Why do human diversity levels vary at a megabase scale? (original) (raw)
Abstract
Levels of diversity vary across the human genome. This variation is caused by two forces: differences in mutation rates and the differential impact of natural selection. Pertinent to the question of the relative importance of these two forces is the observation that both diversity within species and interspecies divergence increase with recombination rates. This suggests that mutation and recombination are either directly coupled or linked through some third factor. Here, we test these possibilities using the recently generated sequence of the chimpanzee genome and new estimates of human diversity. We find that measures of GC and CpG content, simple-repeat structures, as well as the distance from the centromeres and the telomeres predict diversity as well as divergence. After controlling for these factors, large-scale recombination rates measured from pedigrees are still significant predictors of human diversity and human-chimpanzee divergence. Furthermore, the correlation between human diversity and recombination remains significant even after controlling for human-chimpanzee divergence. Two plausible and non-mutually exclusive explanations are, first, that natural selection has shaped the patterns of diversity seen in humans and, second, that recombination rates across the genome have changed since humans and chimpanzees shared a common ancestor, so that current recombination rates are a better predictor of diversity than of divergence. Because there are indications that recombination rates may have changed rapidly during human evolution, we favor the latter explanation.
Levels of nucleotide diversity within humans vary substantially across the genome at the megabase scale (The International SNP MAP Working Group 2001). This variation is of great interest because it may reflect the action of natural selection (Kaplan et al. 1989; Charlesworth et al. 1995; Hudson and Kaplan 1995; Simonsen et al. 1995; Nordborg et al. 1996). However, there are also other factors that affect levels of diversity, and these need to be corrected in order to interpret patterns of variation.
To begin with, if mutation rates vary, so will levels of diversity. Moreover, for neutrally evolving regions, the divergence rate (total divergence between two species divided by the divergence time) is equal to the mutation rate (Li 1997) and based on divergence, mutation rates do seem to vary along the genome (Wolfe et al. 1989; Ebersberger et al. 2002; Lercher and Hurst 2002; Malcom et al. 2003). Although the average divergence at noncoding regions between humans and chimpanzees is 1.23%, estimates vary from 0.99% to 1.53% (25th-75th-quartile range), which is more than expected by chance under a neutral model in which mutations occur according to a Poisson process (The Chimpanzee Sequencing and Analysis Consortium 2005).
Some of this variation in mutation rates is due to sequence properties. On a local scale, only CpG dinucleotides have a strong impact on mutation rates (Hwang and Green 2004). At a larger scale, the comparison of mouse and human sequences has revealed that divergence correlates with GC and CpG content as well as with recombination rates (Mouse Genome Sequencing Consortium 2002; Hardison et al. 2003). In addition, we would expect mutation rates to vary according to genomic features such as gene expression and the density of genes and repeats because these features are linked either directly or indirectly to the occurrence of DNA damage and/or repair mechanisms (Holmquist 1992; Surralles et al. 2002).
Natural selection may also lead to variation in diversity levels. Indeed, while the rate of divergence at neutral sites does not depend on selection at linked sites, diversity levels do. In particular, both selective sweeps, that is, positive selection driving one allele to fixation (Smith and Haigh 1974; Kaplan et al. 1989; Hudson 1994; Przeworski 2002), and background selection, that is, the removal of deleterious alleles (Charlesworth et al. 1993; Begun and Aquadro 1994; Hudson and Kaplan 1995; Nordborg et al. 1996; Kim and Stephan 2000; Andolfatto 2001; Aquadro et al. 2001), reduce diversity at linked neutral sites. Since the impact of selection on diversity depends on how quickly linkage breaks down, which, in turn, is determined by the recombination rate, variation-reducing selection has a stronger effect on diversity in regions of lower recombination (Nachman 1997; Nachman et al. 1998; Przeworski et al. 2000). On this basis, it was originally hypothesized that the correlation between diversity and recombination may be evidence for widespread natural selection in humans (Nachman 2001).
As an alternative to this selective explanation, Lercher and Hurst (2002) suggested that recombination may be mutagenic. Indeed, we recently demonstrated that recombination rates are a predictor of human-chimpanzee, as well as human-baboon, divergence (Hellmann et al. 2003), implying that mutation and recombination are linked. Furthermore, we found that the correlation of diversity and recombination was no longer significant given divergence, suggesting that the correlation of diversity and recombination may be fully explained by the association between recombination and mutation.
Although the association of recombination and mutation can be explained if recombination is mutagenic (and there is some evidence to support this; see, e.g., Strathern et al. 1995; Rattray et al. 2001), such an association may also be noncausal and mediated by factors that influence both recombination and mutation. Indeed, there are many sequence motifs that show a correlation with both recombination and mutation rates, including GC, CpG, and gene content (Kong et al. 2002), as well as various repeat structures (Jensen-Seaman et al. 2004).
In this study, we examine which factors might contribute to variation in diversity rates across the human genome using the recently assembled chimpanzee genome and human diversity estimates from the recent shotgun re-sequencing effort at the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) and the Broad Institute (The International HapMap Consortium 2003; The Chimpanzee Sequencing and Analysis Consortium 2005).
Results
Sequence features, diversity, and divergence
It has been known for some time that mutation rates vary across the genome and that this variation is not because of pure stochasticity, but due to inherent differences of genome structure. For example, in a previous study we showed an association of diversity and divergence with recombination rates. Recombination rates, however, also show a significant correlation with 12 out of 18 other sequence features that we consider here as predictors of divergence (Supplemental Table 1). In an attempt to tease apart the effects of recombination from those of other genome properties, we analyzed all these factors jointly using multiple linear regression implemented in a stepwise procedure (see Methods). This technique can help unravel relationships that are masked by other factors and only keeps factors that remain significant after inclusion of other variables, that is, we were asking questions like: Do we find a significant correlation between recombination rates and divergence just because both features vary with gene content?
In order to exclude variance that is due to differing selective constraints, we tried to measure divergence and diversity for neutrally evolving sites only. Thus, we used two mutually exclusive measures of intergenic divergence and diversity. First, we counted only fixed differences or polymorphisms outside of interspersed repeats, and as a second measure, only inside interspersed repeats. Using the first measure, there might still be a non-negligible fraction of functionally important sites. However, although we are most confident that interspersed repeats are not functional, the second measure might not be as representative of the genomic region (Gu et al. 2000; Chen and Li 2001). In this section, we discuss 19 sequence properties that might be linked to variations in mutation rates as described above: (1) in intergenic nonrepetitive sequences and (2) in intergenic interspersed repeats.
We found that 12 sequence features explain 53% of the variance of our nonrepeat divergence and 11 sequence features explain 32% of the variance in diversity (Table 1). Recombination rates, GC content, CpG content, poly(A/T) content, simple-repeat content, poly(R/Y) content, CpG-island content, and poly(G/C) content, as well as distance to telomeres and centromeres are significant predictors of human diversity and human-chimpanzee divergence (Table 1; Fig. 1; Supplemental Fig. 1). For divergence, although not for diversity, poly(CA) and gene content also improve the model significantly. Similarly, SINE count improves the prediction of diversity, but not divergence. Poly(CA) content and SINE count are, however, only weak predictors, whereas gene content is a rather strong one. The single best predictor for divergence is simple-repeat content (_R_2 = 0.209); for diversity, it is recombination rates (_R_2 = 0.164). For both divergence and diversity, most of the variance is explained by only three predictors, namely, simple-repeat content, recombination rates, and poly(R/Y) content.
Table 1.
Linear regression models for human-chimpanzee divergence and human diversity
| Human-chimp divergence | Human diversity | ||||
|---|---|---|---|---|---|
| _R_2 | Slope | τ | _R_2 | Slope | τ |
| Recombination rate (cM/Mb) | 0.311* | 0.291* | 0.239* | 0.328* | |
| Poly(R/Y) | −0.380* | −0.031 | −0.151* | 0.020 | |
| Simple repeat content | 0.255* | 0.357* | 0.143* | 0.221* | |
| The top three predictors | 0.380 | 0.241 | |||
| CpG content | 2.021* | 0.046** | 1.105* | 0.125* | |
| CpG islands | −0.504* | −0.076*** | −0.224*** | 0.042 | |
| Distance to centromere | −0.103* | 0.046** | −0.121* | 0.032 | |
| Distance to telomeres | −0.145* | −0.339* | −0.159* | −0.283* | |
| GC content | −2.664* | 0.009 | −1.474* | 0.094* | |
| Gene content | −0.107* | −0.187* | N.S. | −0.066*** | |
| Poly(A/T) | −1.465* | −0.047** | −0.875* | −0.115* | |
| Poly(CA) | −0.079** | 0.227* | N.S. | 0.196* | |
| Poly(G/C) | −0.202* | −0.062*** | −0.253* | 0.024 | |
| SINE count | N.S. | 0.013 | 0.087*** | 0.051** | |
| Full model | 0.526 | 0.324 |
Figure 1.

Scatterplots of the residuals from the full multiple linear regression of non-repeat human-chimpanzee divergence on each significant predictor variable. To assess which factors influence mutation rates, we focus here on the divergence data, which is more precise and extensive than our diversity estimates. The analogous plots for diversity are in Supplemental Figure 1.
When divergence and diversity are measured in interspersed repeats, the overall amount of variance (divergence, _R_2 = 0.45; diversity, _R_2 = 0.26) explained by the 13 and eight significant predictors is lower than if nonrepetitive sequences are considered (Table 2). This might be explained by the higher levels of noise due to the fact that less sequence per window is scored. The finding that the best set of predictors for repeat diversity contains three predictors less than when using the nonrepeat measure, might also be attributed to more noise. For repeat divergence, two additional factors are significant: LINE count and male germ-line expression. Both are only marginally significant predictors of repeat divergence and are nearly significant when divergence is measured using intergenic, nonrepetitive sites. Thus, this model difference is of a quantitative rather than of a qualitative nature. Another difference between the two measures of divergence (diversity) is the importance of poly(R/Y) content. Poly(R/Y) is one of the three best predictors of intergenic divergence, whereas it is not significant for repeat divergence. In this respect, it is important to mention that there is considerable overlap among the various simple-repeat motifs that are significant predictors of divergence (diversity): poly(R/Y) contains part of poly(A/T) and poly(G/C), and the majority of all three is contained in the simple-repeat category. Thus, since these various repeat structures are highly correlated, the fact that different measures of divergence (diversity) retain different repeat structures should not be overinterpreted. Since the two measures of divergence (diversity) do not lead to differences in our conclusions, we only discuss the results from the measure excluding repeats.
Table 2.
Linear regression models to predict human-chimpanzee divergence and human diversity
| Human-chimp divergence | Human diversity | ||||
|---|---|---|---|---|---|
| _R_2 | Slope | τ | _R_2 | Slope | τ |
| CpG islands | −0.486* | −0.142* | −0.337* | −0.027 | |
| Recombination rate (cM/Mb) | 0.189* | 0.248* | 0.268* | 0.270* | |
| Simple repeat content | 0.156* | 0.299* | N.S. | 0.161* | |
| The top three predictors | 0.242 | 0.150 | |||
| CpG content | 2.007* | −0.023 | 1.623* | 0.050** | |
| Distance to centromere | −0.123* | 0.017 | −0.100*** | 0.013 | |
| Distance to telomeres | −0.123* | −0.280* | −0.094** | −0.229* | |
| Fraction of genes expressed in testes | −0.041** | −0.121* | N.S. | −0.065** | |
| GC content | −2.445* | −0.076*** | −2.217* | 0.006 | |
| LINE count | −0.053** | −0.136* | N.S. | −0.111* | |
| Gene content | −0.133* | −0.207* | N.S. | −0.073*** | |
| Poly(A/T) | −1.023* | 0.053** | −1.044* | −0.016 | |
| Poly(CA) | −0.100** | 0.147* | N.S. | 0.013 | |
| Poly(G/C) | −0.149*** | −0.113* | −0.184*** | −0.031 | |
| Full model | 0.452 | 0.258 |
We also tried several predictors related to gene expression, namely, average expression breadth, the average expression strength across 63 tissues or expression strength in the germ line (testes and/or ovaries), and the average number of genes expressed the germ line in a given window. None of these variables explains any additional part of the variance in divergence or diversity, except male germ-line expression for divergence measured in interspersed repeats only. The rank correlation of these predictors with human diversity and human-chimpanzee divergence is listed in Supplemental Table 2. The nonsignificance of these predictors does not prove that they have no effect on mutation rate, since predictors may fail to be significant if the magnitude of their effect is swamped by measurement error.
Recombination, diversity, and divergence
Recombination rates are one of the most powerful predictors of human-chimpanzee divergence and are the single most powerful predictor of human diversity (Fig. 2) (_R_2 = 0.164, p <10-6). Even after controlling for a variety of sequence motifs that could mediate the observed correlation, divergence and diversity increase with increasing recombination rates (Tables 1 and 2). Thus, the correlation between divergence and recombination suggests an association between mutation and recombination.
Figure 2.

Human recombination rates are the strongest predictor of (A) human diversity (_R_2 = 0.164, p <10-15) and are also correlated with (B) chimpanzee diversity (_R_2 = 0.0379, p <10-7). After correcting for the effects of recombination on mutation rate (using human-chimpanzee divergence), we still observe a positive correlation (C) between the residuals of human diversity and recombination rates (_R_2 = 0.035, p <10-6), but not (D) between the residuals of chimpanzee diversity and human recombination rates (_R_2 = 0.0022, p = 0.10).
It has been hypothesized that recombination could be mutagenic (Lercher and Hurst 2002; Hellmann et al. 2003), and, at least in yeast, there is some evidence to support this (see, e.g., Strathern et al. 1995; Rattray et al. 2001). Based on the model developed here (Table 1), we can estimate the fraction of mutations that might be due to recombination. For this purpose, we assume a divergence time of 6 million years and a generation time of 20 years. We then use the median values across the 3-Mb windows for the model parameters other than recombination rates. Given these parameters, the mutation rate increases by ∼1.5 × 10-9/base pair per generation with a 1 cM/Mb increase in the recombination rate. In other words, roughly three of every 20 recombination events introduce a mutation.
Since the results suggest a link between mutation and recombination, this raises the possibility that the correlation between recombination and diversity is simply the result of this association, rather than of natural selection. To examine this, we assessed the relationship between diversity and human recombination rates in a multiple linear regression, including divergence as a predictor (Fig. 2B). This effectively asks if recombination predicts diversity beyond what is expected from the relationship of recombination and divergence. In contrast to our previous findings (Hellmann et al. 2003), we find that recombination is still a significant predictor of human diversity after correction for human-chimpanzee divergence (Fig. 2B) (_R_2 = 0.035, p <10-6), although the proportion of the variation that it explains is small.
This observation reopens the question of whether the variation in diversity levels could be, in part, due to variation-reducing selection. In order to gain a sense of how plausible a selective explanation would be, we assessed the effect of different selective models on the relationship between recombination and diversity using parameters that seem realistic for humans. In particular, we ran simulations to determine how three models of selection influence diversity at neutral loci: (1) recurrent selective sweeps, (2) background selection, and (3) the combination of these two. The results of the simulations were then compared to the relationship between the residuals of diversity and recombination rates after a regression on divergence.
Based on a comparison of the standardized regression slopes for different sets of selection parameters, we cannot rule out any of the three scenarios (Supplemental Table 3), and any conclusions can only be tentative as the models are simplistic (see Methods). This said, the selective sweep model predicts a logarithmic relationship between diversity and recombination, while the relationship of our data appears to be linear. In particular, for the selective sweep model, we would expect much lower diversity in regions of low recombination than we observe. Furthermore, it predicts little effect on levels of diversity for all but the lowest rates of recombination (Fig. 3A). On the other hand, the model of background selection, as well as the model including both background selection and selective sweeps, predict an approximately linear relationship between diversity and recombination rates (Fig. 3B,C).
Figure 3.
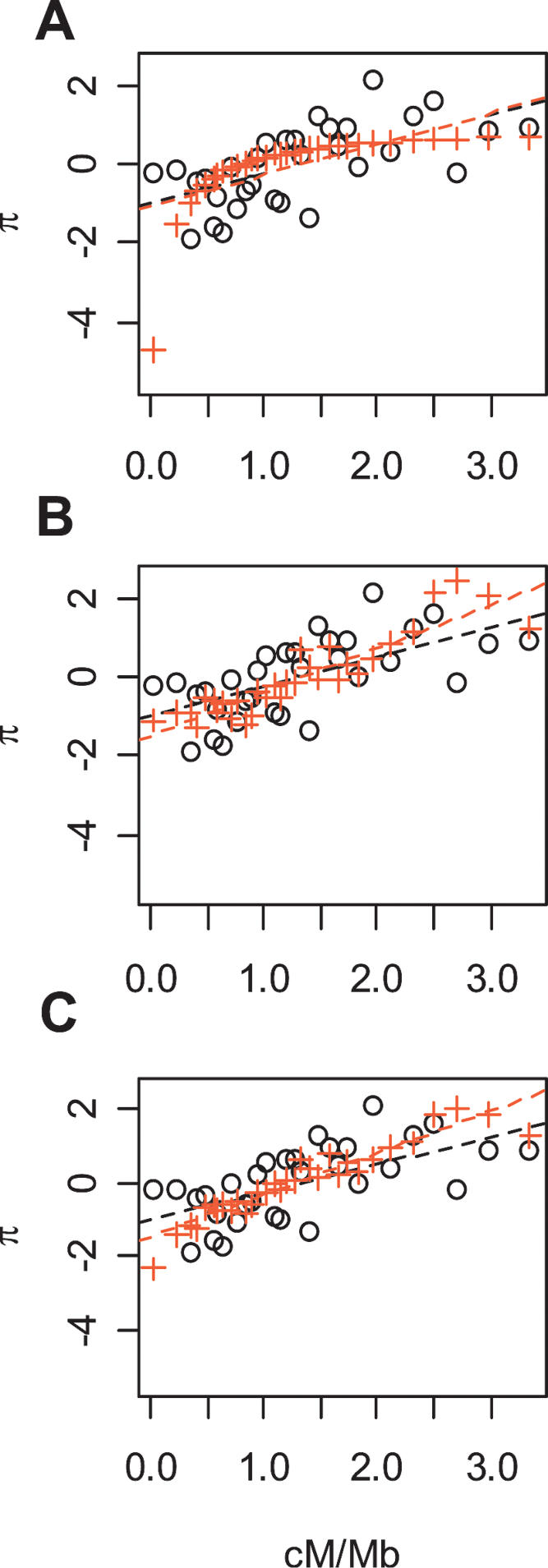
Correlation of recombination and the residuals of diversity after regression on divergence for the observed data (black dots), for simulated data (red crosses) under (A) a recurrent selective sweep model with s = 0.02 and μS =2 × 10-10, (B) a background selection model with μd =2 × 10-10 and t = 0.02, and (C) a model with both background selection and recurrent selective sweeps. We chose the parameter values for each of these three models that appeared to best fit the observed data. The points on all three curves represent means over 25 windows. To make the simulated data more comparable to the residuals of diversity, they were transformed to approximate mean 0 and variance 1.
However, we note that the frequency of positively and negatively selected alleles in a given window increases with the number of functionally important sites. Thus, we might expect gene-rich regions to show lower diversity. Thus, in addition to recombination rates, gene content should remain a significant predictor of diversity after correction for divergence. This is not the case (p = 0.114).
Moreover, assuming that similar selective forces (i.e., similar frequencies and strength of selective events) acted to shape human and chimpanzee diversity, we would expect to find a correlation between chimpanzee diversity and recombination as well. Yet human recombination rates are not a significant predictor of chimpanzee diversity, after correction for human-chimpanzee divergence (Clint-diversity: _R_2 = 0.0016, p = 0.13; Central-Western diversity: _R_2 = 0.0022, p = 0.10) (Fig. 2D). This could reflect lack of power, as the chimpanzee diversity estimates are based on less data and possibly have more errors than the human estimates. However, when the amount of human diversity data is reduced to what is available for the chimpanzee, the relationship of recombination rates with human diversity only becomes as weak as observed for chimpanzee diversity if unrealistic amounts of error are added (Table 3). Thus, we should see a significant relationship in chimpanzees if the putative effect of selection on diversity were as strong as it is in humans.
Table 3.
Percentage of bootstraps over 3-Mb windows for which recombination remains a stronger predictor of the residuals of human diversity with errors added than chimpanzee diversity after regression on divergence
| P (βmin-human ≥ βchimp) | ||
|---|---|---|
| λ | Clint diversity | Central-Western diversity |
| 1/100,000 | 99.24 | 99.19 |
| 1/50,000 | 93.33 | 99.65 |
| 1/25,000 | 71.28 | 98.45 |
| 1/10,000 | 0.10 | 29.20 |
In summary, while models including background selection are roughly consistent with the data, the absence of a significant relationship of diversity with gene content (controlling for recombination rates and divergence) and the absence of a similar relationship in chimpanzees cast doubt on their plausibility.
Discussion
Predictors of mutation rates
Altogether, we considered 19 features of DNA sequences that could have an influence on mutation rates. Of these, 12 and 11 were significant predictors of divergence and diversity, respectively (Table 1; Methods). The sequence features are also correlated with each other. Out of the 171 possible pairwise comparisons, 117 show a significant correlation (Supplemental Table 1). Nevertheless, the fact that 12 (11) predictors are retained in the regression model indicates that each explains a unique aspect of divergence (diversity). The use of multiple linear regression allows us to gauge the extent to which the relationship between divergence (or diversity) and a second factor is confounded by the fact that they both correlate with additional variables. For example, GC content shows a significant pairwise correlation with 16 of the 18 other traits that might affect divergence and/or diversity. In fact, the variation in GC content has even been suggested to be causatively linked to recombination via biased gene conversion (Meunier and Duret 2004). However, once we have corrected for the covariates of GC content and recombination rate, divergence decreases with increasing GC content.
At first, the finding of a linear and negative relationship of divergence (diversity) and GC content in the full model seems to contradict previous reports: The International SNP Map Working Group (2001) found a positive relationship between GC content and diversity, while Hardison et al. (2003) reported a quadratic function between GC content and mouse-human divergence. However, if we consider a model only with GC content, then we also see a positive correlation between diversity and GC content (Table 1), as well as a quadratic relationship with GC content as a predictor of human-chimpanzee divergence (Fig. 4). Moreover, when we add CpG content to the model, then we obtain the negative relationship between GC content and divergence (diversity). Thus our finding of a linear, negative relationship in the full model is not contradictory; it just reflects the importance of CpGs.
Figure 4.

The relationship between human-chimpanzee divergence and GC content is quadratic (red line) rather than linear. However, the contribution of the quadratic term becomes insignificant if CpG content is added to the model. The fitted values of this model are indicated by the blue circles.
An intuitive explanation for the quadratic relationship between divergence and GC content is that the probability of observing a CpG is a function of GC-squared. Since CpGs are mutational hotspots (Shen et al. 1994), one expects CpG content to be positively correlated with divergence, which, indeed, is the case. However, the strong positive correlation between CpG content and divergence in the full model remains even if one measures divergence excluding CpGs (Supplemental Table 4). Thus, the influence of CpG content on divergence extends beyond CpGs being mutational hotspots.
As with CpG dinucleotides, it is not clear whether the other significant predictors covary with mutation rate or together predict a higher-level feature such as chromatin structure or replication timing, which might be more directly linked to mutation rates (Goldman et al. 1984; Holmquist and Caston 1986). In particular, Holmquist (1992) suggested a distinction between five chromatin “flavors” that were classified based on their chromosome band association, GC content, and Alu content. He showed that these chromatin flavors differ in their gene content and the frequency with which chromosomal breakpoints occur. There is also evidence to suggest that sequence motifs capture information about replication timing, as GC content, gene content, and repeat content have been demonstrated to correlate with replication timing (Woodfine et al. 2004). Furthermore, transcription-coupled repair complexes have been found to be enriched in early replicating gene-rich bands (Surralles et al. 2002). We find a relatively strong pairwise correlation between germ-line expression and divergence (Supplemental Table 2), but no or only a weak association in our full model (Tables 1 and 2), which is most likely caused by higher error rates in the expression measures as compared to the sequence motifs. Furthermore, if there is an association between open chromatin and mutation beyond the suggested mechanism of transcription-coupled repair, gene density might be more informative than gene expression (Gilbert et al. 2004). Thus, it is conceivable that the DNA sequence motifs found to predict mutation rates may be proxies for chromatin structure and/or replication timing, which may affect mutation rates in the germ line.
The distances from centromeres and telomeres also predict sequence divergence and diversity. Telomeres are unusual genomic areas in many respects. They are relatively GC-rich, have a high gene density, and exhibit higher recombination rates. However, even with these features added to the model, distance to the telomeres is retained as a predictor of sequence divergence (Table 1). Furthermore, unlike in most other regions of the genome, open chromatin is not necessarily associated with replication timing in telomeres (Gilbert et al. 2004). Thus, one possibility is that sequence motifs in telomeres predict mutation rates differently than in nontelomeric regions.
Altogether, we find that the 12 parameters explain 53% of the variance in human-chimpanzee divergence and the 11 parameters explain 32% of the variation in human diversity (Table 1). Most of the variance for both divergence and diversity is explained by just three predictors: simple-repeat content, poly(R/Y) content, and recombination rate. Of these parameters, 10 are in common to divergence and diversity. Moreover, the pairwise correlations of those parameters with divergence and diversity are not dissimilar, suggesting that the difference in predictors kept is quantitative rather than qualitative (Tables 1 and 2).
Recombination, divergence, and diversity
Interestingly, recombination rate still remains a significant positive predictor of both diversity and divergence after controlling for various sequence motifs (Table 1). One possible explanation is that recombination is mutagenic. Based on our model, we would expect roughly three in 20 recombination events to cause a mutation. It is difficult to find support for this estimate from laboratory experiments. We only know that the repair of double-strand breaks in yeast leads to more errors than repair associated with genome duplication (Strathern et al. 1995). However, we do not know how well this result extends to humans, the size of the region affected by double-strand repair, nor what proportion of double-strand breaks in a region leads to actual recombination events. Nevertheless, although it does not seem implausible that three in 20 recombination events cause a mutation, we still cannot exclude the possibility that recombination and mutation are linked only indirectly.
Given that divergence and recombination are positively correlated, we wanted to ask whether this could explain the correlation between diversity and recombination. Contrary to our previous study (Hellmann et al. 2003), we find that recombination and diversity remain correlated even after the effect of human-chimpanzee divergence has been eliminated (Fig. 2). The remaining relationship is quite weak, so that the difference in the two studies is presumably due to the increased power of this study (75 vs. 834 data points).
One longstanding hypothesis for the positive relationship between diversity and recombination is that it reflects a signature of variation-reducing selection in the human genome. We reexamined the three main models for variation-reducing selection and found that only models including background selection are consistent with the observed data (Fig. 3). Moreover, two lines of evidence suggest that selection may not be the best explanation for this correlation. First, windows with a high gene density should be more likely to experience selection, yet gene density does not explain a significant portion of the variance in diversity levels (after correction for recombination rates). Second, we would have to postulate that selection is more frequent or stronger in humans than in chimpanzees, since we find no correlation between chimpanzee diversity and human recombination rates (Fig. 2). Thus, it seems unlikely that selection alone explains our results.
An alternative explanation for our finding is that large-scale recombination rates across the genome changed during primate evolution so that they differ between humans and chimpanzees. Unfortunately, this hypothesis is difficult to evaluate, as we do not have a genetic map for chimpanzees. However, it seems possible that recombination rates could have changed rapidly during human and chimpanzee evolution. Heritable variation for recombination does exist (see Brooks 1988 and references within), including for humans (Kong et al. 2004). Moreover, the genetic map of baboons (Papio hamadryas) is 30 cM shorter than for humans (Rogers et al. 2000), and genetic maps from different human populations also seem to differ slightly (Jorgenson et al. 2005). Furthermore, two human hotspots are absent in chimpanzees (Ptak et al. 2004a) and rhesus macaques (Macaca mulatta) (Wall et al. 2003), respectively. In fact, recent data suggest that the fine-scale recombination landscape of humans and chimpanzees differ dramatically (Ptak et al. 2005). If recombination rates are thus rapidly evolving, the correlation of human recombination rates would be strongest with human diversity, weaker for human-chimpanzee divergence, and weakest for the correlation with chimpanzee diversity. Qualitatively, this is what we find.
Obviously, the above explanation does not exclude the action of natural selection. In fact, we know that natural selection has acted on particular regions of the genome (e.g., Harding et al. 2000; Makova et al. 2001; Enard et al. 2002; Hamblin et al. 2002; Sabeti et al. 2002). The open question is whether it has been frequent and strong enough to shape genome-wide patterns of variability.
In conclusion, we demonstrate that a wide variety of sequence motifs are correlated with human diversity and human-chimpanzee divergence and are likely to influence mutation rates. Many of these motifs had not been previously implicated as potential factors in explaining variation in mutation rates. Despite the inclusion of these additional factors, recombination remains an important predictor of both diversity and divergence. Moreover, the correlation between recombination and diversity cannot be explained solely by the link between mutation and recombination. This suggests some salient feature is missing from our model, such as natural selection and/or the rapid evolution of large-scale recombination rates. Given our data and recent comparisons of fine-scale recombination rates between humans and chimpanzees, the latter explanation seems more plausible.
Methods
Recombination, diversity, and divergence
We determined the average human-chimpanzee divergence and human diversity within 834 consecutive 3-Mb windows across autosomes for which sex-averaged recombination rates could be estimated (Kong et al. 2002). A window size of 3 Mb seemed to be a reasonable choice, given the accuracy of recombination rate estimates from the original study, which limits us from going smaller (Kong et al. 2002). We also examined our full model for divergence as presented in Table 1 at window sizes of 6, 10, and 20 Mb, but these differences were minor and did not change our qualitative conclusions (data not shown). We calculated recombination rates as the slope of a regression of genetic and physical distances of markers within a 3-Mb window. The windows were set to obtain the maximal number of 3-Mb windows. Estimates of genetic distances came from Michael L. Frigge, who kindly provided us with an updated version of the genetic map published in Kong et al. (2002), while the physical distances came from the mapping of the markers to human genome build 34.
Estimates of human diversity for the windows were taken from the alignment of sequences from whole-genome shotgun libraries of eight African-American individuals generated at the BCM-HGSC and sequenced at the BCM-HGSC and the Broad Institute (http://www.cardiogene.org/bpr/background.htm; http://www.hapmap.org) to the human genome (build 34). Only high-quality bases were taken into account (Altshuler et al. 2000), and in order to obtain comparable diversity estimates across the genome, we used only pairwise comparisons. That is, if there were more than two sequences (chromosome) sampled at a position, we picked two at random from all sequences including the human consensus of build 34. Most of the time, we compared one of the 16 different chromosomes represented in the shotgun library to the human genome sequence. Hence the diversity π is the number of sites with two alleles in two randomly chosen sequences divided by the number of sites for which we had at least two high-quality reads. We interrogate a median of 815 kb/window, excluding genic regions and simple repeats. Of these, 437 kb fall into nonrepetitive regions and 378 kb into interspersed repeats.
Similarly, we estimated human-chimpanzee divergence from the alignment of shotgun reads to the human genome, using the same quality criteria as for the alignments of the human reads. Again, if multiple reads were aligned and disagreed with respect to a substitution, we picked one read at random. This approach provided us with the number of differences and the total number of base pairs compared. Since humans and chimpanzees are very similar at the nucleotide level, we did not apply a correction for multiple substitutions per site.
Estimates of chimpanzee diversity were obtained from two sources. One was based on overlapping alignments of shotgun reads from the different alleles of one chimpanzee (Clint) to the human genome. A second was obtained by comparing any of the two other Western Chimpanzees with any of the three Central Chimpanzees sequenced to 0.1× coverage each for the chimpanzee genome project (The Chimpanzee Sequencing and Analysis Consortium 2005).
We generated two sets of divergence and diversity estimates: (1) unique sequence outside of genes and (2) interspersed repeats outside of genes as annotated in the UCSC Genome Browser (http://genome.ucsc.edu). In both cases, we excluded regions annotated in the human genome as duplicated regions (Bailey et al. 2002) and as simple repeats (UCSC Genome Browser).
Potential linking factors between recombination and diversity and divergence
As potential predictors of both diversity or divergence and recombination for our 3-Mb windows, we considered GC, CpG, poly(A/T) (W_n_ ≥ 4), simple repeats, polypurine/pyrimidine (R_n_ ≥ 30/Y_n_ ≥ 30), poly(G/C) (S_n_ ≥ 20), poly(CA)n ≥ 20 content, the count of CpG islands, SINEs, LINEs, AluYs, and the percentage of sequence within genes (gene content) as estimated from the annotation of the human genome build 34 given in the UCSC Genome Browser. We chose a smaller minimal run length for poly(A/T), because this run length has previously been shown to be highly correlated with recombination rates (Kong et al. 2002). We also looked at the distance from the middle of a given window to telomeres and centromeres in humans, also given in the UCSC Genome Browser. Distance to telomeres was always measured as the distance to the telomere of the same chromosomal arm.
To consider the effect of gene expression on divergence, we took the count of expressed genes in testis germ cells or ovaries (Su et al. 2004), where a gene was counted as expressed if one probe set associated with the gene had a detection _p_-value ≤ 0.05. We also measured expression breadth, using 63 non-cancer, non-cell-line tissues from the same data set, as the median number of tissues in which a gene was detected. Finally, we calculated the median RMA-values as a measure of absolute mRNA levels (Bolstad et al. 2003) for each window in all 63 tissues, as well as in testis germ cells and ovaries.
Multiple linear regression
Divergence and diversity were log-transformed to be roughly normally distributed. All data were further transformed according to the Cochrane and Orcutt method (Montgomery et al. 2001) to account for effects of spatial autocorrelation in the data. For easier comparisons, all parameters were scaled to have mean 0 and variance 1. Thus, the slopes given for the various parameters are directly comparable measures of the strength of the relationship between explanatory and response variables. To determine which parameters significantly improve the fit of the model, we used a stepwise procedure (predictors were kept or removed according to the criterion of minimizing AIC as implemented in R; http://www.r-project.org). We then refined the model using standard regression diagnostics to evaluate the validity of the model and the influence of outliers, which finally led to a model with 12 (11) predictors for nonrepeat divergence (diversity) and a model with 13 (8) predictors for repeat divergence (diversity). As part of these diagnostics, we examined the plots of residuals for each predictor against divergence (Fig. 1) and diversity (Supplemental Fig. 1) to check whether the assumption of linearity was violated. Since both our response and our explanatory variables have measurement errors, the use of multiple regression for these data results in slope estimates that are biased downward. Furthermore, the explanatory variables investigated here are correlated with each other. In particular, the slope estimates for GC, CpG, and poly(A/T) are inflated because these three are highly correlated with one another. In addition, the correlation among our predictors makes predictions outside our parameter space extremely unreliable. However, since we are considering whole-genome data, this is not a problem here.
Modeling recurrent selective sweeps
There are 834 windows with human diversity estimates. To characterize the effects of repeated (but nonoverlapping) fixations of beneficial alleles at nearby sites, we simulated each window as a neutral locus of 10 kb, with a recombination rate estimated as detailed above. The neutral mutation rate was set as 2 × 10-8/base pair and generation (Nachman and Crowell 2000), and the effective population size was assumed to be 15,000 (Ptak et al. 2004b). We used the program msHH kindly provided by M. Przeworski, which assumes a constant rate of favorable fixations per unit time, constant recombination rates, and an infinite sites mutation model at the neutral locus (Przeworski 2002). We assumed selection coefficients of 0.02 or 0.002 and that 1% or 0.1% of mutations are advantageous, equivalent to an advantageous mutation rate of 2 × 10-10 and 2 × 10-11 per generation, which is constant across the genome. For each recombination value, we calculated the average diversity from 100,000 simulations.
Modeling background selection
We assumed that the effect of background selection is a reduction in Ne, which is realistic as long as purifying selection is not weak and the deleterious mutation rate is high (Nordborg et al. 1996). We calculated this reduction, fb, from equation 10 in Kim and Stephan (2000), which assumes fitness effects to be multiplicative. Hence, N̂e = fb × 15,000. fb depends on five parameters: μ_d_, the deleterious mutation rate per base pair per generation; t, the deleterious selection coefficient; _L_L and _L_R, the length of segment (to the left and right, respectively) undergoing deleterious selection; and r(x), the recombination rate along the chromosome (x indicates position). The recombination rate was allowed to vary piecewise according to the 3-Mb estimates. Note that this includes variation in recombination rate at the 3-Mb scale, but not finer-scale variation, for example, recombination hotspots. For _L_L and L_R, we used the distance from the midpoint of a window to the two telomeres. For t, we assumed the same coefficients as for the selective sweep model, namely, 0.02 and 0.002. To gain a rough sense of the magnitude of μ_d, we assumed ∼1%-10% of sites to be under strong constraint as estimated from the comparison of the rat, mouse, and human genomes (Cooper et al. 2004). This yields a deleterious mutation rate of 2 × 10-9 to 2 × 10-10, which we assume to be constant along the genome.
To model both background selection and recurrent sweeps, we applied this reduction (fb) to two parameters in the selective sweep program: ρ and θ, where ρ is the population recombination rate and θ is the population mutation rate (Kim and Stephan 2000).
Accounting for possible errors in the chimpanzee sequence
Quality scores and neighbor quality standards were applied similarly to chimpanzee and human shotgun reads. However, to estimate chimpanzee diversity, we compared unfinished sequence, that is, shotgun reads, to unfinished sequence, while for the human data, unfinished sequence is compared to finished human sequence in most cases. Furthermore, one of our two estimates for chimpanzee diversity comes from the comparison of reads from the same individual, and roughly half of the time, we compared the same chromosome. In this case, apparent diversity for this region will, in fact, reflect sequencing errors alone. As a result, the measured diversity will be approximately halved relative to its true value, and the impact of sequencing errors will be relatively stronger than for human diversity. To compare the Clint-Clint-SNPs to the human diversity estimates, we therefore halved human diversity. In order to estimate the error rate in the chimpanzee SNP data, we estimated diversity levels for Clint's X-chromosome (Supplemental Figure 2). Since Clint is male (and thus has only one X chromosome), these “diversity estimates” provide an estimate of the rate of sequencing errors. We modeled this rate of false positives as a Γ-distribution, with a shape similar to the observed errors as estimated from the X-chromosome (i.e., shape-parameter α = 2) and a mean equal to the observed mean error rate (i.e., λ = 1/100,000). In addition, we also used a series of Γ-distribution with a higher mean error rate (Table 3).
To test how much sequencing errors decrease the underlying relationship of diversity and recombination rates, we drew an error rate from the Γ-distribution and multiplied it by the number of base pairs compared in chimpanzees. This number of simulated sequence errors was then added to the number of SNPs expected given the human diversity estimates. Finally, we bootstrapped 100,000 times over the windows for which we had divergence and chimpanzee diversity estimates, calculating each time the slope of the partial regression of recombination rates on human and chimpanzee diversity after correcting for divergence. We then tabulated how often the slope for chimpanzee diversity would remain smaller than the one observed for human diversity (Table 3).
Supplementary Material
[Supplemental Research Data]
[ [The Chimpanzee Genome]]
Acknowledgments
We thank Molly Przeworski for many helpful suggestions, critical reading of the manuscript, and for helping us with her software msHH; Laurent Duret for helpful comments; Michael L. Frigge for providing an updated human genetic map; and all members of the chimpanzee genome consortium for helpful discussions. Furthermore, we thank the members of the Baylor College of Medicine Human Genome Sequencing Center and the Broad Institute for generation and early release of the DNA sequence data used to derive the estimates of human diversity in this study. We also thank the Max Planck Society and the Bundesministerium für Forschung for financial support.
Footnotes
[Supplemental material is available online at www.genome.org.\]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3461105\. Freely available online through the Genome Research Immediate Open Access option.
References
- Altshuler, D., Pollara, V.J., Cowles, C.R., Van Etten, W.J., Baldwin, J., Linton, L., and Lander, E.S. 2000. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407**:** 513-516. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P. 2001. Adaptive hitchhiking effects on genome variability. Curr. Opin. Genet. Dev. 11**:** 635-641. [DOI] [PubMed] [Google Scholar]
- Aquadro, C.F., Bauer DuMont, V., and Reed, F.A. 2001. Genome-wide variation in the human and fruitfly: A comparison. Curr. Opin. Genet. Dev. 11**:** 627-634. [DOI] [PubMed] [Google Scholar]
- Bailey, J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002. Recent segmental duplications in the human genome. Science 297**:** 1003-1007. [DOI] [PubMed] [Google Scholar]
- Begun, D.J. and Aquadro, C.F. 1994. Evolutionary inferences from DNA variation at the 6-phosphogluconate dehydrogenase locus in natural populations of Drosophila: Selection and geographic differentiation. Genetics 136**:** 155-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad, B.M., Irizarry, R.A., Astrand, M., and Speed, T.P. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19**:** 185-193. [DOI] [PubMed] [Google Scholar]
- Brooks, L.D. 1988. The evolution of recombination rates. In The evolution of sex: An examination of current ideas (eds. R.E. Michod and B.R. Levin), pp. 87-105. Sinauer Associates, Sutherland, MA.
- Charlesworth, B., Morgan, M.T., and Charlesworth, D. 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134**:** 1289-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D., Charlesworth, B., and Morgan, M.T. 1995. The pattern of neutral molecular variation under the background selection model. Genetics 141**:** 1619-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F.C. and Li, W.H. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68**:** 444-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequencing of the chimpanzee genome and comparison with the human genome. Nature (in press).
- Cooper, G.M., Brudno, M., Stone, E.A., Dubchak, I., Batzoglou, S., and Sidow, A. 2004. Characterization of evolutionary rates and constraints in three mammalian genomes. Genome Res. 14**:** 539-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger, I., Metzler, D., Schwarz, C., and Paabo, S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70**:** 1490-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard, W., Przeworski, M., Fisher, S.E., Lai, C.S., Wiebe, V., Kitano, T., Monaco, A.P., and Paabo, S. 2002. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418**:** 869-872. [DOI] [PubMed] [Google Scholar]
- Gilbert, N., Boyle, S., Fiegler, H., Woodfine, K., Carter, N.P., and Bickmore, W.A. 2004. Chromatin architecture of the human genome; gene-rich domains are enriched in open chromatin fibers. Cell 118**:** 555-566. [DOI] [PubMed] [Google Scholar]
- Goldman, M.A., Holmquist, G.P., Gray, M.C., Caston, L.A., and Nag, A. 1984. Replication timing of genes and middle repetitive sequences. Science 224**:** 686-692. [DOI] [PubMed] [Google Scholar]
- Gu, Z., Wang, H., Nekrutenko, A., and Li, W.H. 2000. Densities, length proportions, and other distributional features of repetitive sequences in the human genome estimated from 430 megabases of genomic sequence. Gene 259**:** 81-88. [DOI] [PubMed] [Google Scholar]
- Hamblin, M.T., Thompson, E.E., and Di Rienzo, A. 2002. Complex signatures of natural selection at the Duffy blood group locus. Am. J. Hum. Genet. 70**:** 369-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, R.M., Healy, E., Ray, A.J., Ellis, N.S., Flanagan, N., Todd, C., Dixon, C., Sajantila, A., Jackson, I.J., Birch-Machin, M.A., et al. 2000. Evidence for variable selective pressures at MC1R. Am. J. Hum. Genet. 66**:** 1351-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison, R.C., Roskin, K.M., Yang, S., Diekhans, M., Kent, W.J., Weber, R., Elnitski, L., Li, J., O'Connor, M., Kolbe, D., et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13**:** 13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann, I., Ebersberger, I., Ptak, S.E., Paabo, S., and Przeworski, M. 2003. A neutral explanation for the correlation of diversity with recombination rates in humans. Am. J. Hum. Genet. 72**:** 1527-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist, G.P. 1992. Chromosome bands, their chromatin flavors, and their functional features. Am. J. Hum. Genet. 51**:** 17-37. [PMC free article] [PubMed] [Google Scholar]
- Holmquist, G.P. and Caston, L.A. 1986. Replication time of interspersed repetitive DNA sequences in hamsters. Biochim. Biophys. Acta 868**:** 164-177. [DOI] [PubMed] [Google Scholar]
- Hudson, R.R. 1994. How can the low levels of DNA sequence variation in regions of the Drosophila genome with low recombination rates be explained? Proc. Natl. Acad. Sci. 91**:** 6815-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R.R. and Kaplan, N.L. 1995. Deleterious background selection with recombination. Genetics 141**:** 1605-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, D.G. and Green, P. 2004. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl. Acad. Sci. 101**:** 13994-14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2003. The International HapMap Project. Nature 426**:** 789-796. [DOI] [PubMed] [Google Scholar]
- The International SNP MAP Working Group. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409**:** 928-933. [DOI] [PubMed] [Google Scholar]
- Jensen-Seaman, M.I., Furey, T.S., Payseur, B.A., Lu, Y., Roskin, K.M., Chen, C.F., Thomas, M.A., Haussler, D., and Jacob, H.J. 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 14**:** 528-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson, E., Tang, H., Gadde, M., Province, M., Leppert, M., Kardia, S., Schork, N., Cooper, R., Rao, D.C., Boerwinkle, E., et al. 2005. Ethnicity and human genetic linkage maps. Am. J. Hum. Genet. 76**:** 276-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, N.L., Hudson, R.R., and Langley, C.H. 1989. The “hitchhiking effect” revisited. Genetics 123**:** 887-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. and Stephan, W. 2000. Joint effects of genetic hitchhiking and background selection on neutral variation. Genetics 155**:** 1415-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, A., Gudbjartsson, D.F., Sainz, J., Jonsdottir, G.M., Gudjonsson, S.A., Richardsson, B., Sigurdardottir, S., Barnard, J., Hallbeck, B., Masson, G., et al. 2002. A high-resolution recombination map of the human genome. Nat. Genet. 31**:** 241-247. [DOI] [PubMed] [Google Scholar]
- Kong, A., Barnard, J., Gudbjartsson, D.F., Thorleifsson, G., Jonsdottir, G., Sigurdardottir, S., Richardsson, B., Jonsdottir, J., Thorgeirsson, T., Frigge, M.L., et al. 2004. Recombination rate and reproductive success in humans. Nat. Genet. 36**:** 1203-1206. [DOI] [PubMed] [Google Scholar]
- Lercher, M.J. and Hurst, L.D. 2002. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 18**:** 337-340. [DOI] [PubMed] [Google Scholar]
- Li, W.H. 1997. Molecular evolution. p. 49. Sinauer, Sunderland, MA.
- Makova, K.D., Ramsay, M., Jenkins, T., and Li, W.H. 2001. Human DNA sequence variation in a 6.6-kb region containing the melanocortin 1 receptor promoter. Genetics 158**:** 1253-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcom, C.M., Wyckoff, G.J., and Lahn, B.T. 2003. Genic mutation rates in mammals: Local similarity, chromosomal heterogeneity, and X-versus-autosome disparity. Mol. Biol. Evol. 20**:** 1633-1641. [DOI] [PubMed] [Google Scholar]
- Meunier, J. and Duret, L. 2004. Recombination drives the evolution of GC-content in the human genome. Mol. Biol. Evol. 21**:** 984-990. [DOI] [PubMed] [Google Scholar]
- Montgomery, D.C., Peck, E.A., and Vining, G.G. 2001. Introduction to linear regression analysis. John Wiley, New York.
- Mouse Genome Sequencing Consortium. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420**:** 520-562. [DOI] [PubMed] [Google Scholar]
- Nachman, M.W. 1997. Patterns of DNA variability at X-linked loci in Mus domesticus. Genetics 147**:** 1303-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 2001. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 17**:** 481-485. [DOI] [PubMed] [Google Scholar]
- Nachman, M.W. and Crowell, S.L. 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156**:** 297-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman, M.W., Bauer, V.L., Crowell, S.L., and Aquadro, C.F. 1998. DNA variability and recombination rates at X-linked loci in humans. Genetics 150**:** 1133-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., Charlesworth, B., and Charlesworth, D. 1996. The effect of recombination on background selection. Genet. Res. 67**:** 159-174. [DOI] [PubMed] [Google Scholar]
- Przeworski, M. 2002. The signature of positive selection at randomly chosen loci. Genetics 160**:** 1179-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., Hudson, R.R., and Di Rienzo, A. 2000. Adjusting the focus on human variation. Trends Genet. 16**:** 296-302. [DOI] [PubMed] [Google Scholar]
- Ptak, S.E., Roeder, A.D., Stephens, M., Gilad, Y., Paabo, S., and Przeworski, M. 2004a. Absence of the TAP2 human recombination hotspot in chimpanzees. PLoS Biol. 2**:** 849-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak, S.E., Voelpel, K., and Przeworski, M. 2004b. Insights into recombination from patterns of linkage disequilibrium in humans. Genetics 167**:** 387-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak, S.E., Hinds, D.A., Koehler, K., Nickel, B., Patil, N., Ballinger, D.G., Przeworski, M., Frazer, K.A., and Paabo, S. 2005. Fine-scale recombination patterns differ between chimpanzees and humans. Nat. Genet. 37**:** 429-434. [DOI] [PubMed] [Google Scholar]
- Rattray, A.J., McGill, C.B., Shafer, B.K., and Strathern, J.N. 2001. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: A role for SAE2/COM1. Genetics 158**:** 109-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, J., Mahaney, M.C., Witte, S.M., Nair, S., Newman, D., Wedel, S., Rodriguez, L.A., Rice, K.S., Slifer, S.H., Perelygin, A., et al. 2000. A genetic linkage map of the baboon (Papio hamadryas) genome based on human microsatellite polymorphisms. Genomics 67**:** 237-247. [DOI] [PubMed] [Google Scholar]
- Sabeti, P.C., Reich, D.E., Higgins, J.M., Levine, H.Z., Richter, D.J., Schaffner, S.F., Gabriel, S.B., Platko, J.V., Patterson, N.J., McDonald, G.J., et al. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419**:** 832-837. [DOI] [PubMed] [Google Scholar]
- Shen, J.C., Rideout III, W.M., and Jones, P.A. 1994. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 22**:** 972-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, K.L., Churchill, G.A., and Aquadro, C.F. 1995. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics 141**:** 413-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J.M. and Haigh, J. 1974. The hitch-hiking effect of a favourable gene. Genet Res. 23**:** 23-35. [PubMed] [Google Scholar]
- Strathern, J.N., Shafer, B.K., and McGill, C.B. 1995. DNA synthesis errors associated with double-strand-break repair. Genetics 140**:** 965-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, A.I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K.A., Block, D., Zhang, J., Soden, R., Hayakawa, M., Kreiman, G., et al. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. 101**:** 6062-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surralles, J., Ramirez, M.J., Marcos, R., Natarajan, A.T., and Mullenders, L.H. 2002. Clusters of transcription-coupled repair in the human genome. Proc. Natl. Acad. Sci. 99**:** 10571-10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J.D., Frisse, L.A., Hudson, R.R., and Di Rienzo, A. 2003. Comparative linkage-disequilibrium analysis of the β-globin hotspot in primates. Am. J. Hum. Genet. 73**:** 1330-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K.H., Sharp, P.M., and Li, W.H. 1989. Mutation rates differ among regions of the mammalian genome. Nature 337**:** 283-285. [DOI] [PubMed] [Google Scholar]
- Woodfine, K., Fiegler, H., Beare, D.M., Collins, J.E., McCann, O.T., Young, B.D., Debernardi, S., Mott, R., Dunham, I., and Carter, N.P. 2004. Replication timing of the human genome. Hum. Mol. Genet. 13**:** 191-202. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://genome.ucsc.edu/; UCSC human genome browser.
- http://www.cardiogene.org/bpr/background.htm; Baylor Polymorphism Resource.
- http://www.hapmap.org/; International HapMap Project.
- http://www.r-project.org/; The R project for statistical computing.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Research Data]
[ [The Chimpanzee Genome]]