermore, misplacements of alignment gaps may cause regions to appear more or less conserved than they really are. It will be important to recompute predictions of conserved elements continually as multiple aligners improve.
A survey of conserved elements is of interest in its own right, by helping to shed light on the evolutionary forces that have shaped eukaryotic genomes, but it is only a first step toward exhaustively characterizing the diverse functional elements in these genomes. Much work needs to be done to establish which conserved elements are functional and to work out what their functions are (ENCODE Project Consortium 2004). We hope that genome-wide predictions of conserved elements and visualization devices such as the conservation track in the UCSC Genome Browser will be helpful resources toward this end.
Methods
Sequence data and multiple alignments
The most recent assemblies displayed in the UCSC Genome Browser as of Dec. 1, 2004 were used for the human, mouse, rat, chicken, D. melanogaster, D. yakuba, D. pseudoobscura, A. gambiae, C. elegans, C. briggsae, and S. cerevisiae genomes. Contigs for S. castelli, S. kluyveri, and S. kudriavzevii were obtained from http://www.genetics.wustl.edu/saccharomycesgenomes/Contigs and contigs for S. mikatae, S. bayanus, and S. paradoxus were obtained from http://www.broad.mit.edu/ftp/pub/annotation/fungi/comp_yeasts. Additional details are given in Supplemental Table S1.
Multiple alignments for each species group were prepared using version 10 of the MULTIZ program. MULTIZ builds a multiple alignment from local pairwise alignments of a designated reference genome with each other genome of interest (Blanchette et al. 2004). Pairwise alignments were obtained by using BLASTZ (Schwartz et al. 2003), then were passed through the alignment “chaining” and “netting” pipeline described by Kent et al. (2003), which ensures that each base of the reference genome is aligned to at most one base in each other genome, with the selection procedure being guided by considerations of synteny.
The multiple alignments consisted of blocks of local alignment covering 40.0% of the human genome, 86.9% of D. melanogaster, 43.8% of C. elegans, and 96.6% of S. cerevisiae (Supplemental Table S2). The alignment coverage in known coding regions was considerably higher than the overall coverage (95.4% for human, 99.5% for D. melanogaster, 80.6% for C. elegans, and 99.5% for S. cerevisiae).
Annotations
Annotations for only the reference genome of each species group were considered. The sets of known human coding regions, 5′ and 3′ UTRs and introns were based on the UCSC “Known Genes” track as of 11/17/2004 and the set of “other mRNAs” was based on the “human mRNAs” and “human spliced ESTs” tracks as of the same date. The “other trans” set was based on data from Phase 2 of the Affymetrix/NCI Human Transcriptome project (Cheng et al. 2005). The transcriptional fragments for the SK-N-AS cell line only were used; coverage may increase when additional cell lines become available. Results were extrapolated from chromosomes 6, 7, 13, 14, 19, 20, 21, 22, X, and Y to the entire genome. Ancestral repeats (ARs) were defined as sequences in the human genome annotated by RepeatMasker (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) as belonging to any of a large number of repeat families previously identified as having been active prior to the eutherian radiation (Mouse Genome Sequencing Consortium 2002; Hardison et al. 2003). Putative RNA genes were identified by extracting 451 human entries from the hand-curated “seed” portion of Rfam (Griffiths-Jones et al. 2003) and mapping them to the genome using BLAT (Kent 2002). Only exact matches were retained, but many of these genes are short and some tend to match repeats, so some false matches are undoubtedly included in this set. (The 451 sequences mapped perfectly to 561 positions in the genome.)
All base-level coverage statistics were computed in the coordinate system of the reference genome using the featureBits program (http://www.soe.ucsc.edu/~kent/src/unzipped/hg/featureBits). The CDS, 5′ UTR, 3′ UTR, other mRNA, other transcribed, and intron sets were kept disjoint by giving priority to annotation classes in the order listed; e.g., if a base was annotated as both CDS and 5′ UTR, then it was counted as belonging to the CDS class. Statistics describing overlapping features (e.g., the fraction of exons overlapped by conserved elements) were based on a version of the annotations containing only unique, nonoverlapping instances of each feature type (e.g., only one of several CDS exons associated with different isoforms of the same gene). The longest member of each set of overlapping features was selected.
Similar procedures were used with the other species groups. For D. melanogaster, known protein-coding genes from FlyBase were used (release 3.2 annotations) (FlyBase Consortium 2003), and for S. cerevisiae, known genes from the Saccharomyces Genome Database (SGD) (Christie et al. 2004) were used. (ORFs classified in SGD as “verified” and “uncharacterized” were included and those classified as “dubious” were excluded.) For C. elegans, known protein-coding genes from WormBase (Harris et al. 2004) and RefSeq (Pruitt et al. 2003) were combined, in order to maximize data on UTRs.
PhastCons
PhastCons is based on a two-state phylogenetic hidden Markov model (phylo-HMM), with a state for conserved regions and a state for nonconserved regions (Fig. 1). Phylo-HMMs are hidden Markov models whose states are associated with probability distributions over alignment columns, as defined by phylogenetic models (Yang 1995; Felsenstein and Churchill 1996; Siepel and Haussler 2005). In the model used by phastCons, the phylogenetic models associated with the two states are identical except for a scaling parameter ρ (0 ≤ ρ ≤ 1), which is applied to the branch lengths of the conserved phylogeny and represents the average rate of substitution in conserved regions as a fraction of the average rate in nonconserved regions. The model is defined more precisely in the Supplemental material.
The free parameters of the model were estimated from a multiple alignment by maximum likelihood, using an expectation maximization (EM) algorithm. The algorithm alternates between an expectation (E) step and a maximization (M) step until convergence. The E step involves computing posterior expected counts of the number of times each distinct alignment column is “emitted” by each state of the HMM, and posterior expected counts of the number of times each possible state transition occurs. The M step involves updating the parameters of the model to maximize a quantity called the expected complete log likelihood, which is based on the posterior expected counts. This is a completely “unsupervised” learning procedure—i.e., the parameters of the model and a separation between “conserved” and “nonconserved” sequences are learned directly from the data, without relying on annotations of known conserved elements. Details are given in the Supplemental material.
For practical reasons, exact maximum likelihood estimates (m.l.e.'s) of free parameters were not obtained for entire genome-wide data sets, but instead, the genome-wide alignments were divided into fragments of, at most, about 1 Mb in length (in the coordinate system of the reference genome), and parameters were estimated separately for each fragment. (Wherever possible, the “cuts” between fragments were made in regions of no crossspecies alignment, e.g., in transposon insertions in the reference genome.) These separately estimated parameters were then averaged to obtain a single approximate m.l.e. for each genome-wide data set. Fragments with sparse data (<1000 sites aligned) were excluded from this procedure, but the amount of data discarded in this way was negligible, and the average parameter estimates can reasonably be said to represent the entire genome-wide data sets. The individual parameter estimates were fairly consistent and their averages should be close to true genome-wide m.l.e.'s (see Supplemental material).
Using these average parameter estimates, conserved elements were then predicted, and conservation scores were generated for each alignment fragment in a second genome-wide pass. Conserved elements were predicted using the Viterbi algorithm (Durbin et al. 1998). Conservation scores—posterior probabilities that each site was generated by the conserved state—were computed using the forward/backward algorithm (Durbin et al. 1998). Each predicted conserved element was assigned a log-odds score, indicating how much more likely it is under the conserved phylogenetic model than under the nonconserved model (see Supplemental material).
For purposes of both parameter estimation and prediction, missing data in the alignments (e.g., from large-scale insertions and deletions, assembly gaps, or extreme sequence divergence) and smaller scale alignment gaps (from micro-indels) were handled by marginalizing over missing bases when computing emission probabilities. Predicted vertebrate elements were discarded if they did not fall on the syntenic net between human and mouse (Kent et al. 2003), and predicted insect elements were discarded if they did not fall on either the D. melanogaster/D. yakuba syntenic net or the D. melanogaster/D. pseudoobscura syntenic net. See the Supplemental material for additional details.
Constraints on coverage and smoothness
The parameters of the model were estimated by maximum likelihood subject to two constraints: a coverage constraint, determining how much of the target genome is predicted to be conserved and a smoothness constraint, determining how similar the conservation scores are from one site to the next and how fragmented the conserved elements are. As noted above, the coverage constraint was that some target fraction of the bases in known coding regions must be covered by predicted conserved elements, after adjusting for alignment coverage in coding regions. A target of 65% was used for the main analysis, but we also experimented with targets of 55% and 75% (see Supplemental material). The smoothness constraint was that a quantity called the phylogenetic information threshold (PIT) must be the same for all species groups. This constraint was designed to ensure that predicted conserved elements were supported by similar (minimal) amounts of phylogenetic evidence, taking into consideration differences in phylogenetic information per site.
Briefly, let L_min be the expected minimum length of a sequence of conserved sites (in the midst of a stretch of nonconserved sites) required for a conserved element to be predicted. Assuming conserved and nonconserved sites are drawn independently from the distributions associated with ψ_c and ψ_n_, respectively, _L_min is given by:
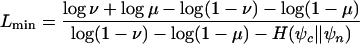 |
(1) |
|---|
where H(ψ_c_∥ψ_n_) is the relative entropy of the distribution associated with ψ_c_ with respect to the distribution associated with ψ_n_. The PIT is defined as the product of L_min and the relative entropy H(ψ_c_∥ψ_n); it can be interpreted as the expected minimum amount of phylogenetic information required to predict a conserved element. The PIT was constrained to be equal to 9.8 bits for all data sets. More details are given in the Supplemental material.
These constraints were met by iteratively adjusting two tuning parameters as follows: γ, defined as the expected coverage by conserved elements, and ω, defined as the expected length of a conserved element. These tuning parameters are related to the state-transition parameters μ and ν by the equations
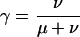
and
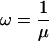
(see Supplemental material). Because μ and ν are completely determined by γ and ω, they need not be estimated by maximum likelihood when γ and ω are set by the user to satisfy coverage and smoothing constraints.
Secondary structure
The folding potential score (FPS) is based on two phylo-SCFGs, one with a stem-pairing and a nonstem-pairing component (θ_sp_) and one with only a nonstem-pairing component (θ_nsp_). Given a multiple alignment x, the FPS is given by s(x) = log P(x|θ_sp_) – log P(x|θ_nsp_), where P(x|θ_i_) is the total probability of x under model θ_i_ (a sum over all allowable structures). The phylogenetic models in θ_sp_ and θ_nsp_ for stem-pairing and nonpairing positions were trained on known RNA structures from Rfam (Griffiths-Jones et al. 2003), but were forced to have equal expected rates of substitution to avoid biases related to conservation level. In addition, the rate matrix in θ_nsp_ is a marginalized version of the rate matrix in θ_sp_, so that the two models make use of the same nucleotide distribution. Possible biases related to alignment gaps and missing data were avoided by constraining the structures such that alignment columns with >50% gaps or missing data had to fall in nonpairing regions. See the Supplemental material for additional details.
The FPSs for both the HCE and the null data sets were computed from local multiple alignments extracted from the genome-wide MULTIZ alignments. To avoid length biases, we did not score whole UTRs, but instead, computed the FPS locally in a sliding window of 150 bp (step size 50 bp). The distribution of FPSs for 150-bp windows in UTR HCEs were simply compared with the distributions of FPSs for 150-bp windows in randomly selected non-HCE UTRs—i.e., the scores were not combined per UTR. Similarly, the distributions of FPSs for intronic, intergenic, and coding HCEs represented individual windows only. These distributions reflect the potential for local structural features (e.g., stem loops) in each set, rather than the potential for global structures. Distributions of FPSs were compared using the Wilcoxon rank sum test. For the purposes of statistical testing, only the FPSs for nonoverlapping windows were considered. Using different window sizes, relaxing the constraints on columns with missing data and using the Kolmogorov-Smirnov test instead of the Wilcoxon test did not produce significant differences in our results.
Functional enrichment
The observed number of genes in each set of interest (e.g., genes overlapped by HCEs in coding regions) was compared with the number that would be expected if genes were assigned to categories randomly in the relative frequencies observed for all known genes in the species in question. _P_-values were computed using the tail of the hypergeometric distribution. No correction for multiple testing was performed, but there were fewer than 1000 individual tests, so P < 5 e-5 can be considered significant. At most, one isoform of each gene was included in both the background and test sets.
Acknowledgments
We thank Tom Gingeras for permission to use the Affymetrix transcriptional fragments for the SK-N-AS cell line prior to publication; Doug Smith for permission to use preliminary D. virilis and D. mojavensis assemblies, and Karin Remington for permission to use a preliminary D. ananassae assembly to check our insect results; Krishna Roskin for preparing Figure 6; Shan Yang and John Karro for looking at correlations of conserved elements with local rates of neutral human/mouse substitutions; Elliott Margulies for sharing his manuscript with us ahead of publication; Ivan Ovcharenko, Chuck Sugnet, and Ross Hardison for helpful conversations; Stefan Bekiranov for comments on the manuscript; and the anonymous reviewers for their especially thorough reading of the manuscript and their insightful suggestions. Funding was provided by the Howard Hughes Medical Institute, the National Human Genome Research Institute (grants IP41HG02371 and HG02238), the Achievements Rewards for College Scientists (ARCS) foundation, the University of California Biotechnology Research and Education Program (Graduate Research and Education in Adaptive Biotechnology fellowship), and the Danish Research Council (Grant 21-04-0444).
[Supplemental material is available online at www.genome.org. The multiple alignments, predicted conserved elements, and base-by-base conservation scores presented here can be downloaded from http://www.cse.ucsc.edu/\~acs/conservation. Up-to-date versions of these data sets are displayed in the “Conservation” and “Most Conserved” tracks in the UCSC Genome Browser (http://genome.ucsc.edu). The phastCons program is part of a software package called PHAST (PHylogenetic Analysis with Space/Time models), which is available by request from acs@soe.ucsc.edu.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3715005\. Article published online before print in July 2005.
References
- Aruscavage, P.J. and Bass, B.L. 2000. A phylogenetic analysis reveals an unusual sequence conservation within introns involved in RNA editing. RNA 6**:** 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al. 2000. Gene ontology: Tool for the unification of biology. Nat. Genet. 25**:** 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano, G., Haussler, D., and Blanchette, M. 2004a. Into the heart of darkness: Large-scale clustering of human non-coding DNA. Bioinformatics 20**:** I40–I48. [DOI] [PubMed] [Google Scholar]
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W., Mattick, J., and Haussler, D. 2004b. Ultraconserved elements in the human genome. Science 304**:** 1321–1325. [DOI] [PubMed] [Google Scholar]
- Bergman, C.M. and Kreitman, M. 2001. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res. 11**:** 1335–1345. [DOI] [PubMed] [Google Scholar]
- Bergman, C.M., Pfeiffer, B.D., Rincon-Limas, D.E., Hoskins, R.A., Gnirke, A., Mungall, C.J., Wang, A.M., Kronmiller, B., Pacleb, J., Park, S., et al. 2002. Assessing the impact of comparative genomic sequence data on the functional annotation of the Drosophila genome. Genome Biol. 3**:** research0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, D.L. 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72**:** 291–336. [DOI] [PubMed] [Google Scholar]
- Blanchette, M., Kent, W.J., Riemer, C., Elnitski, L., Smit, A.F.A., Roskin, K.M., Baertsch, R., Rosenbloom, K., Clawson, H., Green, E.D., et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14**:** 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffelli, D., McAuliffe, J., Ovcharenko, D., Lewis, K.D., Ovcharenko, I., Pachter, L., and Rubin, E.M. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299**:** 1391–1394. [DOI] [PubMed] [Google Scholar]
- Bray, N. and Pachter, L. 2004. MAVID: Constrained ancestral alignment of multiple sequences. Genome Res. 14**:** 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten, R.J. 1997. Mobile elements inserted in the distant past have taken on important functions. Gene 205**:** 177–182. [DOI] [PubMed] [Google Scholar]
- Brudno, M., Do, C., Cooper, G., Kim, M.F., Davydov, E., Green, E.D., Sidow, A., and Batzoglou, S. 2003. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13**:** 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, M.A., Donaldson, I.J., Gilbert, J., Grafham, D., Rogers, J., Green, A.R., and Gottgens, B. 2004. Analysis of multiple genomic sequence alignments: A web resource, online tools, and lessons learned from analysis of mammalian SCL loci. Genome Res. 14**:** 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, J., Kapranov, P., Drenkow, J., Dike, S., Brubaker, S., Patel, S., Long, J., Stern, D., Tammana, H., Helt, G., et al. 2005. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308**:** 1149–1154. [DOI] [PubMed] [Google Scholar]
- Chiaromonte, F., Weber, R.J., Roskin, K.M., Diekhans, M., Kent, W.J., and Haussler, D. 2003. The share of human genomic DNA under selection estimated from human–mouse genomic alignments. Cold Spring Harbor Symp. Quant. Biol. 68**:** 245–254. [DOI] [PubMed] [Google Scholar]
- Christie, K.R., Weng, S., Balakrishnan, R., Costanzo, M.C., Dolinski, K., Dwight, S.S., Engel, S.R., Feierbach, B., Fisk, D.G., Hirschman, J.E., et al. 2004. Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 32**:** D311–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A.G. 2001. The search for meaning in noncoding DNA. Genome Res. 11**:** 1319–1320. [DOI] [PubMed] [Google Scholar]
- Cooper, G.M., Brudno, M., Stone, E.A., Dubchak, I., Batzoglou, S., and Sidow, A. 2004. Characterization of evolutionary rates and constraints in three mammalian genomes. Genome Res. 14**:** 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahary, D., Elroy-Stein, O., and Sorek, R. 2005. Naturally occurring antisense: Transcriptional leakage or real overlap? Genome Res. 15**:** 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis, E.T., Reymond, A., Lyle, R., Scamuffa, N., Ucla, C., Deutsch, S., Stevenson, B.J., Flegel, V., Bucher, P., Jongeneel, C.V., et al. 2002. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 420**:** 578–582. [DOI] [PubMed] [Google Scholar]
- Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. 1998. Biological sequence analysis: Probabilistic models of proteins and nucleic acids. Cambridge University Press, UK.
- Duret, L., Dorkeld, F., and Gautier, C. 1993. Strong conservation of non-coding sequences during vertebrates evolution: Potential involvement in post-transcriptional regulation of gene expression. Nucleic Acids Res. 21**:** 2315–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H., Smith, N.G.C., and Webster, M.T. 2003. Mutation rate variation in the mammalian genome. Curr. Opin. Genet. Dev. 13**:** 562–568. [DOI] [PubMed] [Google Scholar]
- Elnitski, L., Giardine, B., Shah, P., Zhang, Y., Riemer, C., Weirauch, M., Burhans, R., Miller, W., and Hardison, R.C. 2005. Improvements to GALA and dbERGE II: Databases featuring genomic sequence alignment, annotation and experimental results. Nucleic Acids Res. 33**:** D466–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. 2004. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306**:** 636–640. [DOI] [PubMed] [Google Scholar]
- Enright, A.J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D.S. 2003. MicroRNA targets in Drosophila. Genome Biol. 5**:** R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J. and Churchill, G.A. 1996. A hidden Markov model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 13**:** 93–104. [DOI] [PubMed] [Google Scholar]
- FlyBase Consortium. 2003. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 31**:** 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer, K.A., Sheehan, J.B., Stokowski, R.P., Chen, X., Hosseini, R., Cheng, J.-F., Fodor, S.P.A., Cox, D.R., and Patil, N. 2001. Evolutionarily conserved sequences on human chromosome 21. Genome Res. 11**:** 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer, K.A., Tao, H., Osoegawa, K., de Jong, P.J., Chen, X., Doherty, M.F., and Cox, D.R. 2004. Noncoding sequences conserved in a limited number of mammals in the SIM2 interval are frequently functional. Genome Res. 14**:** 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazov, E.A., Pheasant, M., McGraw, E.A., Bejerano, G., and Mattick, J.S. 2005. Ultraconserved elements in insect genomes: A highly conserved intronic sequence implicated in the control of homothorax mRNA splicing. Genome Res. 15**:** 800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A., and Eddy, S.R. 2003. Rfam: An RNA family database. Nucleic Acids Res. 31**:** 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzybowska, E.A., Wilczynska, A., and Siedlecki, J.A. 2001. Regulatory functions of 3′ UTRs. Biochem. Biophys. Res. Co. 288**:** 291–295. [DOI] [PubMed] [Google Scholar]
- Hardison, R.C., Oeltjen, J., and Miller, W. 1997. Long human–mouse sequence alignments reveal novel regulatory elements: Reasons to sequence the mouse genome. Genome Res. 7**:** 959–966. [DOI] [PubMed] [Google Scholar]
- Hardison, R.C., Roskin, K.M., Yang, S., Diekhans, M., Kent, W.J., Weber, R., Elnitski, L., Li, J., O'Connor, M., Kolbe, D., et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13**:** 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, T.W., Chen, N., Cunningham, F., Tello-Ruiz, M., and Antoshechkin, I. 2004. WormBase: A multi-species resource for nematode biology and genomics. Nucleic Acids Res. 32**:** D411–D417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier, L.W., Graves, T.A., Fulton, R.S., Fulton, L.A., Pepin, K.H., Minx, P., Wagner-McPherson, C., Layman, D., Wylie, K., Sekhon, M., et al. 2005. Generation and annotation of the DNA sequences of human chromosomes 2 and 4. Nature 434**:** 724–731. [DOI] [PubMed] [Google Scholar]
- Hoopengardner, B., Bhalla, T., Staber, C., and Reenan, R. 2003. Nervous system targets of RNA editing identified by comparative genomics. Science 301**:** 832–836. [DOI] [PubMed] [Google Scholar]
- Hwang, D. and Green, P. 2004. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl. Acad. Sci. 101**:** 13994–14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Chicken Genome Sequencing Consortium. 2004. Sequencing and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432**:** 695–716. [DOI] [PubMed] [Google Scholar]
- John, B., Enright, A.J., Aravin, A., Tuschl, T., Sander, C., and Marks, D.S. 2004. Human microRNA targets. PLoS Biol. 2**:** e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone, I.L., 2000. Cuticle collagen genes: Expression in Caenorhabditis elegans. Trends Genet. 16**:** 21–27. [DOI] [PubMed] [Google Scholar]
- Jordan, I.K., Rogozin, I.B., Glazko, G.V., and Koonin, E.V. 2003. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 19**:** 68–72. [DOI] [PubMed] [Google Scholar]
- Karolchik, D., Baertsch, R., Diekhans, M., Furey, T.S., Hinrichs, A., Lu, Y.T., Roskin, K.M., Schwartz, M., Sugnet, C.W., Thomas, D.J., et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res. 31**:** 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., Patterson, N., Endrizzi, M., Birren, B., and Lander, E.S. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423**:** 241–254. [DOI] [PubMed] [Google Scholar]
- Kent, W.J., 2002. BLAT: The BLAST-like alignment tool. Genome Res. 12**:** 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J., Baertsch, R., Hinrichs, A., Miller, W., and Haussler, D. 2003. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. 100**:** 11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura-Yoshida, C., Kitajima, K., Oda-Ishii, I., Tian, E., Suzuki, M., Yamamoto, M., Suzuki, T., Kobayashi, M., Aizawa, S., and Matsuo, I., et al. 2004. Characterization of the pufferfish Otx2 _cis_-regulators reveals evolutionarily conserved genetic mechanisms for vertebrate head specification. Development 131**:** 57–71. [DOI] [PubMed] [Google Scholar]
- King, D.C., Taylor, J., Elnitski, L., Chiaromonte, F., Miller, W., and Hardison, R.C. 2005. Evaluation of regulatory potential and conservation scores for detecting _cis_-regulatory modules in aligned mammalian genome sequences. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Knudsen, B. and Hein, J., 1999. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 15**:** 446–454. [DOI] [PubMed] [Google Scholar]
- Krek, A., Grun, D., Poy, M., Wolf, R., Rosenberg, L., Epstein, E., Macmenamin, P., da Piedade, I., Gunsalus, K., Stoffel, M., et al. 2005. Combinatorial microRNA target predictions. Nat. Genet. 37**:** 495–500. [DOI] [PubMed] [Google Scholar]
- Lavorgna, G., Dahary, D., Lehner, B., Sorek, R., Sanderson, C.M., and Casari, G. 2004. In search of antisense. Trends Biochem. Sci. 29**:** 88–94. [DOI] [PubMed] [Google Scholar]
- Lewis, B.P., Burge, C.B., and Bartel, D.P. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120**:** 15–20. [DOI] [PubMed] [Google Scholar]
- Lipman, D.J. 1997. Making (anti)sense of non-coding sequence conservation. Nucleic Acids Res. 25**:** 3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots, G.G., Locksley, R.M., Blankespoor, C.M., Wang, Z.E., Miller, W., Rubin, E.M., and Frazer, K.A. 2000. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288**:** 136–140. [DOI] [PubMed] [Google Scholar]
- Margulies, E.H., Blanchette, M., NISC Comparative Sequencing Program, Haussler, D., and Green, E.D. 2003. Identification and characterization of multi-species conserved sequences. Genome Res. 13**:** 2507–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor, C., Brudno, M., Schwartz, J.R., Poliakov, A., Rubin, E.M., Frazer, K.A., Pachter, L., and Dubchak, I. 2000. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16**:** 1046–1047. [DOI] [PubMed] [Google Scholar]
- Mignone, F., Gissi, C., Liuni, S., and Pesole, G. 2002. Untranslated regions of mRNAs. Genome Biol. 3**:** reviews0004.1–0004.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420**:** 520–562. [DOI] [PubMed] [Google Scholar]
- Nobrega, M.A., Ovcharenko, I., Afzal, V., and Rubin, E.M. 2003. Scanning human gene deserts for long-range enhancers. Science 302**:** 413. [DOI] [PubMed] [Google Scholar]
- Nobrega, M.A., Zhu, Y., Plajzer-Frick, I., Afzal, V., and Rubin, E.M. 2004. Megabase deletions of gene deserts result in viable mice. Nature 431**:** 988–993. [DOI] [PubMed] [Google Scholar]
- Ovcharenko, I., Loots, G.G., Hardison, R.C., Miller, W., and Stubbs, L. 2004. zPicture: Dynamic alignment and visualization tool for analyzing conservation profiles. Genome Res. 14**:** 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko, I., Loots, G.G., Giardine, B.M., Hou, M., Ma, J., Hardison, R.C., Stubbs, L., and Miller, W. 2005a. Mulan: Multiple-sequence local alignment and visualization for studying function and evolution. Genome Res. 15**:** 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko, I., Loots, G.G., Nobrega, M.A., Hardison, R.C., Miller, W., and Stubbs, L. 2005b. Evolution and functional classification of vertebrate gene deserts. Genome Res. 15**:** 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, J.S., Meyer, I.M., Forsberg, R., Simmonds, P., and Hein, J. 2004. A comparative method for finding and folding RNA secondary structures within protein-coding regions. Nucleic Acids Res. 32**:** 4925–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt, K.D., Tatusova, T., and Maglott, D.R. 2003. NCBI Reference Sequence project: Update and current status. Nucleic Acids Res. 31**:** 34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, L., Bliskovski, V., Kaye, F.J., and Zajac-Kaye, M. 2004. Evolutionary conservation of a 2-kb intronic sequence flanking a tissue-specific alternative exon in the PTBP2 gene. Genomics 83**:** 76–84. [DOI] [PubMed] [Google Scholar]
- Rat Genome Sequencing Project Consortium. 2004. Genome sequence of the Brown Norway Rat yields insights into mammalian evolution. Nature 428**:** 493–521. [DOI] [PubMed] [Google Scholar]
- Rivas, E. and Eddy, S.R. 2001. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2**:** 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskin, K.M., Diekhans, M., and Haussler, D. 2003. Scoring two-species local alignments to try to statistically separate neutrally evolving from selected DNA segments. In Proc. 7th Annual Int'l Conf. on Research in Computational Molecular Biology pp. 257–266.
- Ross, J. 1996. Control of messenger RNA stability in higher eukaryotes. Trends Genet. 12**:** 171–175. [DOI] [PubMed] [Google Scholar]
- Rubin, G.M., Yandell, M.D., Wortman, J.R., Gabor Miklos, G.L., Nelson, C.R., Hariharan, I.K., Fortini, M.E., Li, P.W., Apweiler, R., Fleischmann, W., et al. 2000. Comparative genomics of the eukaryotes. Science 287**:** 2204–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, S., Zhang, Z., Frazer, K., Smit, A., Riemer, C., Bouck, J., Gibbs, R., Hardison, R., and Miller, W. 2000. PipMaker: A web server for aligning two genomic DNA sequences. Genome Res. 10**:** 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, S., Kent, W.J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003. Human–mouse alignments with BLASTZ. Genome Res. 13**:** 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina, S.A. and Kondrashov, A.S. 1999. Pattern of selective constraint in C. elegans and C. briggsae genomes. Genet. Res. 74**:** 23–30. [DOI] [PubMed] [Google Scholar]
- Shabalina, S.A., Ogurtsov, A.Y., Kondrashov, V.A., and Kondrashov, A.S. 2001. Selective constraint in intergenic regions of human and mouse genomes. Trends Genet. 17**:** 373–376. [DOI] [PubMed] [Google Scholar]
- Shendure, J. and Church, G.M. 2002. Computational discovery of sense-antisense transcription in the human and mouse genomes. Genome Biol. 3**:** research0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel, A. and Haussler, D. 2004. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 21**:** 468–488. [DOI] [PubMed] [Google Scholar]
- ———. 2005. Phylogenetic hidden Markov models. In Statistical methods in molecular evolution (ed. R. Nielsen), pp. 325–351. Springer, New York.
- Smith, N.G.C., Brandstrom, M., and Ellegren, H. 2004. Evidence for turnover of functional noncoding DNA in mammalian genome evolution. Genomics 84**:** 806–813. [DOI] [PubMed] [Google Scholar]
- Sorek, R. and Ast, G. 2003. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res. 13**:** 1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. 2003. The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biol. 1**:** E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovic, N., Florea, L., Riemer, C., Gumucio, D., Slightom, J., Goodman, M., Miller, W., and Hardison, R. 1999. Comparison of five methods for finding conserved sequences in multiple alignments of gene regulatory regions. Nucleic Acids Res. 27**:** 3899–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugnet, C.W., Kent, W.J., Ares, M., and Haussler, D. 2004. Transcriptome and genome conservation of alternative splicing events in humans and mice. In Proc. 9th Pacific Symp. on Biocomputing, pp. 66–77. [DOI] [PubMed]
- Thomas, J.W., Touchman, J.W., Blakesley, R.W., Bouffard, G.G., Beckstrom-Sternberg, S.M., Margulies, E.H., Blanchette, M., Siepel, A.C., Thomas, P.J., McDowell, J.C., et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424**:** 788–793. [DOI] [PubMed] [Google Scholar]
- Uchikawa, M., Takemoto, T., Kamachi, Y., and Kondoh, H. 2004. Efficient identification of regulatory sequences in the chicken genome by a powerful combination of embryo electroporation and genome comparison. Mech. Dev. 121**:** 1145–1158. [DOI] [PubMed] [Google Scholar]
- van de Lagemaat, L.N., Landry, J.-R., Mager, D.L., and Medstrand, P. 2003. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 19**:** 530–536. [DOI] [PubMed] [Google Scholar]
- Wolfe, K.H., Sharp, P.M., and Li, W.-H. 1989. Mutation rates differ among regions of the mammalian genome. Nature 337**:** 283–285. [DOI] [PubMed] [Google Scholar]
- Woolfe, A., Goodson, M., Goode, D., Snell, P., McEwen, G., Vavouri, T., Smith, S., North, P., Callaway, H., Kelly, K., et al. 2005. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3**:** e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, X., Lu, J., Kulbokas, E.J., Golub, T.R., Mootha, V., Lindblad-Toh, K., Lander, E.S., and Kellis, M. 2005. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434**:** 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. 1995. A space-time process model for the evolution of DNA sequences. Genetics 139**:** 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekta, S., Shih, I.-H., and Bartel, D.P. 2004. MicroRNA-directed cleavage of HOXB8 mRNA. Science 304**:** 594–596. [DOI] [PubMed] [Google Scholar]
- Yelin, R., Dahary, D., Sorek, R., Levanon, E.Y., Goldstein, O., Shoshan, A., Diber, A., Biton, S., Tamir, Y., Khosravi, R., et al. 2003. Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 21**:** 379–386. [DOI] [PubMed] [Google Scholar]
Web site references
- http://www.cse.ucsc.edu/~acs/conservation; Supplemental data for this study.
- http://genome.ucsc.edu; UC Santa Cruz Genome Browser.
- http://genome.ucsc.edu/cgi-bin/hgTables; UC Santa Cruz Table Browser.
- http://www.genetics.wustl.edu/saccharomycesgenomes/Contigs; download page for yeast sequence data, Washington University, St. Louis.
- http://www.broad.mit.edu/ftp/pub/annotation/fungi/comp_yeasts; download page for yeast sequence data, Broad Institute.
- http://ftp.genome.washington.edu/cgi-bin/RepeatMasker; RepeatMasker home page.
- http://www.soe.ucsc.edu/~kent/src/unzipped/hg/featureBits; featureBits source code.
- http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM; Online Mendelian Inheritance in Man home page.