Liposome reconstitution of a minimal protein-mediated membrane fusion machine (original) (raw)
Abstract
Biological membrane fusion is dependent on protein catalysts to mediate localized restructuring of lipid bilayers. A central theme in current models of protein-mediated membrane fusion involves the sequential refolding of complex homomeric or heteromeric protein fusion machines. The structural features of a new family of fusion-associated small transmembrane (FAST) proteins appear incompatible with existing models of membrane fusion protein function. While the FAST proteins function to induce efficient cell–cell fusion when expressed in transfected cells, it was unclear whether they function on their own to mediate membrane fusion or are dependent on cellular protein cofactors. Using proteoliposomes containing the purified p14 FAST protein of reptilian reovirus, we now show via liposome–cell and liposome–liposome fusion assays that p14 is both necessary and sufficient for membrane fusion. Stoichiometric and kinetic analyses suggest that the relative efficiency of p14-mediated membrane fusion rivals that of the more complex cellular and viral fusion proteins, making the FAST proteins the simplest known membrane fusion machines.
Keywords: FAST proteins, fusion proteins, liposomes, membrane fusion, reovirus
Introduction
Biological membrane fusion requires protein catalysts to overcome the thermodynamic barriers that prevent spontaneous merger of membranes. Studies of viral and cellular fusion proteins have provided detailed structural and functional insights into the protein complexes that mediate membrane merger (Skehel and Wiley, 2000; Ungar and Hughson, 2003). In the case of enveloped viruses, the fusion catalyst is provided by multimeric glycoproteins that exist as metastable structures embedded in the virus envelope (Earp et al, 2005). Following structural rearrangements, a previously sequestered hydrophobic fusion peptide motif is extended toward, and inserts into, the target cell membrane, anchoring the viral fusion protein in both membranes (Tamm et al, 2002). In contrast, intracellular fusion of transport vesicles is dependent on the pairing of a preformed vesicle (v)-SNARE (soluble _N_-ethylmaleimide-sensitive fusion protein attachment protein receptor) complex with a cognate target membrane (t)-SNARE to form a _trans_-acting heteromeric complex (Ungar and Hughson, 2003). In spite of extensive structural diversity in these fusion protein complexes, there are striking similarities in the structural rearrangements that accompany the fusion reaction (Skehel and Wiley, 1998; Weber et al, 1998). However, a clear understanding of the energetics and lipid rearrangements involved in protein-mediated membrane fusion remains incomplete.
A new family of viral proteins involved in cell–cell membrane fusion has recently been described, whose features are incongruent with existing models of protein-mediated membrane fusion. The fusion-associated small transmembrane (FAST) proteins are encoded by the fusogenic reoviruses, an unusual group of nonenveloped viruses that induce syncytium formation (Duncan et al, 2004). The FAST protein family is comprised of the p10 proteins of avian reovirus and Nelson Bay reovirus, the p14 protein of reptilian reovirus, and the p15 protein of baboon reovirus (Shmulevitz and Duncan, 2000; Dawe and Duncan, 2002; Corcoran and Duncan, 2004). Unlike enveloped virus fusion proteins, the FAST proteins are nonstructural viral proteins and are not involved in virus entry. The sole defined function of the FAST proteins is the induction of cell–cell fusion following their expression in reovirus-infected cells and trafficking through the ER–Golgi pathway to the plasma membrane (Shmulevitz et al, 2004a).
Structural and functional properties of the FAST proteins distinguish them from both enveloped virus fusion proteins and SNARE proteins, although they share certain features with each of these distinct groups of membrane fusion proteins. The FAST proteins assume a bitopic Nexoplasmic/Ccytoplasmic (Nexo/Ccyt) membrane topology with small, approximately equal-sized ecto- and endodomains (Shmulevitz and Duncan, 2000; Corcoran and Duncan, 2004; Dawe et al, 2005). At 95–140 residues in size, the FAST proteins are more similar in size to SNARE proteins than enveloped virus fusion proteins. They also function, in effect, as ‘cellular' fusion proteins mediating cell–cell rather than virus–cell membrane fusion. However, the FAST proteins lack the heptad repeat sequences responsible for formation of the helical bundles characteristic of the SNARE proteins and of the class I enveloped virus fusion proteins (Skehel and Wiley, 1998). Similarly, the p10 and p14 FAST proteins contain short regions (18–20 residues) of moderate hydrophobicity in their ectodomains that may function in an analogous manner as the enveloped virus fusion peptide or fusion-loop motifs, serving to destabilize lipid bilayers (Tamm et al, 2003; Gibbons et al, 2004; Modis et al, 2004). However, the fusion peptide motifs of the p10 and p14 FAST proteins are considerably less hydrophobic than typical fusion peptides, they comprise approximately half of the ectodomains of these proteins, and the p14 fusion peptide motif has an obligate requirement for myristoylation (Corcoran et al, 2004; Shmulevitz et al, 2004b). The unusual structural features of the FAST proteins are difficult to reconcile with existing models of protein-mediated membrane fusion, all of which invoke significant energy input derived from extensive rearrangement of homo- or heteromeric complexes (Kozlov and Chernomordik, 1998; Bentz, 2000; Bentz and Mittal, 2003).
Expression of individual FAST proteins in transfected cells induces efficient syncytium formation in different cell types. In addition, numerous point mutations eliminate cell–cell fusion activity without altering FAST protein topology or trafficking, clearly indicating that the FAST proteins are intimately involved in the membrane fusion reaction and are the only reovirus proteins required for syncytium formation (Shmulevitz and Duncan, 2000; Shmulevitz et al, 2003; Corcoran et al, 2004). The remarkable physical attributes of the FAST protein family, however, questioned whether the FAST proteins are membrane fusion proteins per se and can function by themselves to induce membrane fusion, or whether they function through unidentified cellular fusion factors that serve as the actual membrane fusion complex. Using proteoliposomes containing the purified p14 FAST protein, we now show that p14 is both necessary and sufficient for membrane fusion, and that the activity of this minimal protein-mediated membrane fusion machine rivals that of the more complex viral and cellular fusion complexes.
Results
p14 does not form homomeric trans-acting complexes
The p14 FAST protein of reptilian reovirus (Figure 1) has an ∼36-residue ectodomain containing an N-terminal myristate moiety and a 20-residue moderately hydrophobic patch that includes a loop structure (residues 5–13). Both the myristate moiety and the hydrophobic patch are essential for p14 membrane fusion activity (Corcoran et al, 2004). The 68-residue endodomain contains a membrane-proximal polybasic region (nine residues of 18 in this region are basic) of no known function and a nonessential C-proximal polyproline motif. To determine whether p14 must be present in both membranes undergoing fusion, p14-transfected cells were monitored for fusion to nontransfected target cells using a fluorescent heterotypic cell–cell fusion assay. As indicated by the presence of numerous large syncytia, all of which contained both donor (red) and target (blue) cell nuclei (Supplementary Figure 1), p14 does not need to form _trans_-acting homomeric complexes to effect membrane fusion.
Figure 1.
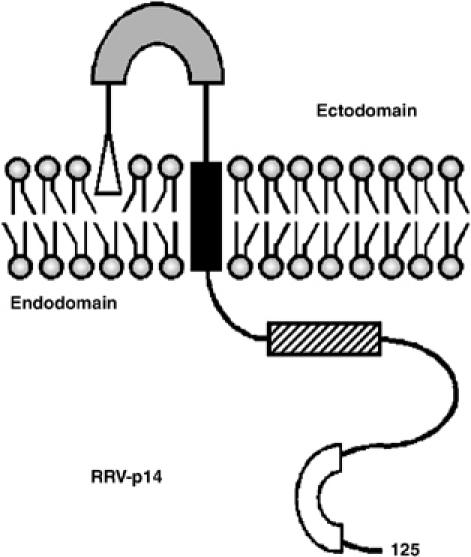
Structural motifs in the p14 FAST protein. The 125-residue p14 protein is depicted in its functional Nexo/Ccyt membrane topology. Structural motifs include an N-terminal myristate (open triangle) that may interact with the external leaflet of the bilayer, a moderately hydrophobic patch (shaded rectangle) that shares certain features with fusion peptide motifs, a transmembrane domain (black rectangle), a membrane-proximal polybasic region (hatched rectangle), and a C-proximal polyproline motif (open rectangle).
Reconstitution of p14-proteoliposomes
Baculovirus-expressed p14 was purified to near homogeneity in the presence of _n_-octyl β-D-glucopyranoside (OG) (Supplementary Figure 2). The purified protein was reconstituted into large (400 nm diameter) unilamellar vesicles (LUVs) by mixing detergent-suspended p14 with LUVs presaturated with detergent, followed by removal of the detergent. Sucrose gradient fractionation and SDS–PAGE analyses of the resulting protein–lipid mixtures revealed inefficient incorporation of p14 into liposomes when the detergent was removed by dialysis at 4°C or room temperature (Figure 2B, lanes 1–6), as evidenced by the presence of p14 in the protein–lipid aggregate (20–30% sucrose) and free protein (30% sucrose) fractions of the gradient. However, stepwise dilutions at 4°C, but not at room temperature, to gradually lower the OG concentration in 0.1% increments resulted in efficient incorporation of p14 into liposomes (Figure 2B, lanes 7–9 versus 10–12). Apparently, the rate of detergent depletion and temperature influences on membrane fluidity, protein–detergent, and/or lipid–detergent interactions influence the efficiency of p14 incorporation into proteoliposomes.
Figure 2.
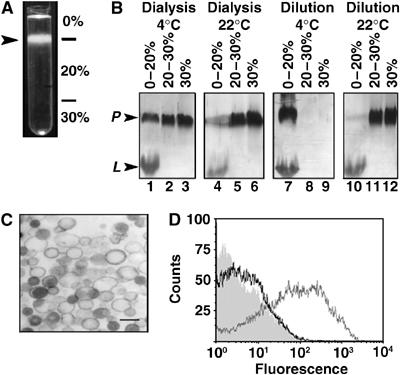
Reconstitution of p14 into liposomes. (A) Suspensions of p14-liposomes were fractionated by centrifugation through a discontinuous sucrose gradient. The liposomes migrated primarily to the 0–20% sucrose interface (arrowhead). (B) p14-liposomes were prepared using the dilution or dialysis approaches at room temperature or 4°C. Fractions obtained from the indicated sucrose gradient interfaces were analyzed by SDS–PAGE and silver staining to detect p14 (P) and lipid (L). (C) Analysis of p14-proteoliposomes by electron microscopy indicated the presence of LUVs. Scale bar=0.5 μm. (D) Flow cytometric analysis of p14-liposomes immunostained using normal rabbit serum (shaded histogram), antibody against the C-terminal enterokinase tag (black line tracing), or anti-p14 ectodomain antibody (gray line tracing) and fluorescent secondary antibody.
Electron microscopy confirmed that the p14-liposomes consisted of LUVs (Figure 2C). Immunofluorescent staining of the p14-liposomes, using antisera specific for the N-terminal ectodomain or for the C-terminal enterokinase tag, revealed that a significant proportion of p14 resides in the correct Nout/Cin topology (Figure 2D). The low level of staining observed with the anti-enterokinase antibody was consistently above background levels, suggesting that at least a proportion of p14 may reside in the inverse Nin/Cout topology, as previously reported for other membrane proteins inserted into detergent-saturated liposomes (Rigaud and Levy, 2003). Calculations based on quantitative analysis of phospholipid and protein concentrations in the isolated proteoliposomes estimated an average protein:lipid molar ratio of ∼1:300 (i.e. 3–3.5 p14 molecules per thousand lipid molecules).
Reconstituted p14-liposomes mediate liposome–cell lipid mixing at the plasma membrane
Lipid mixing between p14-liposomes and cell membranes was assessed using a fluorescence resonance energy transfer assay (Struck et al, 1981). Flow cytometry, rather than fluorimetry, was used to quantify increased cell-associated NBD fluorescence, indicative of transfer of the fluorescent lipids from liposomes to cell membranes. Flow cytometry provided a clearer indication of the range of fusion events (e.g. cells that have fused to multiple liposomes fluoresce more intensely and can be distinguished from cells that have fused to fewer liposomes), and allowed a clear distinction between fluorescent cells and background fluorescence due to residual liposomes adhered to cells.
The fluorescence intensity of cells incubated with p14-liposomes at 37°C was compared to control samples representing p14-liposomes incubated at 4°C (to prevent fusion) or liposomes lacking p14 incubated at 37°C. Time-course analysis revealed a progressive increase in cellular fluorescence when p14-liposomes were incubated with target cells at 37°C (Figure 3). Within 5–10 min after the shift to 37°C, fluorescence intensity increased, as indicated by the rightward shift of the histogram, and continued to increase to maximal levels by 30–40 min. As indicated by the distribution of the histogram (Figure 3A), cell fluorescence intensity varied over two orders of magnitude, suggesting a wide variation in the number of fusion events per target cell. A graphical representation of the time points (Figure 3B), plotting the percent of cells in the target population with fluorescence intensities above background (_t_=0 intensity), indicated that lipid mixing reached maximal levels of ∼35–40% of the cells fluorescing above background. Using quantitative video microscopy, a similar situation was observed with fusion of HA-expressing cells to target red blood cells, where 25–30% of cells undergo full lipid mixing, with 70–80% of cells showing any level of lipid mixing (Mittal et al, 2003). Increased cell fluorescence over time was not observed when LUVs lacking p14 (i.e. standard liposomes) were incubated with cells at 37°C or when cells treated with p14-liposomes were incubated at 4°C (Figure 3B), suggesting that purified p14 mediates lipid mixing and, by inference, the hemifusion phase of membrane fusion.
Figure 3.
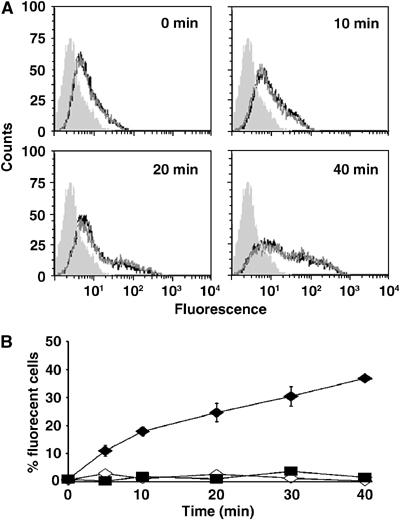
p14 induces liposome–cell lipid mixing. (A) Time-course analysis by flow cytometry of lipid mixing at 37°C between fluorescent p14-proteoliposomes and target QM5 cells. The line tracings indicate that fluorescence intensity increases over the indicated durations, in triplicate. The shaded histogram represents cell autofluorescence. (B) Overton subtraction analysis of the flow cytometry results obtained with p14-liposomes incubated with target cells at 37°C (filled diamonds) or 4°C (open diamonds), and standard liposomes lacking p14 incubated with target cells at 37°C (filled squares). Results are presented as the percent of cells fluorescing above background autofluorescence over time, and are the mean±standard error of three separate experiments in duplicate.
The increase in fluorescence over time observed with p14-liposomes could have resulted from increased p14-liposome adherence to target cells, followed by ‘spontaneous' lipid transfer or endocytic disruption of the adhered liposomes. Analysis by atomic force microscopy revealed that p14-liposomes do exhibit increased adhesive properties compared to standard liposomes (Supplementary Figure 3). Cell-binding analysis also indicated that p14-liposomes adhere more strongly to target cells (Figure 4A). To exclude that this increased adherence was responsible for the observed difference in lipid mixing between p14-liposomes and standard liposomes, the dose of input liposomes was adjusted to yield equivalent numbers of liposomes adhered to cells. Under such conditions, standard liposomes exhibited no increase in fluorescence while p14-liposomes underwent a time-dependent increase in lipid mixing (Figure 4B), suggesting that the fluorescence increase observed in the lipid-mixing assay was specifically due to p14-mediated lipid mixing. Additional support for this conclusion was obtained by demonstrating that lysophosphatidylcholine (LPC), a monoacylated fatty acid known to inhibit membrane fusion (Chernomordik and Kozlov, 2003), effectively inhibited lipid mixing between p14-liposomes and target cells in a dose-dependent manner (Supplementary Figure 4A). Furthermore, endocytosis inhibitors reduced p14-induced lipid mixing by only 20% (Supplementary Figure 4B), suggesting that endocytic entry pathways make only a minor contribution to the observed lipid-mixing. We conclude that p14 induces liposome–cell lipid mixing, indicative of the hemifusion phase of membrane fusion, and that membrane fusion occurs primarily at the plasma membrane, independent of cellular endocytic pathways.
Figure 4.
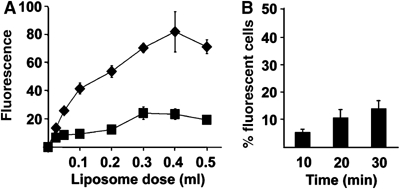
Adherence properties of p14-liposomes. (A) Fluorescently labeled p14-liposomes (diamonds) or standard liposomes (squares) at the indicated doses were bound to target cell monolayers at 4°C, and the relative binding efficiency (in arbitrary fluorescence units) was determined by fluorimetry. Results are the mean±s.d. from a representative experiment conducted in triplicate. (B) Target QM5 cell monolayers were incubated with 50 μl of p14-liposomes or 300 μl of standard liposomes (liposome doses that gave equivalent moles of liposomes bound to cells as shown in panel A) and the time course of liposome–cell lipid mixing was followed by flow cytometry as described in Figure 3. Results are presented as the percent of cells treated with p14-liposomes that fluoresced above levels observed in cells treated with standard liposomes, as determined by Overton subtraction. Results are the mean±s.d. from a representative experiment conducted in triplicate.
Reconstituted p14-liposomes mediate content mixing
The ability of p14 to mediate pore formation was assessed using the aqueous fluor calcein to monitor delivery of liposome contents to the cytosol of target cells. As with the lipid-mixing assay, cells treated with calcein-containing p14-liposomes at 37°C showed increased fluorescence over time, reaching maximal levels by 20 min that corresponded to ∼25% of the target cell population (Figure 5A). There was no increase in cellular fluorescence when cells were incubated at 37°C with standard liposomes or with p14-liposomes incubated at 4°C. Addition of similar levels of soluble calcein to cell monolayers resulted in no increased cell fluorescence (data not shown), implying that the cell fluorescence observed with calcein-containing p14-liposomes was not due to leakage of calcein out of the liposomes and into cells. Unlike the rapid onset of lipid mixing in <5 min, the intracellular delivery of calcein was not detected until after 10 min (Figure 5A). Whether this apparent lag simply reflects a decreased sensitivity of the content-mixing assay (i.e. the extent of calcein that must be delivered to cells before increased cell fluorescence is detectable) or something more interesting, such as the uncoupling of the lipid mixing and pore formation phases of the p14 fusion reaction, is unclear.
Figure 5.
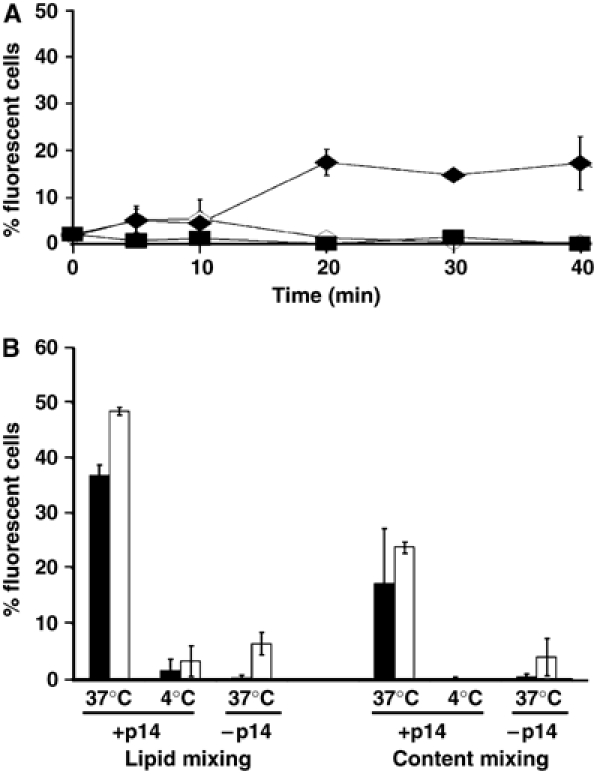
p14 induces liposome–cell content mixing. (A) Target QM5 cells were incubated with calcein-loaded p14-liposomes at 37°C (filled diamonds) or 4°C (open diamonds), or with standard liposomes lacking p14 (open squares). At the indicated durations, cell fluorescence was quantified by flow cytometry and the percent cells fluorescing above background cell autofluorescence was determined by Overton subtraction. Results are the mean±standard error of three separate experiments. (B) Lipid mixing and content mixing assays were conducted as described in Figure 3 and above, using Vero cells (white bars) or QM5 cells (black bars) incubated for 20 min with p14-liposomes at 37 or 4°C, or with standard liposomes at 37°C. Cell fluorescence was quantified by flow cytometry and the percent cells fluorescing above background cell autofluorescence was determined by Overton subtraction. Results are the mean±s.d. from a representative experiment conducted in triplicate.
Repeating these assays with Vero epithelial cells indicated levels of lipid mixing and content mixing comparable to the results obtained with QM5 fibroblasts (Figure 5B). Using fluorescence microscopy, cellular fluorescence was observed for both the aqueous and lipidic fluors when Vero cells were incubated with p14-liposomes at 37°C, but not when cells were incubated at 4°C or with liposomes that lacked p14 at 37°C (Figure 6). Examination of the _z_-angles revealed an asymmetric distribution of the two fluors, with the red lipidic fluor associated preferentially with the extremity of cells and the green aqueous fluor with the cell interior, consistent with lipid mixing at the plasma membrane and cytosolic delivery of the aqueous liposome contents. Therefore, p14 mediates liposome–cell membrane fusion and intracellular content delivery to different cell types.
Figure 6.
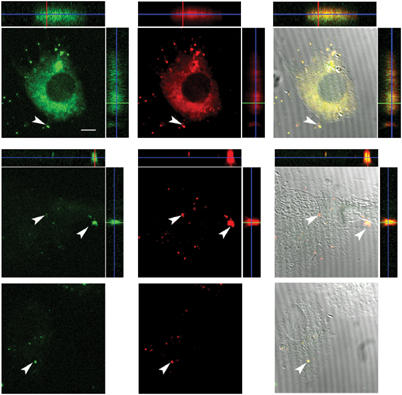
Fluorescence microscopy analysis of lipid mixing and content mixing. Vero cells were treated with p14-liposomes at 37°C (top row) or 4°C (middle row), or with liposomes lacking p14 (bottom row). Liposomes were labeled with calcein (green, left columns) to detect content mixing and with a lipidic fluor (red, middle columns) to monitor lipid mixing. The right-hand columns are an overlay of the DIC image and the lipid and content mixing of a single cell. The flanking panels are _z_-angles showing the asymmetric distribution of the lipidic fluor near the apical surface of the cell (top and right) and the aqueous calcein fluor in the interior of the cell (bottom and left). Arrows indicate residual liposomes adhered to cells. Size bar=10 μm.
p14 is both necessary and sufficient for membrane fusion
Liposome–liposome fusion assays were conducted to exclude the possibility that p14 might de dependent on formation of a _trans_-acting complex with a protein cofactor present in the target cell membrane. Initial experiments using our neutral liposomes did not detect p14-induced lipid mixing (data not shown). While p14-liposomes adhered to target cells (Figure 4), we suspected that the same might not be true for p14-liposome adherence to artificial phospholipid bilayers, and that the lack of liposome–liposome fusion was due to the absence of sufficiently close liposome–liposome interactions. Therefore, p14 was reconstituted into the anionic liposome formulation (POPC (1-palmitoyl-2-oleoyl-phosphatidylcholine):DOPS (dioleoyl-phosphatidylserine) at 85:15 mol%) widely used to assess SNARE-mediated liposome fusion (Weber et al, 1998; Parlati et al, 1999; McNew et al, 2000; Chen et al, 2004), and liposome–liposome interactions were promoted using divalent cations. In the absence of divalent cations, p14-induced lipid mixing barely exceeded the low level of spontaneous lipid mixing (Figure 7A and B, curve c). The addition of divalent cations resulted in a dose-dependent increase in lipid mixing for p14-liposomes, but had little to no effect on lipid mixing in the absence of p14 (Figure 7A and B, curves a and b). This situation applied to both Ca2+ and Mg2+, with Ca2+ exerting a more pronounced effect (Figure 7C).
Figure 7.
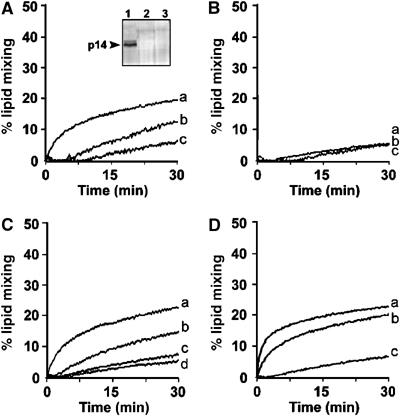
p14 mediates liposome–liposome fusion. (A) Unlabeled anionic (POPC:DOPS at 85:15 mol%) p14-liposomes were prepared using the standard detergent depletion protocol, and were mixed with fluorescently labeled target anionic liposomes in the presence of 10, 1, or 0 mM Ca2+ (curves a, b, and c, respectively). Increased NBD fluorescence due to lipid mixing was monitored by fluorimetry over time at 37°C. Results are reported as the percent maximal fluorescence, as determined by detergent solubilization of the liposomes. Inset: The 0–20% (lane 1), 20–30% (lane 2), and 30% (lane 3) fractions from a sucrose gradient were analyzed by SDS–PAGE and silver staining (see Figure 2B legend) to reveal efficient incorporation of p14 into the POPC–DOPS liposomes by the detergent dilution approach. (B) The same experimental conditions as described in panel A were performed using POPC-DOPS liposomes lacking p14. (C) Unlabeled p14-liposomes (curves a and b) or unlabeled liposomes lacking p14 (curves c and d) were mixed with fluorescently labeled target liposomes in the presence of 10 mM Ca2+ (curves a and c) or 10 mM Mg2+ (curves b and d) and lipid mixing was quantified as described in panel A. (D) Liposome–liposome fusion assays were conducted as described in panel A in the presence of 10 mM Ca2+, under conditions where p14 was present in both donor and target liposomes (curve a), donor liposomes only (curve b), or absent from both liposomes (curve c).
Under optimal conditions for p14-induced lipid mixing, there was a sharp increase in NBD fluorescence in the first few minutes, which leveled off by 5–7 min at ∼15–20% of the theoretical maximum lipid mixing followed by a slow progressive increase in fluorescence from 10 to 30 min that paralleled the low rate of spontaneous lipid mixing observed between liposomes lacking p14 (Figure 7A). As with liposome–cell fusion, additional studies indicated that p14 need only be present in the donor membrane to effect lipid mixing between liposomes (Figure 7D). A modest increase in fusion activity was noted when p14 was present in both membranes; however, this effect was most likely the result of the 25% increase in the total amount of p14 in the system (i.e. a 4:1 ratio of donor:target membranes) rather than the need for p14 in both membranes, as suggested by the ability of p14-transfected cells to fuse to nontransfected cells (Supplementary Figure 1). The ability of p14 to induce liposome–liposome fusion suggests that the p14 FAST protein is both necessary and sufficient to induce the lipid rearrangements required for merger of two lipid bilayers, making p14 the least complex protein-based membrane fusion machine described to date.
Intracellular delivery of a proapoptotic peptide by fusogenic p14-liposomes
To further examine the range of conditions under which p14 can function, p14 was formulated into cationic multilamellar vesicles (DOPC (1,2-dioleoyl-sn-glycerol-3 phosphatidylcholine):DOPE (1,2-dioleoyl-sn-glycerol-3 phosphatidylethanolamine):cholesterol:DC-cholesterol, molar ratios of 60:30:8:2, respectively), and assessed for its ability to mediate intracellular delivery of a proapoptotic peptide, lactoferricin (Lfcin). The Lfcin peptide is related to penetratins and defensins, and penetrates cell membranes to trigger the mitochondrial-induced apoptotic pathway (Vogel et al, 2002; Mader et al, 2005). Soluble Lfcin induced an apoptotic response in Jurkat cells resulting in ∼80% cell death (Figure 8). Treatment of Jurkat cells with liposome-encapsulated Lfcin, using liposomes lacking p14, resulted in low levels of cell death (∼5–10%) that were comparable to spontaneous apoptosis in these cells, indicating that the cationic Lfcin peptide remained entrapped within the cationic liposomes, and that such nonfusogenic liposomes are incapable of efficient intracellular cargo delivery. In contrast, identical liposome formulations containing p14 resulted in ∼80% cell death. The supernatant obtained following incubation of Lfcin-containing p14-liposomes or normal liposomes in cell culture medium for 24 h induced limited cell killing (Figure 8), indicating that p14 did not induce detectable leakage of the Lfcin from the liposomes. Although we cannot exclude the possibility that p14-liposomes may become leaky when interacting with cells, the results from the calcein studies (Figure 5) indicate that p14 mediates intracellular content delivery independent of liposome leakage. Therefore, based on several lines of evidence, we conclude that p14 can mediate liposome fusion to a diversity of cell types, resulting in intracellular delivery of different aqueous cargoes in a manner that is relatively tolerant to wide variations in the donor membrane lipid composition.
Figure 8.
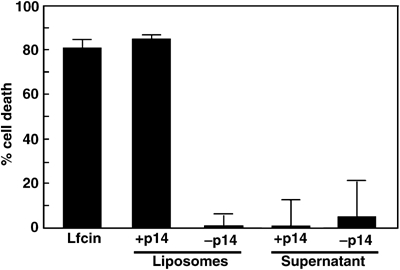
p14-liposomes mediate intracellular delivery of a proapoptotic peptide. Jurkat cells were treated with 20 μg of the proapoptotic lactoferricin peptide, either in solution (Lfcin) or entrapped within p14-liposomes (+p14) or standard liposomes (−p14). Cells were also treated with the supernatant obtained from ±p14-liposomes containing lactoferricin that were incubated in tissue culture medium for 24 h at 37°C to detect leakage of the peptide cargo from liposomes. The percent apoptosis was quantified by analysis of DNA degradation. Results are the mean±s.d. from a representative experiment conducted in quadruplicate.
Discussion
The exceptional structural features of the FAST protein family raised the question of whether these proteins function through indirect means to effect cell–cell membrane fusion, possibly by activating or modulating cellular fusion factors. We now show that purified p14 reconstituted into an artificial lipid bilayer mediates membrane fusion independent of any _cis_- or _trans_-acting protein cofactors. These results were confirmed using liposome–liposome and liposome–cell fusion assays, with both adherent and nonadherent cell lines, using different liposome cargoes (a fluorescent aqueous probe and a proapoptotic peptide), and in the presence of different lipid formulations (neutral, cationic, or anionic). The p14 FAST protein, therefore, is a promiscuous fusogen that functions to mediate complete membrane fusion. We also conclude that p14 is both necessary and sufficient to mediate membrane fusion, making the FAST proteins the least complex protein-mediated membrane fusion machines currently known. The reconstitution of fusogenic p14-liposomes has important implications on proposed models of FAST protein-mediated membrane fusion and on the available approaches to investigate the mechanism of FAST protein function.
The protein:lipid molar ratio of p14-liposomes (∼1:300) corresponds to 6–7 × 103 p14 molecules per 400 nm LUV and surface densities of 1.2–1.4 × 104 p14 molecules/μm2 (see Supplementary data). These p14 densities are similar to those reported for influenza HA monomers on the surface of transfected cells or in virions, and for v-SNAREs in reconstituted liposomes or synaptic vesicles, which range from 0.3 to 9 × 104 protein molecules/μm2 (Jahn and Sudhof, 1994; Danieli et al, 1996; Markovic et al, 2001; Chen et al, 2004), and are approximately five-fold less than the densities of synaptic SNAREs in reconstituted liposomes (6 × 104) (Weber et al, 1998). Consequently, approximately the same membrane density of p14 proteins is required to induce membrane fusion as reported for other, more complex membrane fusion complexes. The possibility of p14 clustering in the liposome membrane precludes speculation on the actual number of p14 molecules required at a given fusion site.
Kinetic analysis indicated that p14-liposomes initiated liposome–cell lipid mixing within 5 min following the shift to the fusion-permissive temperature of 37°C, reaching maximal levels by 20–30 min with a half-time of ∼10 min. While lipid mixing between influenza HA- or HIV env-expressing cells and target cell membranes exhibits faster kinetics, going to completion in 3–5 min (Danieli et al, 1996; Markosyan et al, 2003; Mittal et al, 2003), other enveloped virus fusion proteins, either expressed in transfected cells or present in virus particles or reconstituted lipid vesicles, follow similar kinetics as p14-liposomes (Eidelman et al, 1984; Blumenthal et al, 1987; Earp et al, 2003; Kolokoltsov and Davey, 2004). In the case of p14-mediated liposome–liposome fusion, the kinetics and extent of lipid mixing closely mirror results reported for SNARE-mediated membrane fusion. For example, neuronal SNAREs mediate liposome liposome lipid mixing with half-times of 4–10 min (Parlati et al, 1999; Melia et al, 2002), while yeast SNAREs fuse liposomes with a half-time of 20–30 min, reaching ∼20% of the theoretical maximum by ∼60 min (Chen et al, 2004). Although these are qualitative comparisons between different systems and different assays, based on membrane protein density and the kinetics of lipid mixing (both in liposome–cell and liposome–liposome fusion assays), the results suggest that the relative efficiency of the p14 membrane fusion machine may rival that of the more complex homomeric or heteromeric enveloped virus and cellular fusion machinery.
The relative efficiency of p14 as a fusogen is a remarkable observation in view of the minimal size of the FAST protein family members. The simplicity of this fusion machine contrasts markedly with the complex homomeric or heteromeric membrane fusion machinery of the enveloped viruses or SNARE proteins. Unlike SNARE proteins that form _trans_-acting multimeric complexes to draw the donor and target membranes into close proximity (Weber et al, 1998), results from liposome–liposome and/or cell–cell fusion assays indicate that this is not the case for the p14 (Supplementary Figure 1 and Figure 7D) and p10 FAST proteins (Shmulevitz et al, 2004b). While enveloped virus fusion proteins also function from only one of the two membranes, they do so using complex ectodomains that contrast markedly with the ∼20–44-residue ectodomains of the FAST proteins (Earp et al, 2005).
For both the SNARE proteins and enveloped virus fusion proteins, lipid-mixing and content-mixing are tightly coupled and dependent on the coordinated, sequential refolding of the multimeric fusion complexes (Melikyan et al, 2000; Markosyan et al, 2003; Cohen and Melikyan, 2004). These structural rearrangements serve to draw the apposing membranes together and/or provide the energy required for pore formation and expansion. Based on the small size of their ectodomains, it seems unlikely that the FAST proteins drive membrane fusion by undergoing such extensive structural remodeling. The physical stature of the FAST protein ectodomains is also incompatible with models based on triggered conformational release of a fusion peptide previously sequestered within a complex folded ectodomain. In fact, analysis of p10 degradation suggests that the fusion peptide may be solvent accessible (Shmulevitz et al, 2004a), and there is no evidence to indicate that the FAST proteins undergo a transition from a fusion-incompetent precursor to a fusion-active form. It therefore seems that the FAST proteins must utilize some means other than triggered conformational changes in complex, multimeric structures to overcome the thermodynamic barriers that prevent spontaneous membrane fusion.
Based on available evidence, we currently favor a model whereby a combination of membrane-destabilizing modules, working from either side of the donor membrane, may function in concert to alter the water layer and lipid packing, leading to more disordered fusion intermediates and lowered energy requirements. Such a ‘lipid-mixer' model was recently proposed for influenza HA (Tamm, 2003; Tamm et al, 2003), and is more in-line with the structural features of the FAST proteins. The p14 protein has at its disposal several membrane-interaction motifs that may serve to effect the various steps in the membrane fusion reaction, including a fusion peptide motif and N-terminal myristate moiety in its small ectodomain, the transmembrane domain, and a membrane-proximal polybasic region (Corcoran and Duncan, 2004). Discerning the role of these p14 motifs in what may be a novel mechanism of protein-mediated membrane fusion can now be pursued using the fusogenic p14-liposome system.
Materials and methods
Plasmids and cells
The full-length cDNA clone of the RRV p14 gene in pcDNA3 was previously described (Corcoran and Duncan, 2004). A recombinant baculovirus was created for p14 expression (see Supplementary data for details). QM5 fibroblasts and Vero cells were grown and maintained as previously described (Corcoran and Duncan, 2004). Human T-cell leukemia (Jurkat) cells were maintained in RPMI 1640 medium (Sigma) supplemented with 5% fetal bovine serum (FBS) and penicillin/streptomycin. SF21 Spodoptera frugiperda cells were grown and maintained in SF900II insect cell culture medium (Invitrogen) supplemented with 3% FBS using suspension cultures.
Purification of p14
Purified p14 was obtained from recombinant baculovirus-infected insect cells following detergent lysis, binding to Talon metal affinity resin, and HiTrap SP HP ion exchange chromatography in the presence of 1.6% OG (see Supplementary data for details). Analysis by mass spectroscopy confirmed that the purified protein (1.5–2 mg/ml) corresponded to p14. The purity of the protein was >95%, as estimated by SDS–PAGE and densitometry analysis of silver-stained gels, and the protein was myristoylated (Supplementary Figure 2).
Preparation of proteoliposomes
All lipids were from Avanti Polar Lipids. Dried lipid films (DOPC:DOPE:cholesterol:sphingomyelin at molar ratios of 40:20:20:20) were resuspended in HEPES buffered salt solution (HBS; 10 mM HEPES, pH 7.4, 150 mM NaCl) and extruded through a 400 nm polycarbonate filter using a small-volume extrusion apparatus (Avestin, Ottawa, Canada). The rest of the procedure was conducted at 4°C, unless otherwise noted. A 0.5 ml detergent-saturated liposome suspension (13.2 mM lipid and 1% (w/v) OG) was mixed with an equal volume of purified p14 (0.6 mg/ml in 1% (w/v) OG) and rocked for 60 min. The concentration of OG was decreased in 0.1% increments to a final concentration of 0.4% (w/v) by sequential dilution at 4°C with HBS (30 min intervals), followed by extensive dialysis against HBS containing Bio-Beads SM-2 Adsorbent (BioRad) to remove residual detergent. For comparison of protein reconstitution methods, proteoliposomes were made at room temperature (22°C) using the above protocol, the dilution steps were omitted before dialysis, or both the temperature was changed and the dilution omitted. A 500 μl aliquot of the resulting proteoliposome mixture was mixed with an equal volume of 60% sucrose (w/w) in HBS, overlaid with 2 ml of 20% sucrose and 1 ml of HBS, and centrifuged at 165 000 g for 1 h at 4°C. Fractions (500 μl) were collected from the 0–20 and 20–30% sucrose interfaces, and from the 30% sucrose fraction, and analyzed by SDS–PAGE and silver staining.
Immunofluorescent staining of p14-liposomes
The topology of p14 in the membrane of proteoliposomes was determined by immunofluorescent staining using 1/400 dilutions of either polyclonal rabbit antiserum raised against a synthetic peptide representing residues 5–31 of the p14 N-terminal ectodomain (New England Peptide) or a polyclonal antibody that recognized the C-terminal enterokinase tag (Bethyl Laboratories), and Alexa 488-conjugated secondary goat anti-rabbit antibody (1/200 dilution). The liposomes were washed extensively with HBS and fluorescence was quantified by flow cytometry (FACScalibur, Beckton-Dickinson). As controls, liposomes lacking p14 were similarly stained, or p14-proteoliposomes were stained with only the secondary antibody or with normal rabbit serum. Western blotting indicated that both antisera recognized the p14 construct (Supplementary Figure 5) with slightly different affinities (α-ectodomain>α-enterokinase).
Binding and fusion assays
Cell monolayers in 12-well plates were washed with Hanks' buffered salt solution (HBSS) and chilled on ice. Suspensions (500 μl) of p14-proteoliposomes or standard liposomes were added to the target cell monolayers (0.33–1.32 mM lipid) and incubated on ice for 1 h to allow liposome adherence to cells. Unbound liposomes were removed by washing, and the monolayers were analyzed for liposome binding, lipid mixing, and content mixing using the following protocols.
Binding assay. Fluorescent liposomes were prepared using DOPC:DOPE:cholesterol:sphingomyelin:NBD-DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-_N_-(7-nitro-2-1,3-benzoxadiazol-4-yl) at molar ratios of 40:19:20:20:1, respectively. Cells were washed with PBS, detached from the substratum using 10 mM EDTA, and bound liposomes quantified by flow cytometry using excitation and emission wavelengths of 460 and 534 nm, respectively.
Lipid-mixing assay. Lipid mixing between liposome and target cell membranes was quantified using a fluorescence resonance energy transfer assay (Struck et al, 1981). Fluorescent liposomes were prepared using DOPC:DOPE:cholesterol: sphingomyelin:NBD-DOPE:rhodamine-DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-_N_-(lissamine rhodamine B sulfonyl)) at molar ratios of 40:17:20:20:1:2, respectively. After the 1 h binding incubation, cell temperature was either kept at 4°C or rapidly changed to 37°C using prewarmed HBSS, and cells were incubated at the same temperatures for the indicated durations. At the end of each time point, cells were rapidly cooled to 4°C and detached from the substratum by incubation on ice for 15 min with 0.1 mg/ml proteinase K (QiaGen). The extent of lipid mixing was quantified by flow cytometry (see below).
Pore formation assay. Liposomes were prepared in the presence of 20 μM calcein. Analysis of the various liposome preparations indicated no significant difference in the entrapment efficiency of calcein inside p14-liposomes versus standard liposomes (±4%). The calcein-containing liposomes were used to monitor intracellular delivery, as described in the lipid-mixing protocol.
Flow cytometry
Both lipid mixing and content mixing were quantified by flow cytometry (FACScalibur, Beckton-Dickinson) as a function of increasing NBD fluorescence or calcein fluorescence, respectively, measured in the FL1 channel. Flow cytometric measurements of fluorescent cells were conducted using a gate in the forward-side scatter plot that excluded free liposomes that were easily distinguishable from cells. The gating parameters were determined from the forward-side scatter plots of untreated cells and free liposomes. A total of 10 000 events within the cell gate were counted. The percentage of cells with increased fluorescence was calculated using Overton subtractions (Overton, 1988) of the experimental sample against the control sample (e.g. fluorescence at _t_=40 min versus fluorescence at _t_=0).
Fluorescence microscopy of lipid mixing and pore formation
Vero cell monolayers were treated with p14-proteoliposomes or normal liposomes, labeled with rhodamine-DOPE and calcein, as described above. After incubation for 20 min to allow lipid mixing and content mixing, the proteinase K-treated cells were resuspended in 1 ml of FBS, pelleted at 100 g for 5 min at 4°C, and resuspended in 200 μl of Earle's medium containing 10% FBS. Suspended cells were centrifuged onto glass slides using a cytocentrifuge, fixed with 2% (w/v) paraformaldehyde, and mounted using fluorescent mounting media (Dako). Cells were visualized and photographed using a Zeiss LSM510 scanning argon laser confocal microscope and the × 100 objective. Serial sections (0.5 μm) were taken at both the rhodamine and calcein wavelengths.
Liposome–liposome fusion assays
Lipid mixing between liposomes was assessed using the method of Weber et al (1998). POPC and DOPS were resuspended in chloroform/methanol (2:1) at an 85:15 molar ratio, and dried under negative pressure. The lipid film was rehydrated to obtain a 20 mM lipid concentration in HBS, and extruded through 400 nm polycarbonate filters. Fluorescent target liposomes lacking p14 were similarly prepared, using an 82:15:2:1 lipid molar ratio of POPC:DOPS:rhodamine-DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-_N_-(lissamine rhodamine B sulfonyl)):NBD-DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-_N_-(7-nitro-2–1,3-benzoxadiazol-4-yl)), respectively. The purified p14 protein was incorporated into nonfluorescent, detergent-saturated (0.9% OG) liposomes following the standard detergent dilution approach described above. The p14-liposomes were mixed with fluorescent liposomes (4:1 ratio, respectively) and incubated on ice for 5 min. The liposome mixture was then added to a cuvette containing prewarmed HBS and 10 mM MgCl2 or CaCl2 as indicated, and incubated at 37°C with stirring. Increased NBD fluorescence was monitored for 120 min using a Varian spectrofluorimeter with a slit width of 20 nm, using excitation and emission wavelengths of 460 or 535 nm, respectively. Maximum lipid mixing was estimated from liposomes solubilized in the presence of 1% Triton X-100. The lowest fluorescence intensity during the time course was set as minimum lipid mixing.
Lactoferricin peptide delivery
The 25-residue Lfcin-B peptide was entrapped inside cationic p14-liposomes (60:30:8:2 molar ratio of DOPC:DOPE:cholesterol:DC-cholesterol (3β-(_N_-(_N_′,_N_′-dimethylaminoethane)-carbamoyl) cholesterol hydrochloride]) by 10 freeze–thaw cycles using liquid nitrogen and a 37°C water bath. The resulting multilamellar liposomes were washed with HBS and resuspended in RPMI medium to give a final concentration of 33.3 mM lipid. The concentration of entrapped Lfcin, as determined by HPLC analysis of the difference between the starting peptide concentration and the concentration of the residual nonentrapped peptide, was 1.13–1.23 mg/ml and differed by <8% between liposomes and p14-proteoliposomes. Intracellular delivery of Lfcin-B was assessed by DNA degradation analysis of [3H]thymidine-labeled Jurkat cells, as previously described (Matzinger, 1991).
Supplementary Material
Supplementary Methods
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Acknowledgments
We thank Jingyun Shou, Tara Read, David Mader, and Paul Lindgreen for excellent technical assistance. This research was supported by grants to RD from the Canadian Institutes of Health Research (CIHR) and the Natural Sciences and Engineering Research Council of Canada (NSERC), and by grants to DH and to MJ from NSERC. DT was supported by a scholarship from Cancer Care Nova Scotia (CCNS) through the Cancer Research Training Program (CRTP). JS was supported by scholarships from NSERC and CCNS-CRTP. JM was supported by a scholarship from the Nova Scotia Health Research Foundation (NSHRF). RD is the recipient of a CIHR-RPP Investigators Award.
References
- Bentz J (2000) Membrane fusion mediated by coiled coils: a hypothesis. Biophys J 78: 886–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz J, Mittal A (2003) Architecture of the influenza hemagglutinin membrane fusion site. Biochim Biophys Acta 1614: 24–35 [DOI] [PubMed] [Google Scholar]
- Blumenthal R, Bali-Puri A, Walter A, Covell D, Eidelman O (1987) pH-dependent fusion of vesicular stomatitis virus with Vero cells. Measurement by dequenching of octadecyl rhodamine fluorescence. J Biol Chem 262: 13614–13619 [PubMed] [Google Scholar]
- Chen Y, Xu Y, Zhang F, Shin YK (2004) Constitutive versus regulated SNARE assembly: a structural basis. EMBO J 23: 681–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM (2003) Protein–lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem 72: 175–207 [DOI] [PubMed] [Google Scholar]
- Cohen FS, Melikyan GB (2004) The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol 199: 1–14 [DOI] [PubMed] [Google Scholar]
- Corcoran JA, Duncan R (2004) Reptilian reovirus utilizes a small type III protein with an external myristoylated amino terminus to mediate cell–cell fusion. J Virol 78: 4342–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran JA, Syvitski R, Top D, Epand RM, Epand RF, Jakeman D, Duncan R (2004) Myristoylation, a protruding loop, and structural plasticity are essential features of a nonenveloped virus fusion peptide motif. J Biol Chem 279: 51386–51394 [DOI] [PubMed] [Google Scholar]
- Danieli T, Pelletier SL, Henis YI, White JM (1996) Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol 133: 559–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe S, Corcoran JA, Clancy E, Salsman S, Duncan R (2005) An unusual topological arrangement of structural motifs in the baboon reovirus fusion-associated small transmembrane (FAST) protein. J Virol 79: 6216–6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe S, Duncan R (2002) The S4 genome segment of baboon reovirus is bicistronic and encodes a novel fusion-associated small transmembrane protein. J Virol 76: 2131–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R, Corcoran J, Shou J, Stoltz D (2004) Reptilian reovirus: a new fusogenic orthoreovirus species. Virology 319: 131–140 [DOI] [PubMed] [Google Scholar]
- Earp LJ, Delos SE, Netter RC, Bates P, White JM (2003) The avian retrovirus avian sarcoma/leukosis virus subtype A reaches the lipid mixing stage of fusion at neutral pH. J Virol 77: 3058–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earp LJ, Delos SE, Park HE, White JM (2005) The many mechanisms of viral membrane fusion proteins. Curr Top Microbiol Immunol 285: 25–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidelman O, Schlegel R, Tralka TS, Blumenthal R (1984) pH-dependent fusion induced by vesicular stomatitis virus glycoprotein reconstituted into phospholipid vesicles. J Biol Chem 259: 4622–4628 [PubMed] [Google Scholar]
- Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA (2004) Conformational change and protein–protein interactions of the fusion protein of Semliki Forest virus. Nature 427: 320–325 [DOI] [PubMed] [Google Scholar]
- Jahn R, Sudhof TC (1994) Synaptic vesicles and exocytosis. Annu Rev Neurosci 17: 219–246 [DOI] [PubMed] [Google Scholar]
- Kolokoltsov AA, Davey RA (2004) Rapid and sensitive detection of retrovirus entry by using a novel luciferase-based content-mixing assay. J Virol 78: 5124–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov MM, Chernomordik LV (1998) A mechanism of protein-mediated fusion: coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys J 75: 1384–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader JS, Salsman J, Conrad DM, Hoskin DW (2005) Bovine lactoferricin selectively induces apoptosis in human leukemia and carcinoma cell lines: role of reactive oxygen species, mitochondrial damage, and caspases associated with the intrinsic pathway of apoptosis. Mol Cancer Ther 4: 612–624 [DOI] [PubMed] [Google Scholar]
- Markosyan RM, Cohen FS, Melikyan GB (2003) HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol Biol Cell 14: 926–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic I, Leikina E, Zhukovsky M, Zimmerberg J, Chernomordik LV (2001) Synchronized activation and refolding of influenza hemagglutinin in multimeric fusion machines. J Cell Biol 155: 833–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P (1991) The JAM test. A simple assay for DNA fragmentation and cell death. J Immunol Methods 145: 185–192 [DOI] [PubMed] [Google Scholar]
- McNew JA, Weber T, Parlati F, Johnston RJ, Melia TJ, Sollner TH, Rothman JE (2000) Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J Cell Biol 150: 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melia TJ, Weber T, McNew JA, Fisher LE, Johnston RJ, Parlati F, Mahal LK, Sollner TH, Rothman JE (2002) Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J Cell Biol 158: 929–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS (2000) Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol 151: 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A, Leikina E, Chernomordik LV, Bentz J (2003) Kinetically differentiating influenza hemagglutinin fusion and hemifusion machines. Biophys J 85: 1713–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, Harrison SC (2004) Structure of the dengue virus envelope protein after membrane fusion. Nature 427: 313–319 [DOI] [PubMed] [Google Scholar]
- Overton WR (1988) Modified histogram subtraction technique for analysis of flow cytometry data. Cytometry 9: 619–626 [DOI] [PubMed] [Google Scholar]
- Parlati F, Weber T, McNew JA, Westermann B, Sollner TH, Rothman JE (1999) Rapid and efficient fusion of phospholipid vesicles by the alpha-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc Natl Acad Sci USA 96: 12565–12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud JL, Levy D (2003) Reconstitution of membrane proteins into liposomes. Methods Enzymol 372: 65–86 [DOI] [PubMed] [Google Scholar]
- Shmulevitz M, Corcoran J, Salsman J, Duncan R (2004a) Cell–cell fusion induced by the avian reovirus membrane fusion protein is regulated by protein degradation. J Virol 78: 5996–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulevitz M, Duncan R (2000) A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. EMBO J 19: 902–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulevitz M, Epand RF, Epand RM, Duncan R (2004b) Structural and functional properties of an unusual internal fusion peptide in a nonenveloped virus membrane fusion protein. J Virol 78: 2808–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulevitz M, Salsman J, Duncan R (2003) Palmitoylation, membrane-proximal basic residues, and transmembrane glycine residues in the reovirus p10 protein are essential for syncytium formation. J Virol 77: 9769–9779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC (1998) Coiled coils in both intracellular vesicle and viral membrane fusion. Cell 95: 871–874 [DOI] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69: 531–569 [DOI] [PubMed] [Google Scholar]
- Struck DK, Hoekstra D, Pagano RE (1981) Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20: 4093–4099 [DOI] [PubMed] [Google Scholar]
- Tamm LK (2003) Hypothesis: spring-loaded boomerang mechanism of influenza hemagglutinin-mediated membrane fusion. Biochim Biophys Acta 1614: 14–23 [DOI] [PubMed] [Google Scholar]
- Tamm LK, Crane J, Kiessling V (2003) Membrane fusion: a structural perspective on the interplay of lipids and proteins. Curr Opin Struct Biol 13: 453–466 [DOI] [PubMed] [Google Scholar]
- Tamm LK, Han X, Li Y, Lai AL (2002) Structure and function of membrane fusion peptides. Biopolymers 66: 249–260 [DOI] [PubMed] [Google Scholar]
- Ungar D, Hughson FM (2003) SNARE protein structure and function. Annu Rev Cell Dev Biol 19: 493–517 [DOI] [PubMed] [Google Scholar]
- Vogel HJ, Schibli DJ, Jing W, Lohmeier-Vogel EM, Epand RF, Epand RM (2002) Towards a structure–function analysis of bovine lactoferricin and related tryptophan- and arginine-containing peptides. Biochem Cell Biol 80: 49–63 [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–772 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6