Notch Inhibition of E47 Supports the Existence of a Novel Signaling Pathway (original) (raw)
Abstract
E47 is a widely expressed transcription factor that activates B-cell-specific immunoglobulin gene transcription and is required for early B-cell development. In an effort to identify processes that regulate E47, and potentially B-cell development, we found that activated Notch1 and Notch2 effectively inhibit E47 activity. Only the intact E47 protein was inhibited by Notch—fusion proteins containing isolated DNA binding and activation domains were unaffected—suggesting that Notch targets an atypical E47 cofactor. Although overexpression of the coactivator p300 partially reversed E47 inhibition, results of several assays indicated that p300/CBP is not a general target of Notch. Notch inhibition of E47 did not correlate with its ability to activate CBF1/RBP-Jκ, the mammalian homolog of Suppressor of Hairless, a protein that associates physically with Notch and defines the only known Notch signaling pathway in drosophila. Importantly, E47 was inhibited independently of CBF1/RPB-Jκ by Deltex, a second Notch-interacting protein. We provide evidence that Notch and Deltex may act on E47 by inhibiting signaling through Ras because (i) full E47 activity was found to be dependent on Ras and (ii) both Notch and Deltex inhibited GAL4-Jun, a hybrid transcription factor whose activity is dependent on signaling from Ras to SAPK/JNK.
E47 is a basic helix-loop-helix (bHLH) protein that is necessary for B-lymphocyte development (7, 89). When overexpressed in non-B cells, E47 is sufficient to activate transcription of the endogenous immunoglobulin heavy-chain locus and the gene encoding terminal deoxynucleotidyltransferase (15, 73). Thus, E47 possesses certain properties expected of a master regulatory protein such as MyoD. However, unlike MyoD, E47 is widely expressed. B-cell-specific activity is known to be controlled, at least in part, by posttranslational modifications that regulate DNA binding by E47 homodimers (75). One proposal suggests that DNA binding is regulated through disulfide-mediated dimerization (8). Another argues that B-cell-specific DNA binding is a consequence of cell-specific phosphorylation (76). Although these models are not mutually exclusive, it is likely that E47 activity is controlled by additional mechanisms as well. Given the importance of E47 in B-cell development, it is important to know how the protein is regulated and if it might respond to cues provided by the hematopoietic microenvironment.
Notch defines a family of transmembrane receptors found in a variety of organisms including drosophila, Caenorhabditis elegans, xenopus, and higher vertebrates (for a review, see reference 2). In general, Notch signaling appears to restrict the development of particular cell fates. In drosophila, this is exemplified by the segregation of neural and epidermal cells in the ventral ectoderm of the embryo or by the induction of the R7 cell in the compound eye (26). Notch signaling has also been shown to be functionally important in xenopus and chickens (4, 14, 17, 36). In humans, activated forms of Notch are associated with T-cell neoplasias (24, 65) and targeted expression of activated Notch to the T-cell compartment of mice affects the ratio of CD4 versus CD8 single-positive cells and allows CD8-positive cells to develop in the absence of the class I major histocompatibility complex (68). Constitutive expression of Notch inhibits myogenic, neural, and myeloid differentiation in cell culture systems (46, 60, 62). Targeted disruption of the Notch1 gene in mice leads to embryonic lethality, a result that underscores the importance of Notch signaling in early development (18, 78).
Two hallmark features of Notch receptors are the presence of several ankyrinlike repeats on the cytoplasmic face of the protein and a series of tandemly arrayed epidermal growth factor (EGF) repeats on the extracellular surface; these likely interact with Notch ligands expressed on the surface of neighboring cells (67). The ankyrin repeats are essential for signaling in the context of the wild-type receptor. Deletion of the EGF repeats, leaving either a wholly cytoplasmic protein or one that associates with the membrane through the transmembrane domain (TMD), results in a constitutively active Notch signal (24, 50, 67, 69, 77). Such truncated proteins are often used to study Notch signaling in the absence of ligand (17, 26, 46, 60, 62). Indeed, proteolysis may be a necessary component of ligand-dependent signaling. Recent evidence indicates that Notch normally resides on the plasma membrane in a form that is proteolytically cleaved N terminal to the TMD but religated with a bond that is sensitive to reducing agents (9).
Notch signaling can be effected through a number of proteins that have been characterized both genetically and biochemically. In drosophila, Suppressor of Hairless [Su(H)] is known to be a DNA binding protein that interacts physically with Notch (27, 57). Su(H) responds to Notch and, in turn, activates the transcription of a number of genes, including those of the Enhancer of Split [E(Spl)] complex (6, 29, 49). E(Spl) proteins directly inhibit the activity of a number of bHLH transcription factors, including those required for neurogenesis in drosophila. Like Notch, such downstream proteins are also conserved and homologs of Su(H) and E(Spl) have been identified in mammalian cells; these are CBF1 (also known as RBP-Jκ) and HES, respectively (16, 22, 28, 63, 72). When the cytoplasmic domain of Notch is expressed alone, it accumulates in the nucleus, where it forms a ternary complex with CBF1 and this correlates with the activation of the HES-1 promoter (42). However, it is a matter of some debate whether nuclear translocation of Notch is an aspect of signaling by the intact protein (26, 30, 46, 67, 77). An additional protein, Deltex, physically interacts with Notch and can augment the Notch signal (11, 21, 32, 56), but the exact role that Deltex plays in the pathway defined by Su(H) is unknown.
Vertebrate Notch1 can inhibit muscle differentiation by inhibiting the activity of MyoD. This can be observed by transfecting cells with truncated (cytoplasmic) Notch1 (46, 74) or by coculturing myoblasts with cells that express the vertebrate Notch ligand Jagged (52). Given that MyoD functions as a heterodimer with E proteins such as E47 (48), we considered the possibility that E47 itself is a Notch target. We demonstrate here that E47 is inhibited by Notch and propose that the target of Notch is a novel E47 coactivator. In addition, we provide evidence that the pathway that connects Notch and E47 is independent of Su(H) and may involve signaling through Deltex.
MATERIALS AND METHODS
Plasmids and transfections.
The following plasmids were used as reporters: [E5+E2]6TATA-chloramphenicol acetyltransferase (CAT) and [E5+E2+μE3]6TATA-luciferase to measure the activity of E47 and E47-VP16 (12, 76), [GAL4]5E1B-CAT to measure the activity of GAL4-E47 and GAL4-p300 (51), 5xGal4TKcat to measure the activity of Gal4-CBF1 (38); 4xwtCBF1Luc to measure the activity of endogenous CBF1 (39), and retinoic acid receptor (RAR)-thymidine kinase (TK)-luciferase (10, 20) to measure the activity of RAR. Expression vectors for E47 (75), E47-VP16 (15), GAL4-E47 (37), GAL4-p300 (87), Gal4-CBF1 (38), and E1A (82, 83) have been described. The expression vector for murine RARα was obtained from Mitch Lazar (University of Pennsylvania). pFR-Luc, pFA-Jun, pFA-Elk, and pFA-CREB were obtained as part of the PathDetect In Vivo Signal Transduction Pathway Reporter System from Stratagene. Detailed information concerning the cytomegalovirus (CMV)-based expression vectors for the various human Notches and Deltex is available upon request. Transfections of NIH 3T3 and 293T cells were carried out by using calcium phosphate coimmunoprecipitation (33) with the amounts of plasmids indicated in the figure legends. Transfections were normalized to β-galactosidase activities determined with 100 ng of CMV–β-galactosidase. The DNA in each transfection was brought to a total of 7 μg with pBluescript.
Immunofluorescence.
NIH 3T3 cells were transfected with 3 μg of the indicated expression plasmids and processed (88) as follows. At 24 h posttransfection, cells were trypsinized and plated in four-well chamber slides. At 48 h posttransfection, cells were washed in phosphate-buffered saline (PBS) and fixed for 30 min in 3% paraformaldehyde and 250 μl of 1 M NaOH. The cells were washed and then permeabilized for 10 min in 10% goat serum–1% Triton in PBS. Cells were washed and blocked in 10% goat serum–1% Tween 20 in PBS. Primary antibody was added at 1:10 and incubated overnight. Cells were then washed and incubated with secondary antibody at 1:100 (fluorescein isothiocyanate–anti-rat immunoglobulin G; Santa Cruz) and finally washed, mounted, and viewed. pCNVhDx-1, which expresses the Myc epitope-tagged human Deltex protein, is described elsewhere (58). NIH 3T3 cells were transfected by using Lipofectamine (Gibco BRL) in accordance with the manufacturer’s instructions.
Western analysis.
NIH 3T3 cells were transfected with 3 μg of Notch-expressing plasmids, and extracts were harvested after 48 h. Extracts were prepared as described by Damm et al. (19). Briefly, cells were washed, scraped from the plates, and resuspended in 100 μl of 20 mM HEPES (pH 7.9)–400 mM KCl–2 mM dithiothreitol–20% glycerol. Extracts (10 μl) were then mixed with sodium dodecyl sulfate loading buffer (10 μl) and resolved on a sodium dodecyl sulfate–10% polyacrylamide gel.
RESULTS
Vertebrate Notch1 and Notch2 inhibit full-length E47 activity.
To assess the effect of Notch on E47 activity, we employed transient transfections of NIH 3T3 cells. Plasmids expressing E47 (E2-5; see Materials and Methods) from a CMV promoter were transfected together with CAT reporters containing multiple E boxes situated upstream of a simple TATA box. As shown in Fig. 1, the presence of E47 led to a large increase in the amount of CAT activity (Fig. 1A, lanes 1 and 2). However, when the transfections also included plasmids that express the cytoplasmic domain of human Notch1 (N1-IC; Fig. 1A, lanes 3 and 4), activity was repressed. Similar inhibitory effects were found with the cytoplasmic domain of human Notch2 (N2-IC, Fig. 1B) and were observed in transfections of B cells (data not shown).
FIG. 1.

Inhibition of intact E47 by cytoplasmic Notch1 and Notch2. NIH 3T3 cells were transfected with the expression plasmids for the indicated proteins and reporter [μE5+μE2]6TATA-CAT (100 ng; lanes 1 to 8) or [GAL4]5E1bTATA-CAT (250 ng; lanes 9 to 11). CMV-E47 was used to express full-length E47 (30 ng; lanes 2 to 4), CMV-E47-VP16 was used to express the E47-VP16 fusion protein (60 ng; lanes 6 to 8), and CMV-GAL4-E47 was used to express the GAL4-E47 fusion protein (100 ng; lanes 10 and 11). Either 1 (lanes 3 and 7) or 6 (lanes 4, 8, and 11) μg of N1-IC (N lanes in A) or N2-IC (N lanes in B) was included in the transfections, as indicated.
We considered that Notch might inhibit DNA binding by E47 or transcriptional activation per se. We therefore tested Notch’s effect on the behavior of two hybrid proteins. One carries the DNA binding domain of E47, the bHLH domain, fused to the transcriptional activation domain of VP16 (E47-VP16), and the other carries the DNA binding domain of GAL4 linked to the transcriptional activation domain of E47 (GAL4-E47). Like intact E47, both hybrid proteins were expressed from the CMV promoter. Although both stimulated the activity of plasmid reporters (Fig. 1A, lanes 5, 6, 9, and 10), neither hybrid protein was inhibited by N1-IC (Fig. 1A, lanes 7, 8, and 11). The same results were obtained with N2-IC (Fig. 1B). These results indicate that the Notch signal targets an unanticipated activity of E47 that is revealed only with the intact protein. In this regard, E47 cannot be viewed simply as the sum of its component parts; namely, an activation domain linked to a DNA binding domain.
Notch does not inhibit the activity of E47 coactivator CBP/p300.
We considered the possibility that the coactivator CBP/p300 is the target of Notch. Early studies by Weintraub and colleagues argued for a MyoD coactivator that recognizes both the MyoD basic region (part of the bHLH DNA binding domain) and the MyoD activation domain (84). Indeed, MyoD responds to Notch in a fashion similar to that of E47: intact MyoD is inhibited by Notch, whereas GAL4-MyoD and MyoD-VP16 are both resistant (46). Perhaps MyoD and E47 both utilize coactivators that recognize only the intact proteins and these are targeted by Notch signaling. CBP/p300 is an excellent candidate for such a coactivator because it is required for the activity of MyoD and probably that of E47 as well (23, 66, 71, 87). Although the bHLH domains of MyoD and E47 are sufficient for some functional interaction with p300 (23), it has recently been shown that the N-terminal activation domain of MyoD also interacts with p300 (71).
We employed three assays to explore the possibility that Notch targets CBP/p300. The first was done to see if excess p300 could overcome Notch’s inhibition of E47. As shown in Fig. 2A, Notch1 inhibited E47 activity while a p300 expression plasmid stimulated E47 activity. The latter result recapitulated the findings of Eckner et al. (23). In the presence of both Notch and excess p300, Notch inhibition was reduced in level but still apparent. Although these results are consistent with Notch affecting CBP/p300, they are inconclusive. The second assay was done to examine the activity of a GAL4-p300 fusion protein in the presence of Notch. This would allow us to test the effect of Notch on the intrinsic coactivator activity of CBP/p300 (87). As shown in Fig. 2B, GAL4-p300 potently activated the transcription of a luciferase reporter under the control of five GAL4 binding sites. However, activity was not affected in the presence of N1-IC, arguing that Notch does not affect the intrinsic activating potential of p300. The third assay was done to assess the effect of Notch on RAR, a protein that requires CBF/p300 for transcriptional activation (13, 43). This assay allowed us to examine whether Notch affects the ability of CBP/p300 to functionally interact with a known transcription factor. However, Notch had no inhibitory effect on retinoic acid-mediated stimulation of RAR (Fig. 2C). This result indicates that Notch does not prevent CBP/p300 from functionally interacting with proteins other than E47.
FIG. 2.

Notch1 does not inhibit CBP/p300. (A) E47 activity was determined in 293T cells transfected with the [μE5+μE2+μE3]6TATA-luciferase reporter (100 ng) in the presence of either Notch1 (N1; 1 μg), p300 (5.5 μg), or both, as indicated. (B) GAL4-p300 fusion protein activity was determined in 293T cells transfected with the [GAL4]5E1bTATA-luciferase reporter (300 ng) in the presence or absence of Notch1, as indicated. (C) Activity of RAR (0.5 μg) was determined by using the RAR-TK-luciferase reporter (2 μg) in the presence or absence of 1 μM retinoic acid (RA) and in the presence of RA plus Notch1 (1 μg), as indicated.
The adenovirus E1A protein provides another means to probe potential involvements of CBP/p300 (1, 54). E1A is thought to inhibit the activity of many transcription factors by binding to and sequestering CBP/p300 or, in some instances, the retinoblastoma protein, Rb. The CBP/p300 interaction domain of E1A maps to the protein’s N terminus (82, 83), and consequently, N-terminal deletions of E1A fail to sequester CBP/p300. As shown in Fig. 3 (left), we found that wild-type E1A did inhibit the activity of intact E47 and that this was dependent on amino acids residing in the N terminus. Two N-terminal deletion mutants, del2-36 and del37-68, failed to inhibit E47. A third E1A mutant, CxDl, that does not interact with Rb gave a level of inhibition similar to that found for wild-type E1A. These results further support the proposal that CBP/p300 is a necessary coactivator of E47 (23). However, we found that E1A also inhibited the activity of both GAL4-E47 and E47-VP16 and that this was dependent on E1A’s N terminus (Fig. 3, center and right), suggesting that CBP/p300 is used as a coactivator for these hybrid proteins as well. Since Notch did not inhibit the activity of either hybrid protein (Fig. 1), we tentatively concluded that CBP/p300 is not a target of Notch inhibition. It appears that inhibition of only the full-length E47 protein (or of full-length MyoD) is a defining feature of the Notch inhibitory pathway and likely reflects the requirement of a novel coactivator.
FIG. 3.

E1A inhibits activities of intact E47 and E47 fusion proteins. Activities of E47, E47-VP16, and GAL4-E47 were determined in NIH 3T3 cells transfected as indicated in the legend to Fig. 1, in the absence or presence of plasmids (1 μg) expressing the indicated E1A proteins. The p300/CBP binding site on E1A is removed in N-terminal deletions del2-36 and del37-68.
Notch-mediated activation of CBF1/Su(H) does not correlate with its ability to inhibit E47.
It has been proposed that Notch inhibits MyoD through the action of CBF1, activating transcription of the HES genes, mammalian equivalents of drosophila E(Spl) (31, 42). HES proteins apparently form inactive heterodimers with MyoD and other bHLH proteins (72) and can inhibit neuronal differentiation (41). We have confirmed that both HES-1 and HES-2 inhibit the activity of E47 in cotransfection assays (data not shown). We therefore considered the possibility that Notch inhibits E47 through a similar pathway. However, we also reasoned that inhibition would require unusually high levels of HES protein synthesis because of the large amounts of E47 expressed from transfected plasmids. Because HES gene transcription would require the activation of CBF1, we chose to explore this issue by examining the relationship between Notch’s abilities to repress E47 and to activate CBF1.
We tested several human Notch2 derivatives that might be expected to have distinct signaling properties (Fig. 4A). In addition to N2-IC, we generated two other deletion constructs. In one, dubbed N2-TMIC, the entire extracellular domain is absent but the TMD is intact. A similar Notch1 deletion construct was shown to efficiently activate the CBF1 promoter (42). In another, dubbed N2-ICΔ, a conserved domain N terminal to the ankyrin repeats is absent. This domain, sometimes referred to as the RAM domain, helps mediate physical interactions with mammalian Notch1 (39, 53, 80), mammalian Notch2 (40), drosophila Notch (57) and C. elegans GLP-1 (70). We anticipated that N2-ICΔ would activate CBF1 poorly, if at all. We confirmed the expression of all three constructs by Western analysis and for appropriate cellular localization by indirect immunofluorescence of transfected cells (Fig. 4B and C). Full-length Notch1 (N1-FL) and Notch2 (N1-FL) were both excluded from the nucleus and gave rise to a punctate pattern in the cytoplasm that likely reflects association with membranes. N2-TMIC gave similarly punctate cytoplasmic staining but was found in the nucleus as well. Nuclear localization of N2-TMIC is likely the consequence of the transmembrane domain being proteolytic cleaved. Both N2-IC and N2-ICΔ localized primarily to the nucleus, as previously reported for similar truncations of Notch1. These results are consistent with the subcellular localization reported previously for different forms of Notch1 (40, 42, 46, 62) and drosophila Notch (26, 67, 77).
FIG. 4.
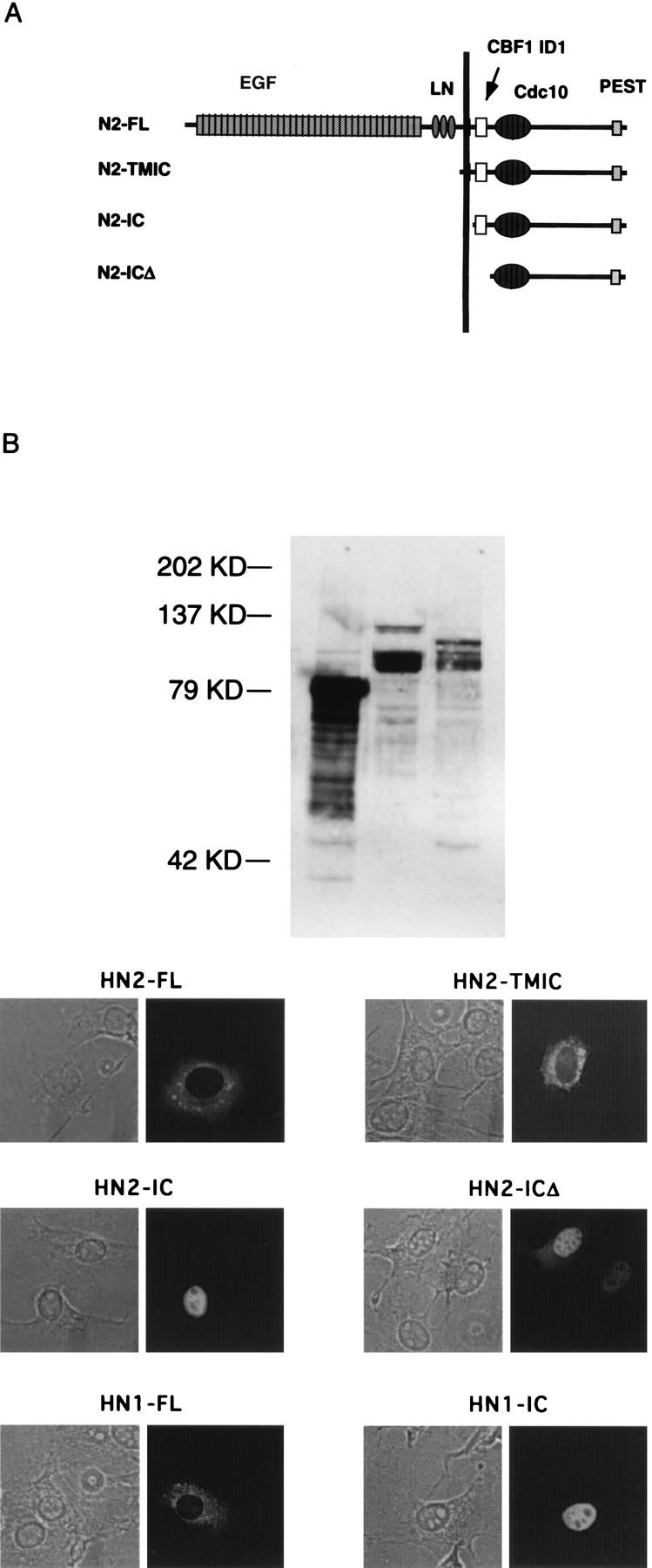
Characterization of Notch2 mutants. (A) Schematic drawing of the various Notch2 proteins. The positions of the extracellular EGF repeats, the cysteine-rich Notch/Lin-12 repeats (LN), the CdC10/Ankyrin repeats (Cdc10), and PEST sequences are indicated. CBF1 ID1 refers to the CBF1 interaction domain, and the position of the membrane domain is indicated by the vertical line. (B) Expression levels of N2-TMIC, N2-IC, and N2-ICΔ determined after transfections of NIH 3T3 cells. Immobilized proteins were probed with Notch-specific antibodies raised against the cytoplasmic domain. KD, kilodaltons. (C) Subcellular location of various Notch proteins. NIH 3T3 cells were transfected with plasmids expressing the indicated Notch proteins and then visualized by phase (left) or fluorescence (right).
We then tested the various Notch2 proteins for their effects on CBF1 and on E47. We employed two assays that measure CBF1 activity. The first assay measured the effect of Notch on a Gal4-CBF1 fusion protein, and the second assay examined the activity of endogenous CBF1 by utilizing a luciferase reporter bearing CBF1 binding sites. In the absence of Notch, Gal-CBF1 represses transcription mediated by a TK promoter linked to CAT, and in the presence of Notch, GAL4-CBF1 activates transcription (39, 40). As shown in Fig. 5A, both N2-TMIC and N2-IC derepressed Gal4-CBF1 (compare lanes 2 and 4 or lanes 2 and 5). N2-ICΔ, which lacks the RAM CBF1 interaction domain, had no apparent effect on Gal4-CBF1 (lane 6), confirming the domain’s functional role in vivo. We reached the same general conclusions when using the second assay for CBF1 activity. In this case, the luciferase reporter responds to the activity of endogenous CBF1 by virtue of multiple CBF1 binding sites in its promoter (39, 40). In the absence of Notch, the reporter exhibited low activity, which was activated 25-fold and 35-fold by N2-TMIC and N2-IC, respectively (Fig. 5B). However, N2-ICΔ gave less-than-10-fold activation of the reporter, despite the fact that it is expressed at higher levels than N2-IC (Fig. 5B). The 10-fold activation may reflect the independent binding of CBF1 to the Notch ankyrin domains seen in vitro (3, 44). When we tested these Notch2 proteins for the ability to inhibit E47, we found that all three were able to inhibit E47 activity with the same efficiency (Fig. 6). Taken together, these results indicate that Notch’s ability to inhibit E47 does not correlate with its ability to activate CBF1. Furthermore, the ability to separate E47 inhibition from CBF1 activation is consistent with the possibility that they are mediated by distinct Notch signaling pathways.
FIG. 5.

The CBF1 interaction domain of Notch is required for efficient activation of CBF1. (A) Effect of Notch proteins on the activity of a GAL4-CBF1 fusion protein. NIH 3T3 cells were transfected with a CAT reporter carrying five GAL4 binding sites upstream of the TK promoter (1 μg) either without (lane 1) or with (lanes 2 to 6) a plasmid expressing GAL4-CBF1 (1 μg). Plasmids expressing the various Notch proteins with deletions (1 μg) were also included, as indicated. The ability of N1-IC to convert GAL4-CBF1 from a repressor to an activator is depicted schematically below. (B) Effect of Notch proteins on the activity of endogenous CBF1. NIH 3T3 cells were transfected with a luciferase reporter carrying four CBF1 binding sites (1 μg of 4xwtCBF1-luciferase; see diagram), and the level of stimulation over basal activity was determined for each Notch protein indicated. SV40, simian virus 40.
FIG. 6.

The CBF1 interaction domain of Notch2 is not required for efficient repression of E47. E47 activity was determined upon transfections of NIH 3T3 cells in the absence or presence of the various Notch proteins (1 μg), as indicated.
Deltex inhibits E47 activity independently of CBF1/Su(H).
Deltex is a widely expressed cytoplasmic protein that augments Notch activity in drosophila and that physically interacts with the Notch ankyrin repeats (11, 21, 32, 56). Although genetic studies suggest that Deltex may function upstream of Notch, virtually nothing is known about the molecular aspects of Deltex activity and how this relates to the known Notch pathway. The human homolog of Deltex has been isolated (58), and we have used it to address potential signaling mechanisms. Mouse Deltex (FXI-T1) was isolated independently as a cDNA induced in fractionated X-irradiated thymoma cells (64). Overexpression of human Deltex had no effect on endogenous CBF1 activity (Fig. 7A), nor did it relieve repression mediated by a Gal4-CBF1 protein (Fig. 7B; note that N1-IC was more potent in these assays than N2-IC). These data indicate that Deltex alone cannot bypass Notch signaling by activating CBF1.
FIG. 7.

Deltex does not activate CBF1. The effects of N1-IC (1 μg) and Deltex (1 μg) on GAL4-CBF1 (A) and on the 4xwtCBF1-luciferase reporter (B) were determined as described in the legend to Fig. 5. Note that the activity of N1-IC in these assays was significantly higher than the activity of N2-IC. SV40, simian virus 40.
When we tested effects on E47, we found that Deltex, like activated Notch, repressed E47 activity (Fig. 8, left). Also, like activated Notch, Deltex was unable to inhibit the activity of either the E47-VP16 hybrid protein (Fig. 8, middle) or the GAL4-E47 hybrid protein (Fig. 8, right). Thus, Deltex behaves similarly to N2-ICΔ and may define a Su(H)-independent pathway for signaling by vertebrate Notch. However, we found that Deltex, unlike N2-ICΔ, localized primarily to the cytoplasm (Fig. 9).
FIG. 8.

Deltex inhibits the activity of intact E47. The effects of Deltex (1 μg) on the activities of full-length E47 (30 ng of CMV-E47; left), an E47-VP16 fusion protein (60 ng of CMV-E47-VP16; center), and a GAL4-E47 fusion protein (100 ng of GAL4-E47; right) were determined as described in the legend to Fig. 1.
FIG. 9.

Subcellular localization of human Deltex. Myc epitope-tagged hDx-1 protein was expressed in NIH 3T3 cells under the control of the CMV promoter. The tagged human Deltex protein was detected with anti-Myc and CY3-conjugated goat anti-mouse antibodies. Nomarski and fluorescence images are shown on the left and right, respectively.
Notch and Deltex inhibit Ras-dependent transcription.
In separate studies of E47 regulation, we found that full E47 activity is dependent on Ras. Cotransfections of E47 with either constitutively active Ras (cHa-ras, Gly12 to Val) (79) or dominant-negative Ras (pASN17, Ser17 to Asn) (25) led to activation or repression of E47, respectively (Fig. 10A). We also found that the E47-VP16 fusion protein was unaffected by Ras (Fig. 10A), as was the GAL4-E47 fusion protein (data not shown). Thus, Ras stimulates an activity of E47 that is dependent on the full-length protein.
FIG. 10.
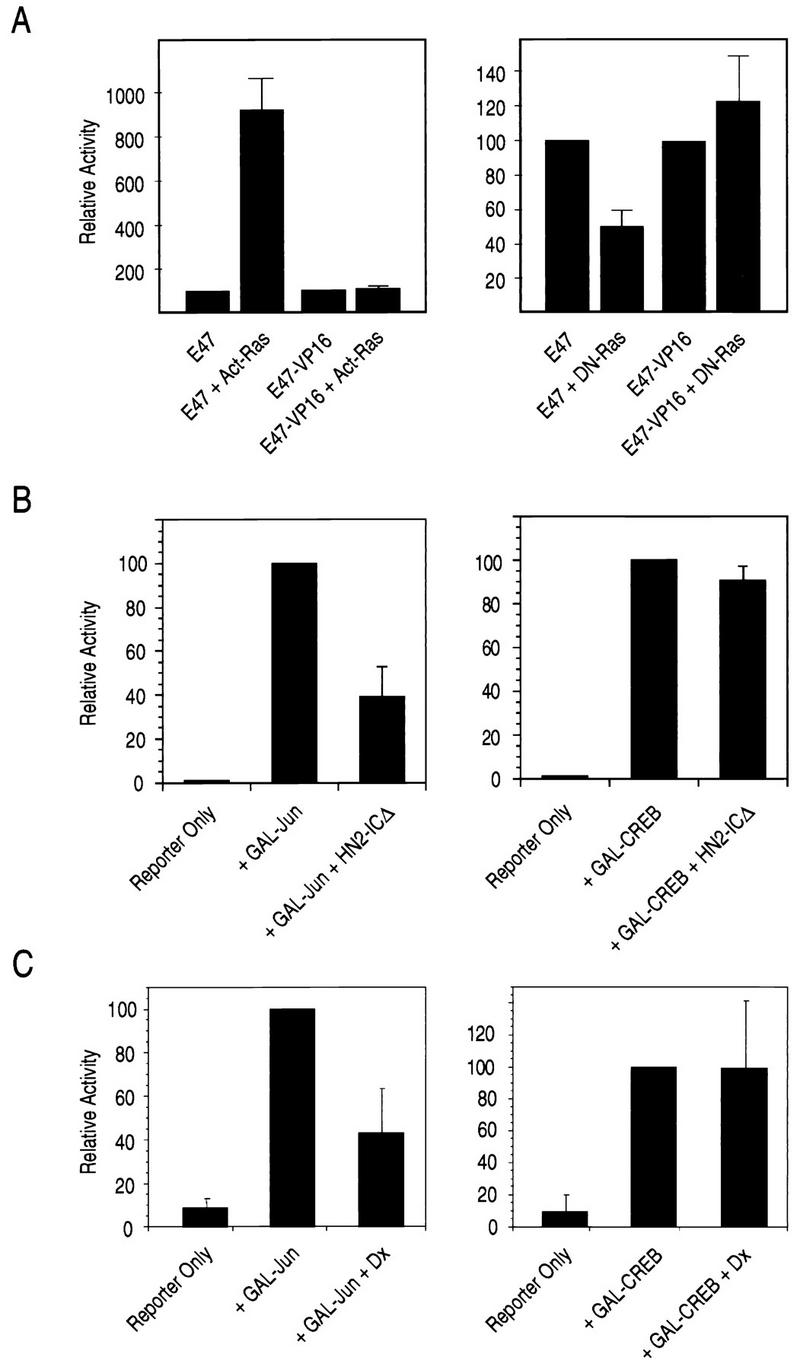
Notch and Deltex inhibit Ras-dependent activation. (A) E47 is stimulated by constitutively active Ras and inhibited by dominant-negative Ras. NIH 3T3 cells were transfected with the expression plasmids for the indicated proteins, and the reporter [μE5+μE2]6TATA-CAT as described in the legend to Fig. 1. Act-Ras refers to 1 μg of constitutively active Ras (cHa-ras) (79), and DN-Ras refers to 1 μg of dominant-negative Ras (pASN17) (25). (B) Notch2 inhibits Jun-mediated transcription. Transfections of NIH 3T3 cells were carried out by using HN2-ICΔ, which lacks the CBF1 interaction domain. GAL-Jun (pFA-Jun; 100 ng) and GAL-CREB (pFA-CREB; 100 ng) were transfected with the reporter GAL5-UAS-luciferase (pFR-luc; 1 μg), as indicated. (C) Deltex inhibits Jun-mediated transcription. Transfections were carried out as for B, except that 5 μg of the Deltex expression plasmid was used in place of HN2-ICΔ).
Our results obtained with Ras and E47 led us to consider the possibility that Notch and Deltex inhibit some aspect of Ras signaling. We therefore examined the responses of a group of reporters whose activities are either dependent on or independent of Ras. The EGR-1 promoter is known to be stimulated by Ras (59) through the action of mitogen-activated protein kinases (MAPKs) on a ternary complex involving ETS proteins (e.g., ELK1) and SRF (81). We found that the activity of a CAT reporter under the control of the EGR-1 promoter was inhibited by Deltex, both in the presence and in the absence of Ras stimulation by platelet-derived growth factor (data not shown). To reduce the complexity of the effects, we employed a series of GAL4 fusions and assessed their abilities to activate a minimal promoter containing GAL4 binding sites. GAL4-Jun includes a portion of the c-Jun protein whose activity is dependent on signaling from Ras to SAPK/JNK (61). GAL4-ELK includes the ELK protein, whose activity can be stimulated by a number of pathways, including Ras to SAPK/JNK and Ras to ERK (55). GAL4-CREB includes the cyclic AMP response element binding (CREB) protein, whose activity is dependent on protein kinase A (34). We found that N2-ICΔ, which lacks the CBF1 interaction domain, inhibited GAL4-Jun activity but had no effect on GAL4-CREB (Fig. 10B). Similarly, Deltex inhibited GAL4-Jun activity and had no effect on GAL4-CREB. The results obtained with the GAL4-ELK protein were less straight forward: N2-ICΔ modestly stimulated activity, while Deltex reproducibly repressed it (data not shown). Although it is likely that N2-ICΔ and Deltex have somewhat different effects on cells, our results clearly show that both Notch and Deltex inhibit signaling by Ras, as measured by the ability to stimulate SAPK/JNK activity. We propose that this is the mechanism by which Notch and Deltex inhibit E47.
DISCUSSION
E47 is a protein essential for the B-lymphocyte lineage. However, unlike most transcription factors that mediate cell type-specific transcription, E47 is widely expressed. Thus, regulation of its activity may be intimately tied to the process of B lymphopoiesis. It is known that one form of regulation is at the level of DNA binding. DNA binding by E47 homodimers is restricted to B cells (75), and it has been proposed that this is controlled by covalent dimerization (8) and/or by phosphorylation (76). However, it is possible that E47 is controlled by other means that do not involve DNA binding per se. Given the potential importance of such regulation in controlling B-cell development, we have begun to identify signaling pathways that affect E47 activity. We have found that signaling by Notch may be one such pathway.
Although activated Notch has been shown to perturb normal T-cell development (24, 65, 68), it is not known if Notch signaling is involved in the normal ontogeny of either T or B lymphocytes. Notch is expressed in both T and B cells, and it is likely that Notch ligands (e.g., Jagged1) are expressed by cells that define the lymphopoietic microenvironments of the thymus, liver, and bone marrow (35). It is interesting that retroviral transfer of activated Notch into hematopoietic cells resulted only in T-cell leukemias (65). The lack of appearance of B-cell neoplasias in the experiment is consistent with the idea that Notch’s effects in B cells do not lead to deregulated growth and may be, instead, to inhibit B-cell differentiation. We are currently generating transgenic mice that will target expression of N2-IC to the B-cell lineage to explore the phenotype of Notch-expressing B cells.
E47 is thought to be a classic bipartite transcription factor with well-defined bHLH DNA binding and transcriptional activation domains. However, our results obtained with Notch indicate that E47 is more complex. We found that Notch inhibited only the full-length protein and did not affect fusion proteins that isolate the E47 DNA binding and transcriptional activation functions. Similar results were noted for Notch inhibition of MyoD, and it was proposed that Notch inhibited a hypothetical MyoD coactivator that simultaneously recognizes the DNA binding and transcriptional activation domains (46). Indirect evidence for such a MyoD coactivator was obtained in the course of a detailed analysis of MyoD activity (84). Given the similarities of E47 and MyoD with respect to inhibition by Notch, we conclude that E47 requires a similar coactivator. Our experiments argue against the coactivator being p300/CBP.
Our results argue that Notch2 can inhibit E47 independently of CBF1 (RBP-Jκ), the mammalian homolog of drosophila Su(H). This result has been confirmed independently by Weinmaster and colleagues, who have shown that neither CBF1 nor HES-1 is involved in Notch-mediated inhibition of myogenesis (74). More recent results from Hayward and colleagues (40) indicate that HES-1 message is induced in cells stably transformed with a truncated form of Notch2 that lacks the major CBF1 interaction domain (40). However, we do not think that Notch-mediated inhibition of E47 results from upregulation of HES-1 because of the large amount of expression that would be needed to tie up the high levels of E47 expressed in transiently transfected cells. Results from Weintraub and colleagues that first showed Notch inhibition of MyoD and of myogenesis were obtained with a truncated Notch protein that lacked part of the N-terminal CBF1 interaction domain (46). However, at the time of those early studies, it was not known that Notch associates with CBF1. Similar conclusions concerning Su(H)-independent signaling have been reported for drosophila Notch (57) and for C. elegans GLP-1 (70). Thus, Su(H)-independent signaling may be a property shared by all members of the Notch family and target transcription factors other than E47. Honjo and coworkers, however, have argued against this (44). Their data indicate that activation of the CBF1 (RBP-J) pathway is sufficient to inhibit myogenic conversion. However, their results do not formally exclude the existence of additional pathways. In drosophila, Notch is inhibited by Wingless signaling through a physical interaction with Dishevelled (5). Whether or not Notch can, conversely, inhibit the Wingless pathway (through Dishevelled) is not known. Arguing against a role for the Wingless pathway in our system was the lack of E47 inhibition with a Notch2 protein that lacks the ankyrin repeats but retains the presumptive Dishevelled interaction domain at the C terminus (data not shown).
It is possible that Notch signaling to E47 involves the Notch-interacting protein Deltex. Our experiments indicate that Deltex expression alone is sufficient to inhibit E47 and that, like Notch, Deltex has no effect on the activity of either GAL4-E47 or E47-VP16. Moreover, Deltex has no effect on the activity of either a transfected GAL4-CBF1 fusion protein or endogenous CBF1. Supporting the idea that Deltex is downstream of Notch is the finding that the Notch proteins that do not bind CBF1/Su(H) well, but do bind Deltex through the ankyrin repeats, can mediate signaling in drosophila (58), C. elegans (70), and vertebrates (74; this work). Although Deltex interacts directly with Notch (21), it is not known if or how this interaction facilitates Notch activity or Deltex activity.
Our results are consistent with a model in which Notch and Deltex act on E47 by inhibiting signaling through Ras. E47 is stimulated by an activated Ras protein and inhibited by a dominant-negative Ras protein. The latter observation suggests that E47 is dependent on Ras activity present in dividing cells and likely involves the action of MAPKs downstream of MEKK and/or Raf (86). Just how MAPKs may affect E47 is not known because, like the effects seen with Notch and Deltex, the effect of Ras requires the intact E47 protein. We have shown here that Deltex can inhibit the activity of the Ras-dependent EGR-1 promoter and that both Notch and Deltex inhibit SAPK/JNK activity, as measured by their abilities to inhibit the activity of a GAL4-Jun fusion protein (61). It is quite possible that the inhibited target in these cases lies upstream of Ras since both drosophila Deltex and human Deltex possess a domain that is conserved in several SH3 domain-interacting proteins (21) and can mediate weak interactions with the adapter protein GRB2 in yeast (58). Such an activity is consistent with Deltex’s cytoplasmic distribution, but it is not apparent how nuclear Notch elicits a similar activity. It has been reported recently that nuclear localization of Notch does not correlate with its ability to activate CBF1 (3), and therefore, Notch’s effects may be entirely cytoplasmic. It is possible that inhibition of Ras also explains the inhibitory effects of Notch on myogenesis (46, 74). Activated Ras typically functions to inhibit myogenesis while having little effect on the ability of MyoD or MRF4 to activate simple promoters (45). However, it appears that inhibition of myogenesis by Ras involves a pathway that is distinct from the one used for Ras-mediated transformation (85). Thus, Notch and Deltex may not be global inhibitors of Ras but may function to inhibit specific aspects of signaling, such as that leading to activation of SAPK/JNK.
REFERENCES
- 1.Arany Z, Newsome D, Oldread E, Livingston D M, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 2.Artavanis-Tsakonas S, Matsuno K, Fortini M E. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 3.Aster J C, Robertson E S, Hasserjian R P, Turner J R, Kieff E, Sklar J. Oncogenic forms of Notch1 lacking either the primary binding site for RPB-Jκ or nuclear localization sequences retain the ability to associate with RBP-Jκ and activate transcription. J Biol Chem. 1997;272:11336–11343. doi: 10.1074/jbc.272.17.11336. [DOI] [PubMed] [Google Scholar]
- 4.Austin C P, Feldman D E, Ida J A, Jr, Cepko C L. Vertebrate retinal ganglion cells are selected from competent progenitors by the action of Notch. Development. 1995;121:3637–3650. doi: 10.1242/dev.121.11.3637. [DOI] [PubMed] [Google Scholar]
- 5.Axelrod J D, Matsuno K, Artavanis-Tsakonas S, Perrimon N. Interaction between Wingless and Notch signaling pathways mediated by Dishevelled. Science. 1996;271:1826–1832. doi: 10.1126/science.271.5257.1826. [DOI] [PubMed] [Google Scholar]
- 6.Bailey A M, Posakony J W. Suppressor of hairless directly activates transcription of enhancer of split complex genes in response to Notch receptor activity. Genes Dev. 1995;9:2609–2622. doi: 10.1101/gad.9.21.2609. [DOI] [PubMed] [Google Scholar]
- 7.Bain G, Maandag E C, Izon D J, Amsen D, Kruisbeek A M, Weintraub B C, Krop I, Schlissel M S, Feeney A J, van Roon M, van der Valk M, te Riele H P J, Berns A, Murre C. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 8.Benezra R. An intermolecular disulfide bond stabilizes E2A homodimers and is required for DNA binding at physiological temperature. Cell. 1994;79:1057–1067. doi: 10.1016/0092-8674(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 9.Blaumueller C M, Qi H, Zagouras P, Artavanis-Tsakonas S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1998;90:281–291. doi: 10.1016/s0092-8674(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 10.Brent G A, Harney J W, Chen Y, Warne R L, Moore D D, Larson P R. Mutations of the rat growth hormone promoter which increase and decrease response to thyroid hormone define a consensus thyroid hormone response element. Mol Endocrinol. 1989;3:1996–2004. doi: 10.1210/mend-3-12-1996. [DOI] [PubMed] [Google Scholar]
- 11.Busseau I, Diederich R J, Xu T, Artavanis-Tsakonas S. A member of the Notch group of interacting loci, deltex, encodes a cytoplasmic basic protein. Genetics. 1994;136:585–596. doi: 10.1093/genetics/136.2.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter R S, Ordentlich P, Kadesch T. Selective utilization of basic helix-loop-helix–leucine zipper proteins at the immunoglobulin heavy-chain enhancer. Mol Cell Biol. 1997;17:18–23. doi: 10.1128/mcb.17.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakravarti D, LaMorte V J, Nelson M C, Nakajima T, Schulman I G, Juguilon H, Montminy M, Evans R M. Role of CBP/P300 in nuclear receptor signaling. Nature. 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- 14.Chitnis A, Henrique D, Lewis J, Ish-Horowicz D, Kintner C. Primary neurogenesis in Xenopus embryos regulated by a homologue of the Drosophila neurogenic gene Delta. Nature. 1995;375:761–766. doi: 10.1038/375761a0. [DOI] [PubMed] [Google Scholar]
- 15.Choi J K, Shen C-P, Radomska H S, Eckhardt L A, Kadesch T. Coordinate activation of the Ig-heavy chain and TdT loci in non-B cells. EMBO J. 1996;15:5014–5021. [PMC free article] [PubMed] [Google Scholar]
- 16.Christensen S, Kodoyianni V, Bosenberg M, Friedman L, Kimble J. lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H) Development. 1996;122:1373–1383. doi: 10.1242/dev.122.5.1373. [DOI] [PubMed] [Google Scholar]
- 17.Coffman C R, Skoglund P, Harris W A, Kintner C R. Expression of an extracellular deletion of Xotch diverts cell fate in Xenopus embryos. Cell. 1993;73:659–671. doi: 10.1016/0092-8674(93)90247-n. [DOI] [PubMed] [Google Scholar]
- 18.Conlon R A, Reaume A G, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;12:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 19.Damm K, Thompson C C, Evans R M. Protein encoded by v-erbA functions as a thyroid-hormone receptor antagonist. Nature. 1989;339:593–597. doi: 10.1038/339593a0. [DOI] [PubMed] [Google Scholar]
- 20.Davis K D, Berrodin T J, Steimach J E, Winkler J D, Lazar M A. Endogenous RXRs can function as hormone receptors in pituitary cells. Mol Cell Biol. 1994;14:7105–7110. doi: 10.1128/mcb.14.11.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diederich R J, Matsuno K, Hing H, Artavanis-Tsakonas S. Cytosolic interaction between deltex and Notch ankyrin repeats implicates deltex in the Notch signaling pathway. Development. 1994;120:473–481. doi: 10.1242/dev.120.3.473. [DOI] [PubMed] [Google Scholar]
- 22.Dou S, Zeng X, Cortes P, Erdjument-Bromage H, Tempst P, Honjo T, Vales L D. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol Cell Biol. 1994;14:3310–3319. doi: 10.1128/mcb.14.5.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckner R, Yao T-P, Oldread E, Livingston D M. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev. 1996;10:2478–2490. doi: 10.1101/gad.10.19.2478. [DOI] [PubMed] [Google Scholar]
- 24.Ellisen L W, Bird J, West D C, Soreng A L, Reynolds T C, Smith S D, Sklar J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 25.Feig L A, Cooper G M. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fortini M E, Rebay I, Caron L A, Artavanis-Tsakonas S. An activated Notch receptor blocks cell-fate commitment in the developing Drosophila eye. Nature. 1993;365:555–557. doi: 10.1038/365555a0. [DOI] [PubMed] [Google Scholar]
- 27.Fortini M E, Artavanis-Tsakonas S. The suppressor of hairless protein participates in notch receptor signaling. Cell. 1994;79:273–282. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- 28.Furukawa T, Maruyama S, Kawaichi M, Honjo T. The Drosophila homolog of the immunoglobulin recombination signal-binding protein regulates peripheral nervous system development. Cell. 1992;69:1191–1197. doi: 10.1016/0092-8674(92)90640-x. [DOI] [PubMed] [Google Scholar]
- 29.Furukawa T, Kobayakawa Y, Tamura K, Kimura K, Kawaichi M, Tanimura T, Honjo T. Suppressor of hairless, the Drosophila homologue of RBP-J kappa, transactivates the neurogenic gene E(spl)m8. Jpn J Genet. 1995;70:505–524. doi: 10.1266/jjg.70.505. [DOI] [PubMed] [Google Scholar]
- 30.Gho M, Lecourtois M, Geraud G, Posakony J W, Schweisguth F. Subcellular localization of Suppressor of Hairless in Drosophila sense organ cells during Notch signaling. Development. 1996;122:1673–1682. doi: 10.1242/dev.122.6.1673. [DOI] [PubMed] [Google Scholar]
- 31.Goodbourn S. Notch takes a short cut. Nature. 1995;377:288–289. doi: 10.1038/377288a0. [DOI] [PubMed] [Google Scholar]
- 32.Gorman M J, Girton J R. A genetic analysis of deltex and its interaction with the Notch locus in Drosophila melanogaster. Genetics. 1992;13:99–112. doi: 10.1093/genetics/131.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graham F L, van der Eb A J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 34.Hagiwara M, Alberts A, Brindle P, Meinkoth J, Feramisco J, Dent T, Karin M, Shenolikar S, Montminy M. Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell. 1992;70:105–113. doi: 10.1016/0092-8674(92)90537-m. [DOI] [PubMed] [Google Scholar]
- 35.Hasserjian R P, Aster J C, Davi F, Weinberg D S, Sklar J. Modulated expression of Notch1 during thymocyte development. Blood. 1996;88:970–976. [PubMed] [Google Scholar]
- 36.Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish-Horowicz D. Expression of a Delta homologue in prospective neurons in the chick. Nature. 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- 37.Henthorn P, Kiledjian M, Kadesch T. Two distinct transcription factors that bind to the immunoglobulin enhancer μE5/kE2 motif. Science. 1990;247:467–470. doi: 10.1126/science.2105528. [DOI] [PubMed] [Google Scholar]
- 38.Hsieh J J-D, Hayward S D. Masking of the CBF1/RBPJκ transcriptional repression domain by Epstein-Barr virus EBNA2. Science. 1995;268:560–563. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- 39.Hsieh J J-D, Henkel T, Salmon P, Robey E, Peterson M G, Hayward S D. Truncated mammalian Notch1 activates CBF1/RBPJκ-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol Cell Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh J J-D, Nofziger D E, Weinmaster G, Hayward S D. Epstein-Barr virus immortalization: Notch2 interacts with CBF1 and blocks differentiation. J Virol. 1997;71:1938–1945. doi: 10.1128/jvi.71.3.1938-1945.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishibashi M, Moriyoshi K, Sasai Y, Shiota K, Nakanishi S, Kageyama R. Persistent expression of helix-loop-helix factor HES-1 prevents mammalian neural differentiation in the central nervous system. EMBO J. 1994;13:1799–1805. doi: 10.1002/j.1460-2075.1994.tb06448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jarriault S, Brou C, Logeat F, Schroeter E H, Kopan R, Israel A. Signaling downstream of activated mammalian Notch. Nature (London) 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 43.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S C, Heyman R A, Roxe D W, Glass C K, Rosenfeld M G. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 44.Kato H, Taniguchi Y, Kurooka H, Minoguchi S, Sakai T, Nomura-Okazaki S, Tamura K, Honjo T. Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development. 1997;124:4133–4141. doi: 10.1242/dev.124.20.4133. [DOI] [PubMed] [Google Scholar]
- 45.Kong Y, Johnson S E, Taparowsky E J, Konieczny S F. Ras p21Val inhibits myogenesis without altering the DNA binding or transcriptional activities of the myogenic basic helix-loop-helix factors. Mol Cell Biol. 1995;15:5205–5213. doi: 10.1128/mcb.15.10.5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kopan R, Nye J S, Weintraub H. The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix-loop-helix region of MyoD. Development. 1994;120:2385–2396. doi: 10.1242/dev.120.9.2385. [DOI] [PubMed] [Google Scholar]
- 47.Kopan R, Schroeter E H, Weintraub H, Nye J S. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci USA. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lassar A B, Davis R L, Wright W E, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell. 1991;66:305–315. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- 49.Lecourtois M, Schweisguth F. The neurogenic suppressor of hairless DNA-binding protein mediates the transcriptional activation of the enhancer of split complex genes triggered by Notch signaling. Genes Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- 50.Lieber T, Kidd S, Alcamo E, Corbin V, Young M W. Antineurogenic phenotypes induced by truncated Notch proteins indicate a role in signal transduction and may point to a novel function for Notch in nuclei. Genes Dev. 1993;7:1949–1965. doi: 10.1101/gad.7.10.1949. [DOI] [PubMed] [Google Scholar]
- 51.Lillie J W, Green M R. Transcription activation by the adenovirus E1a protein. Nature. 1989;338:39–44. doi: 10.1038/338039a0. [DOI] [PubMed] [Google Scholar]
- 52.Lindsell C E, Shawber C J, Boulter J, Weinmaster G. Jagged: a mammalian ligand that activates Notch1. Cell. 1995;80:909–917. doi: 10.1016/0092-8674(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 53.Lu F M, Lux S E. Constitutively active human Notch1 binds to the transcription factor CBF1 and stimulates transcription through a promoter containing a CBF1-responsive element. Proc Natl Acad Sci USA. 1996;93:5663–5667. doi: 10.1073/pnas.93.11.5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lundblad J R, Kwok R P, Laurance M E, Harter M L, Goodman R H. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator. Nature. 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 55.Marais R, Wynne J, Treisman R. The SRF accessory protein ELK-1 contains a growth factor regulated transcription domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- 56.Matsuno K, Diederich R J, Go M J, Blaumueller C M, Artavanis-Tsakonas S. Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development. 1995;121:2633–2644. doi: 10.1242/dev.121.8.2633. [DOI] [PubMed] [Google Scholar]
- 57.Matsuno K, Go M, Sun X, Eastman D, Artavanis-Tsakonas S. Suppressor of Hairless-independent events in Notch signaling imply novel pathway elements. Development. 1997;124:4265–4273. doi: 10.1242/dev.124.21.4265. [DOI] [PubMed] [Google Scholar]
- 58.Matsuno, K., D. S. Eastman, T. Mitsiades, A. M. Quinn, M. L. Carcanciu, P. Ordentlich, T. Kadesch, and S. Artavanis-Tsakonas. Human Deltex: a conserved regulator of Notch signaling. Nature Genet., in press. [DOI] [PubMed]
- 59.McMahon S B, Monroe J G. Activation of the p21ras pathway couples antigen receptor stimulation to induction of the primary response gene egr-1 in B lymphocytes. J Exp Med. 1995;181:417–422. doi: 10.1084/jem.181.1.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Milner L A, Bigas A, Kopan R, Brashem-Stein C, Bernstein I D, Martin D I K. Inhibition of granulocytic differentiation by mNotch1. Proc Natl Acad Sci USA. 1996;93:13014–13019. doi: 10.1073/pnas.93.23.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minden A, Lin A, Claret F X, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 62.Nye J S, Kopan R, Axel R. An activated Notch suppresses neurogenesis and myogenesis but not gliogenesis in mammalian cells. Development. 1994;120:2421–2430. doi: 10.1242/dev.120.9.2421. [DOI] [PubMed] [Google Scholar]
- 63.Oka C, Nakano T, Wakeham A, de la Pompa J L, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak T W, Honjo T. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–3301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- 64.Pampeno C L, Meruelo D. A novel cDNA transcript expressed in fractionated X-irradiation-induced murine thymomas. Cell Growth Differ. 1996;7:1113–1123. [PubMed] [Google Scholar]
- 65.Pear W S, Aster J C, Scott M L, Hasserjian R P, Soffer B, Sklar J, Baltimore D. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996;183:2283–2291. doi: 10.1084/jem.183.5.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Puri P L, Avantaggiati M L, Balsano C, Sang N, Graessmann A, Giordano A, Levrero M. p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 1997;16:369–383. doi: 10.1093/emboj/16.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rebay I, Fehon R G, Artavanis-Tsakonas S. Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell. 1993;74:319–329. doi: 10.1016/0092-8674(93)90423-n. [DOI] [PubMed] [Google Scholar]
- 68.Robey E, Chang D, Itano A, Cado D, Alexander H, Lans D, Weinmaster G, Salmon P. An activated form of Notch influences the choice between CD4 and CD8 T cell lineages. Cell. 1996;87:483–492. doi: 10.1016/s0092-8674(00)81368-9. [DOI] [PubMed] [Google Scholar]
- 69.Roehl H, Kimble J. Control of cell fate in C. elegans by a GLP-1 peptide consisting primarily of ankyrin repeats. Nature. 1993;364:632–635. doi: 10.1038/364632a0. [DOI] [PubMed] [Google Scholar]
- 70.Roehl H, Bosenberg M, Blelloch R, Kimble J. Roles of the RAM and ANK domains in signaling by the C. elegans GLP-1 receptor. EMBO J. 1996;15:7002–7012. [PMC free article] [PubMed] [Google Scholar]
- 71.Sartorelli V, Huang J, Hamamori Y, Kedes L. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol. 1997;17:1010–1026. doi: 10.1128/mcb.17.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sasai Y, Kageyama R, Tagawa Y, Shigemoto R, Nakanishi S. Two mammalian helix-loop-helix factors structurally related to Drosophila hairy and enhancer of split. Genes Dev. 1992;6:2620–2634. doi: 10.1101/gad.6.12b.2620. [DOI] [PubMed] [Google Scholar]
- 73.Schlissel M, Voronova A, Baltimore D. Helix-loop-helix transcription factor E47 activates germ-line immunoglobulin heavy-chain gene transcription and rearrangement in a pre-T-cell line. Genes Dev. 1991;5:1367–1376. doi: 10.1101/gad.5.8.1367. [DOI] [PubMed] [Google Scholar]
- 74.Shawber C, Nofziger D, Hsieh J J-D, Lindsell C, Bogler O, Hayward D, Weinmaster G. Notch signaling inhibits muscle cell differentiation through a CBF1-independent pathway. Development. 1996;122:3765–3773. doi: 10.1242/dev.122.12.3765. [DOI] [PubMed] [Google Scholar]
- 75.Shen C-P, Kadesch T. B-cell-specific DNA binding by an E47 homodimer. Mol Cell Biol. 1995;15:4518–4524. doi: 10.1128/mcb.15.8.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sloan S R, Shen C-P, McCarrick-Walmsley R, Kadesch T. Phosphorylation of E47 as a potential determinant of B-cell-specific activity. Mol Cell Biol. 1996;16:6900–6908. doi: 10.1128/mcb.16.12.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Struhl G, Fitzgerald K, Greenwald I. Intrinsic activity of the LIN-12 and Notch intracellular domains in vivo. Cell. 1993;60:981–990. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- 78.Swiatek P J, Lindsell C E, del Amo F F, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 79.Tabin C, Bradlet S, Bargmann C, Weinberg R, Papageorge A, Scolnick E, Dhar R, Lowy D, Chang E. Mechanism of activation of a human oncogene. Nature. 1982;300:143–149. doi: 10.1038/300143a0. [DOI] [PubMed] [Google Scholar]
- 80.Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jκ/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 81.Treisman R. Ternary complex factors: growth factor regulated transcriptional activators. Curr Opin Genet Dev. 1994;4:96–101. doi: 10.1016/0959-437x(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 82.Wang H-G H, Draetta G, Moran E. E1A induces phosphorylation of the retinoblastoma protein independently of direct physical association between the E1A and retinoblastoma products. Mol Cell Biol. 1991;11:4253–4265. doi: 10.1128/mcb.11.8.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang H-G H, Rikitake Y, Corrigan Carter M, Yaciuk P, Abraham S E, Zerler B, Moran E. Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J Virol. 1993;67:476–488. doi: 10.1128/jvi.67.1.476-488.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weintraub H, Dwarki V J, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscot S J. Muscle-specific transcriptional activation by MyoD. Genes Dev. 1991;5:1377–1386. doi: 10.1101/gad.5.8.1377. [DOI] [PubMed] [Google Scholar]
- 85.Weyman C M, Ramocki M B, Taparowsky E J, Wolfman A. Distinct signaling pathways regulate transformation and inhibition of skeletal muscle differentiation by oncogenic Ras. Oncogene. 1997;14:697–704. doi: 10.1038/sj.onc.1200874. [DOI] [PubMed] [Google Scholar]
- 86.Whitmarsh A J, Davis R J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 87.Yuan W, Condorelli G, Caruso M, Felsani A, Giordano A. Human p300 protein is a coactivator for the transcription factor MyoD. J Biol Chem. 1996;271:9009–9013. doi: 10.1074/jbc.271.15.9009. [DOI] [PubMed] [Google Scholar]
- 88.Zagouras P, Stifani S, Blaumueller C M, Carcangiu M L, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci USA. 1995;92:6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell development. Cell. 1994;79:875–884. doi: 10.1016/0092-8674(94)90076-0. [DOI] [PubMed] [Google Scholar]