Coupling of Cell Growth Control and Apoptosis Functions of Id Proteins (original) (raw)
Abstract
The Id family of helix-loop-helix proteins function as negative regulators of cell differentiation and as positive regulators of G1 cell cycle control. We report here that enforced overexpression of the Id3 gene suppresses the colony-forming efficiency of primary rat embryo fibroblasts. Cotransfection with the antiapoptotic Bcl2 or BclXL gene alleviates this suppression and leads to cell immortalization. Consistent with this, enforced expression of Id genes in isolation was found to be a strong inducer of apoptosis in serum-deprived fibroblast cells. Id3-induced apoptosis was mediated at least in part through p53-independent mechanisms and could be efficiently rescued by Bcl2, BclXL, and the basic helix-loop-helix protein E47, which is known to oppose the functions of Id3 in vivo through the formation of stable heterodimers. Enforced overexpression of Id proteins has previously been shown to promote the cell cycle S phase in serum-deprived embryo fibroblasts (R. W. Deed, E. Hara, G. Atherton, G. Peters, and J. D. Norton, Mol. Cell. Biol. 17:6815–6821, 1997). The extent of apoptosis induced by loss- and gain-of-function Id3 mutants and by wild-type Id3 either alone or in combination with the Bcl2, BclXL, and E47 genes was invariably correlated with the relative magnitude of cell cycle S phase promotion. In addition, Id3-transfected cell populations displaying apoptosis and those in S phase were largely coincident in different experiments. These findings highlight the close coupling between the G1 progression and apoptosis functions of Id proteins and hint at a common mechanism for this family of transcriptional regulators in cell determination.
In mammals, four distinct members of the family of dominant negative helix-loop-helix (HLH) Id proteins, Id1 to Id4, have been identified (5, 6, 9, 36, 45). Accumulating biochemical and genetic evidence implicates the function of these proteins as being in the regulation of cell growth and differentiation in numerous cell lineages. The distinguishing characteristic shared by this family of transcriptional regulators, as originally described for the prototype, Id1 (5), is their lack of a basic DNA-binding domain and their consequent ability to antagonize the functions of DNA-binding basic HLH (bHLH) transcription factors through the formation of nonfunctional Id-bHLH heterodimers (5, 36). In several cell types, exemplified by skeletal myocytes (26) and B lymphocytes (39, 54, 61), Id proteins inhibit bHLH-dependent expression of genes associated with terminal differentiation. Moreover, enforced expression of at least the three well-characterized Id proteins, Id1, Id2, and Id3, has been shown to arrest differentiation in a variety of in vitro cell line models (39, 54, 61), and targeted ectopic expression of the Id1 gene in vivo reportedly arrests early B-lymphocyte development (53). Thus, Id proteins function at a general level as negative regulators of differentiation by opposing the functions of bHLH transcription factors.
In mammalian cell lines, the expression of Id genes is strongly responsive to signalling pathways coupled to mitogenic growth factors (5, 9, 12). Indeed, the mouse and human Id3 genes were identified from cDNAs derived from transcripts encoded by early-response genes (ERGs) (9, 12). Following growth factor withdrawal or in quiescent, G0 senescent, or terminally differentiated cells, Id expression is down-regulated (3, 9, 12, 22, 23). By contrast, Id genes in tumor cells display a general pattern of deregulation (1, 12, 62), consistent with the uncoupling of growth and differentiation controls characteristic of malignancy. During traverse of the cell cycle, Id gene expression is induced during the early G1 phase, followed by a second minor peak of induction coincident with late G1 or early S phase (3, 9, 12, 23). Moreover, partial ablation of Id protein expression or function through antisense oligonucleotide blockade (3, 23) or antibody microinjection (43) strategies results in a delayed entry of cells into the S phase of the cell cycle, implicating Id function in the regulation of G1 progression. Such a role in cell cycle control is further substantiated by the finding that certain bHLH transcriptional regulators such as the E2A-encoded E47 (43) and the muscle-determining MyoD (10, 52) proteins, whose functions are antagonized through heterodimerization with Id proteins, are themselves capable of mediating a G1 cell cycle arrest when ectopically overexpressed in cell line models. In common with several other ERG-encoded proteins implicated in G1 cell cycle control, enforced ectopic expression of Id proteins, at least in some cell types, promotes cell growth, in addition to arresting differentiation (2, 16, 25, 35). Finally, the Id2 protein, but not apparently the Id1 or Id3 family members, is capable of abrogating the growth-suppressive functions of the tumor suppressor, p16, p21, and Rb proteins through a direct interaction with the Rb protein (25, 33), which, in its unphosphorylated state, acts as a pivotal negative regulator of cell cycle S phase commitment (4).
We previously showed that transfection of the Id3 gene into established mouse NIH 3T3 cells induces a morphologically transformed phenotype, but this was not accompanied by any other features of oncogenic transformation (12). In further evaluating its oncogenic potential, we report here that the Id3 gene cooperates with the antiapoptotic Bcl2 and BclXL genes to immortalize primary rat embryo fibroblasts (REF). Consistent with this, enforced expression of Id3 (and of Id1 and Id2) in isolation was found to be a strong inducer of apoptosis in serum-deprived REF cells. Id-mediated apoptosis was mediated at least in part through p53-independent mechanisms and could be efficiently rescued by Bcl2, BclXL, and the bHLH protein E47. In addition, using various loss- and gain-of-function Id3 mutants, we found that the induction of apoptosis was invariably correlated with the ability of Id3 to promote the cell cycle S phase. These findings highlight the close coupling between the G1 progression and apoptosis functions of Id proteins and indicate a common mechanism for this family of transcriptional regulators in cell determination.
MATERIALS AND METHODS
Cell culture.
NIH 3T3 cells were maintained in Dulbecco modified Eagle medium supplemented with 10% newborn calf serum (NBCS). Primary REF were prepared from day 14 rat embryos (32). Primary mouse embryo fibroblasts (MEF) from day 16 to 18 wild-type and p53−/− mice were prepared as described previously (60) and were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum. All primary cells were cryopreserved and used for experiments within two or three passages of isolation. Cos7 cells and RPMI 8225 cells (obtained from the American Type Culture Collection, Rockville, Md.) were maintained in RPMI medium supplemented with 10% fetal calf serum.
Plasmid and vector constructs.
Full-length cDNAs encompassing the respective coding regions of human Id1 (15), Id2 (14), and Id3 (12) together with Id3 Ala5 and Id3 Asp5 mutants (13) were cloned into the vector pcDNA3 (Invitrogen) to generate pcDNAId vectors for use in transient transfections. For some stable transfections, wild-type Id3 cDNA was cloned into the vector pcDNA1 and used in conjunction with pSV2Neo, providing a selectable marker. Additionally, the Id3 cDNA was cloned into the vector pUHD10-1 (20) to generate the tetracycline transactivator (tTA)-responsive vector pUHId3 and was used in conjunction with pJEF33, encoding the tetracycline-regulatable tTA protein (49). Expression constructs carrying c-myc (pDoRG123) (42) and activated ras (pUC EJ6.6) (48) oncogenes were a gift from H. Land (ICRF Laboratory, London, United Kingdom). The Bcl2 expression vector (pMPZenBcl2) (55) was obtained from S. Cory, and the BclXL vector (pSFFV-Bclxl) (7) was obtained from C. Thompson. pCMVE47, encoding the E2A protein E47 (56), was obtained from X.-H. Sun, and the LacZ expression vector, pEQ176 (47), was obtained from M. R. Schleiss.
Transient-transfection analysis.
Subconfluent cultures of embryo fibroblasts in 60-mm-diameter dishes were transfected with a total of 8 μg of DNA by using the standard calcium phosphate procedure, essentially as described previously (12). Typically, transfected DNA comprised 6 μg of Id expression construct and 2 μg of pEQ176 LacZ marker gene. In experiments employing multiple expression constructs (at 3-μg input), the total DNA input was adjusted to 8 μg with empty vector (pcDNA3). After incubation overnight with DNA precipitate, cells were trypsinized and reseeded onto glass slides for a further 24-h culture period. As appropriate, cells were then shifted to low-serum (0.5%) culture medium for a further 18 to 24 h. Cells were pulsed with 10 μM bromodeoxyuridine (BrdU) for 2 h prior to fixation to facilitate analysis of cell cycle S phase. Cells were fixed in 4% paraformaldehyde for 30 min at room temperature and then incubated for a further 1 h at room temperature in blocking solution (5% Marvel) (dried milk in phosphate-buffered saline [PBS]). For immunostaining for LacZ expression, cells were incubated overnight at 4°C with a 1/500 dilution of rabbit LacZ antibody (5 Prime to 3 Prime Inc.). After being washed three times in PBS, slides were treated with a 1/500 dilution of biotin-labelled antirabbit secondary antibody (Dako) for 1 h at room temperature, washed three times in PBS, and then incubated for 1 h with a 1/100 dilution of streptavidin-fluorescein isothiocyanate (FITC) (Dako) at room temperature. In some experiments, cells were immunostained for Id3 expression instead of for LacZ expression (see below). Cells were then treated with 2 M HCl for 20 min at 37°C and immunostained for BrdU incorporation by using a commercial kit (Boehringer) according to the manufacturer’s instructions, except that a Texas red-conjugated antimouse antibody was substituted for the secondary antibody step. Slides were finally mounted in Vectashield mountant containing 4′,6-diamidino-2-phenylindole (DAPI) and were evaluated by fluorescence microscopy. In quantitative analysis, a minimum of 200 LacZ-positive cells were evaluated in each transfection.
Stable transfections.
For focus assays, 50% confluent monolayers of REF cells in 3.5-cm-diameter dishes were transfected with 8 μg of DNA by the calcium phosphate procedure as described above. DNA mixtures comprised 6 μg of each expression construct as appropriate and were normalized to 8 μg with pBluescript carrier DNA (Stratagene). Cells were trypsinized after overnight incubation with precipitates and were seeded into 25-ml flasks (for enumeration of foci) or onto glass slides (for immunostaining). Cells were refed every 3 to 4 days and were evaluated for foci after 3 weeks for Ras- plus Myc-generated foci and after 4 weeks for Id3-generated foci.
Colony-forming efficiency (CFE) assays were performed by transfection of fibroblasts as described above except that 2 μg of pSV2Neo vector was included as a selectable marker. Following transfection, cells were reseeded into 10-cm-diameter dishes. G418 selection (0.15 mg/ml for REF; 0.5 mg/ml for NIH 3T3 cells) was applied the following day, and individual colonies (comprising greater than 100 cells) were scored after a further 2 weeks for NIH 3T3 cells and after 3 weeks for REF cells. Stable transfectant cell lines were established from individual colonies in some experiments. Cells were passaged every 3 days continuously over several months (or until the cells ceased to be viable). Immortalized lines were maintained in 5% NBCS after week 15.
Stable transfectant cell lines were also generated by cotransfecting REF cells either with 3 μg of pUHD10-1 (control vector) (20) or with 3 μg of pUHDId3 together with 3 μg of pJEF33 and 2 μg of pSV2Neo. Cells were trypsinized at 24 h posttransfection and were reseeded into replicate 25-ml flasks and 10-cm-diameter dishes and subjected to G418 selection (0.15 mg/ml) in the presence of 100 ng of anhydrotetracycline (TET) (Janssen Chimica Co.) per ml. After a further 3 weeks, pools of cell clones (derived from at least 200 colonies) were subjected to further analysis. Individual cell colonies were picked from dishes and expanded for analysis of Id3 expression. TET suppression of expression was maintained throughout unless otherwise stated.
Immunostaining and Western analysis of Id3 protein.
Cells grown on microscope slides were fixed in 4% paraformaldehyde for 30 min at room temperature and then incubated with 5% Marvel evaporated milk blocking solution in PBS for a further 1 h. Incubation with primary anti-Id3 rabbit polyclonal antibody, RD1 (11), was performed for 16 h at 4°C with a 1/400 dilution of antiserum (diluted in PBS supplemented with 3% bovine serum albumin). Slides were washed in PBS, incubated with a 1/100 dilution of FITC-conjugated antirabbit secondary antibody (Dako) for 1 h at room temperature, and then washed in PBS prior to evaluation by fluorescence microscopy. To demonstrate the specificity of Id3 staining in selected experiments, a homologous peptide competitor (PIQTAELAPELVISNDKRSF) or heterologous peptide competitor (ISALAAEAACVPADDRIL) at a concentration of 200 μg/ml was used to block the RD1 primary Id3 antibody prior to addition to fixed cells. For Western analysis, cells were harvested by lysis in radioimmunoprecipitation assay buffer (50 mM Tris [pH 8.0], 150 mM sodium chloride, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate), followed by electrophoresis on a 12% polyacrylamide–sodium dodecyl sulfate gel and Western blotting and probing with RD1 anti-Id3 antibody, essentially as described previously (2). Western analysis for Bcl2 and BclXL proteins was performed following electrophoresis on 12% polyacrylamide gels. For Bcl2, a mouse monoclonal antibody (Dako) was used at a dilution of 1/250, followed by incubation with horseradish peroxidase-conjugated rabbit antimouse antibody (Dako) at a dilution of 1/2,000. For BclXL, a rabbit polyclonal antibody (Transduction Labs) was used at a dilution of 1/100, followed by incubation with horseradish peroxidase-conjugated swine antirabbit antibody at a dilution of 1/2,000. Horseradish peroxidase activity was detected by using an ECL chemiluminescence kit (Amersham Life Sciences, Amersham, United Kingdom) as described by the manufacturer.
Analysis of apoptosis and cell cycle S phase in stable transfectants.
Analysis of apoptosis from nuclear morphology (DAPI staining) and determination of cell cycle S phase by BrdU incorporation in individual cell clones was performed exactly as described above for transient transfections except that the step for detection of LacZ marker gene expression was omitted. Pooled cell populations were evaluated for coexpression of exogenous Id3 protein and DAPI-stained nuclear morphology as described for transient transfections.
Apoptotic cells were also monitored by an in situ end-labelling (ISEL) procedure utilizing DNA polymerase I to label DNA ends generated in fragmented nuclei (59). This used digoxin-labelled deoxynucleoside triphosphates and detection with anti-digoxigenin peroxidase and diaminobenzene. Slides were counterstained with hematoxylin.
Detection of early apoptotoic cells (following serum deprivation for 2 h) by monitoring cell surface expression of phosphatidylserine was performed by staining with an FITC conjugate of annexin V (38) by using a commercial kit (Clontech Labs Inc.) essentially as described by the manufacturer. Cells were counterstained with propidium iodide (PI). pUHId3-transfected cells were also incubated in low-serum medium for 24 h prior to PI staining to detect late apoptotic cells and as a positive control for PI staining.
RESULTS
Transformation of primary embryo fibroblasts by Id proteins.
We evaluated the oncogenic potential of Id genes by using the sensitive assay of primary REF cells (32). As shown in Fig. 1, transfection of the pcDNAId3 expression vector alone into REF cells led to the appearance of morphologically distinct foci of transformed cells which immunostained strongly positively for Id3 protein. Similar foci were also induced by the Id1 and Id2 genes (data not shown). To determine whether the Id3 gene could cooperate with other oncogenes in transforming primary cells, the number of foci induced by pcDNAId3 in combination with Ras- or Myc-expressing constructs was evaluated, as shown in Fig. 2. Whereas a combination of Id3 plus Ras led to a marginal (30%) increase in the number of foci compared with that induced by Id3 alone, Id3 plus Myc resulted in a marginal (30%) suppression in focus formation. In both cases the morphology and time of appearance of foci were indistinguishable from those seen with Id3 in isolation. The foci were also clearly distinct from the ill-defined foci induced by Ras or Myc alone (data not shown). These results contrasted markedly with the approximately 10-fold increase in the number of foci induced by a combination of Myc plus Ras (Fig. 2). Consistent with previous studies (32), these foci appeared with a shorter latency period (3 weeks), were faster growing, and displayed a highly refractive morphology comprised of rounded cells which readily detached from the culture dish (data not shown). Numerous attempts to derive established lines from REF cells transfected with Id3 either alone or in combination with Ras or Myc were unsuccessful. Either the cells underwent senescence after several passages or, when stable lines were obtained, they failed to express levels of Id3 mRNA and protein above those seen in controls (data not shown). This finding suggested that ectopic expression of Id3, although capable of stimulating growth to the extent that morphologically distinct foci were induced in monolayer cultures, was not compatible with long-term survival of these primary cells. Moreover, this failure to grow out as stable transformants could not be compensated by cooperative functions supplied by either Ras or Myc oncogenes.
FIG. 1.
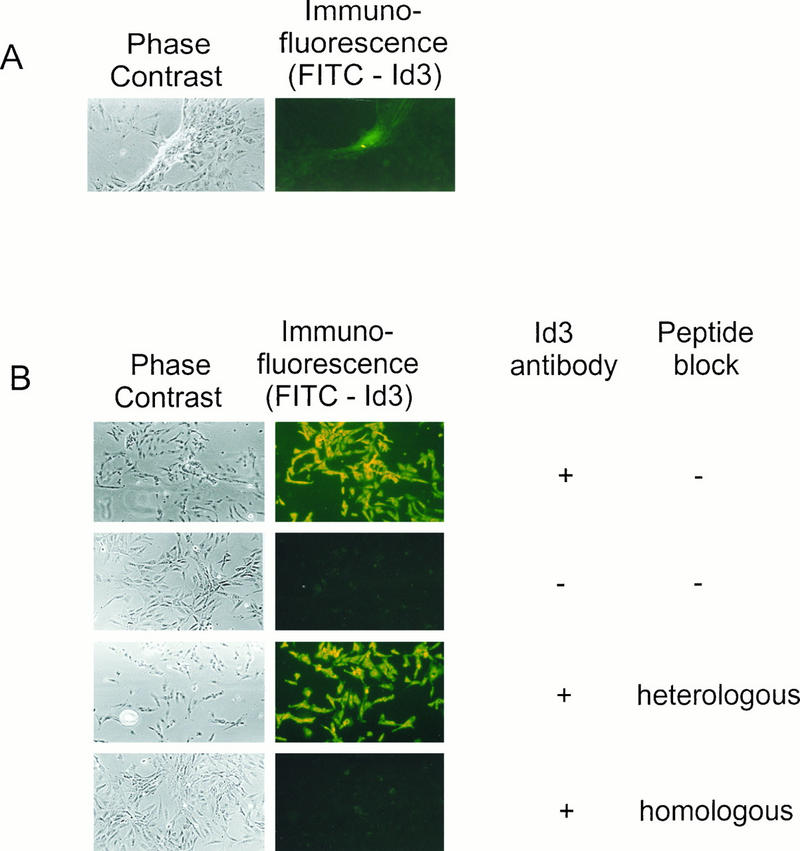
Morphology of foci induced by Id3 transfection of REF cells. (A) Early-passage primary REF cells were transfected with pcDNAId3 vector and scored for morphologically transformed foci after 3 to 4 weeks. The phase-contrast photograph illustrates the morphology of a typical Id3-induced focus of transformed cells, and the immunofluorescence photograph shows high levels of Id3 protein associated with the focus in the same field of view. Cells transfected with a control, empty vector (pcDNA3) did not give rise to such distinctive Id3-expressing foci (data not shown). (B) An individual Id3 focus was picked, and the cells were replated onto slides. To demonstrate the specificity of detection of Id3 protein, the cells were subjected to immunostaining either with or without primary antibody (in combination with FITC-labelled secondary antibody) and with either a homologous or heterologous competitor peptide as shown.
FIG. 2.

Induction of foci by Id3 in combination with ras and c-myc oncogenes. Replicate 3.5-cm-diameter dishes of primary REF cells were transfected with the indicated expression constructs for Id3 (pcDNAId3), Ras (pUC EJ6.6) and c-Myc (pDoRG123). Input DNA was normalized to 8 μg for all transfections with pBluescript carrier DNA. Numbers of foci were scored after 2 to 3 weeks for Ras plus c-Myc or after 5 weeks for all other transfections. The data from replicate analyses are representative of several independent experiments. Error bars indicate standard deviations.
The antiapoptotic Bcl2 and BclXL genes cooperate with Id3.
The apparent poor survival of Id3-expressing cells and/or their intolerance of exogenous Id3 was intriguing, since we and others previously reported that stable transformants of established NIH 3T3 fibroblast lines could be readily generated following transfection with Id genes (12, 43). We further evaluated this issue by employing a colony survival assay with either NIH 3T3 cells or REF cells as recipients for transfection, as shown in Fig. 3. Consistent with previous data (12, 43), transfection of Id3 into established NIH 3T3 cells did not significantly affect the number of surviving colonies (Fig. 3) or the growth rate of transfected cells, although the majority of colonies were comprised of highly refractive cells as reported in our earlier studies (reference 12 and data not shown). By contrast, the number of surviving colonies of REF cells was suppressed approximately 10-fold following transfection with Id3 (Fig. 3), consistent with the apparent intolerance of these cells to exogenous Id3 alluded to above. We also noted that many of the colonies generated from Id3-transfected REF cells grew poorly and had a propensity to detach from the culture dish, with widespread cell death. This observation prompted us to investigate whether cotransfection of the cell survival Bcl2 and BclXL genes (29, 57) could rescue the Id transfectants. However, overexpression of Bcl2 and BclXL family members themselves has previously been shown to be growth suppressive and/or to reduce survival in several cell types (41). Consistent with this, transfection with the Bcl2 expression vector alone suppressed colony formation of REF cells to the same extent as did Id3 (Fig. 3). The suppressive effect of BclXL transfection in isolation was less marked than this, reducing CFE by around 60% (Fig. 3). In marked contrast, when either of these two genes was transfected in combination with the Id3 vector, the CFE was stimulated 10-fold compared with that seen with the Id3 vector alone and indeed was restored to the level seen in cells transfected with control vector (Fig. 3). Moreover, individual colonies from Id3- plus Bcl2- or from Id3- plus BclXL-transfected cells readily established rapidly proliferating immortalized cell lines, in contrast to cells transfected with each of these genes in isolation (Table 1). These stable transformants expressed readily detectable Id3 protein (Fig. 4) and have been maintained continuously in culture for several months.
FIG. 3.

Effect of Id3 transfection on CFE. Replicate 3.5-cm-diameter dishes of subconfluent NIH 3T3 cells or primary REF cells were transfected with pSV2Neo vector in combination with Id3 vector (pcDNA1Id3), Bcl2 vector (pMPZenBcl2), or BclXL vector (pSFFV-Bclxl). All transfections were normalized to 8 μg with pBluescript carrier DNA. At 24 h posttransfection, cells were trypsinized and replated in 10-cm-diameter dishes and G418 selection was applied for 2 to 3 weeks, at which time colonies comprised of greater than 100 cells were enumerated. Error bars indicate standard deviations.
TABLE 1.
Immortalization of primary embryo fibroblasts by Id3 in combination with Bcl2 or BclXLa
| Transfectant | No. of surviving cultures (n = 4) at wk: | |||
|---|---|---|---|---|
| 11 | 13 | 15 | 18 | |
| Control | 2 | 2 | 1 | 0 |
| Id3 | 2 | 1 | 1 | 0 |
| Bcl2 | 2 | 2 | 1 | 0 |
| Id3 + Bcl2 | 4 | 4 | 4 | 4 |
| BclXL | 4 | 4 | 2 | 0 |
| Id3 + BclXL | 4 | 4 | 4 | 4 |
FIG. 4.

Detection of Id3 and Bcl proteins in immortalized REF transfectants. Western blot analysis was performed on cell lysates from two cell clones (cl2 and cl4) transfected with Id3 plus Bcl2 vectors and from two clones (cl6 and cl7) transfected with Id3 plus BclXL vectors. The expression levels of Id3 in transfectants were compared with those of endogenous Id3 protein from control REF cells and from Cos cells either mock transfected (Cos) or transfected with Id3 vector (Cos plus Id3). The expression levels of Bcl2 and BclXL proteins in immortalized clones were similarly compared with endogenous levels of these proteins in control REF cells and in Cos cells either mock transfected or transfected with the respective Bcl vector. An additional positive control from the human B-cell line RPMI 8225 was included in the analysis of Bcl2 protein.
We conclude from these experiments that Id3 cooperates with the Bcl2 and BclXL oncogenes to immortalize primary REF cells. Since the distinguishing property of the Bcl2 and BclXL genes is their ability to rescue cells from apoptosis (29, 57), these data also suggested that our earlier failure to generate established lines with Id3 in isolation was attributable to an ability of this gene, when overexpressed, to promote apoptosis. It should be noted, however, that because of the considerable cell death in control cells that occurred during drug selection in the CFE assay (Fig. 3) and during continual passage of control REF cells in the immortalization assay (Table 1), we were unable to demonstrate directly that Id3 promotes apoptosis under these experimental conditions.
Id genes induce apoptosis in serum-deprived fibroblasts.
Enforced overexpression of all three well-characterized Id genes, Id1, Id2, and Id3, has been reported to promote cell growth in some but not all cell types (2, 13, 16, 25, 35). Typically, this is manifested by the ability of Id-transfected cells to continue cycling on withdrawal of mitogenic growth factors, as evidenced by the percentage of cells in S phase (2, 13). We reasoned that by analogy with other growth-promoting ERGs which also induce apoptosis, such as c-myc (17, 18) and the cyclin D gene (31, 51), these experimental conditions would also be most conducive for observing any apoptotic effects elicited by overexpression of Id genes. As depicted in Fig. 5A, transient transfection of expression constructs carrying the three Id genes into REF cells led to an accumulation of a significant fraction of cells in S phase following serum deprivation. Similarly, when the nuclear morphology of Id transfectants under these conditions was assessed by staining with DAPI, all three Id-transfected lines displayed evidence of apoptotic nuclei (Fig. 5B). Significantly, the S phase and presumptive apoptotic cell populations in these experiments were largely coincident (Fig. 5C). By contrast, no significant apoptosis above the level in control transfectants was observed in the presence of serum (data not shown).
FIG. 5.
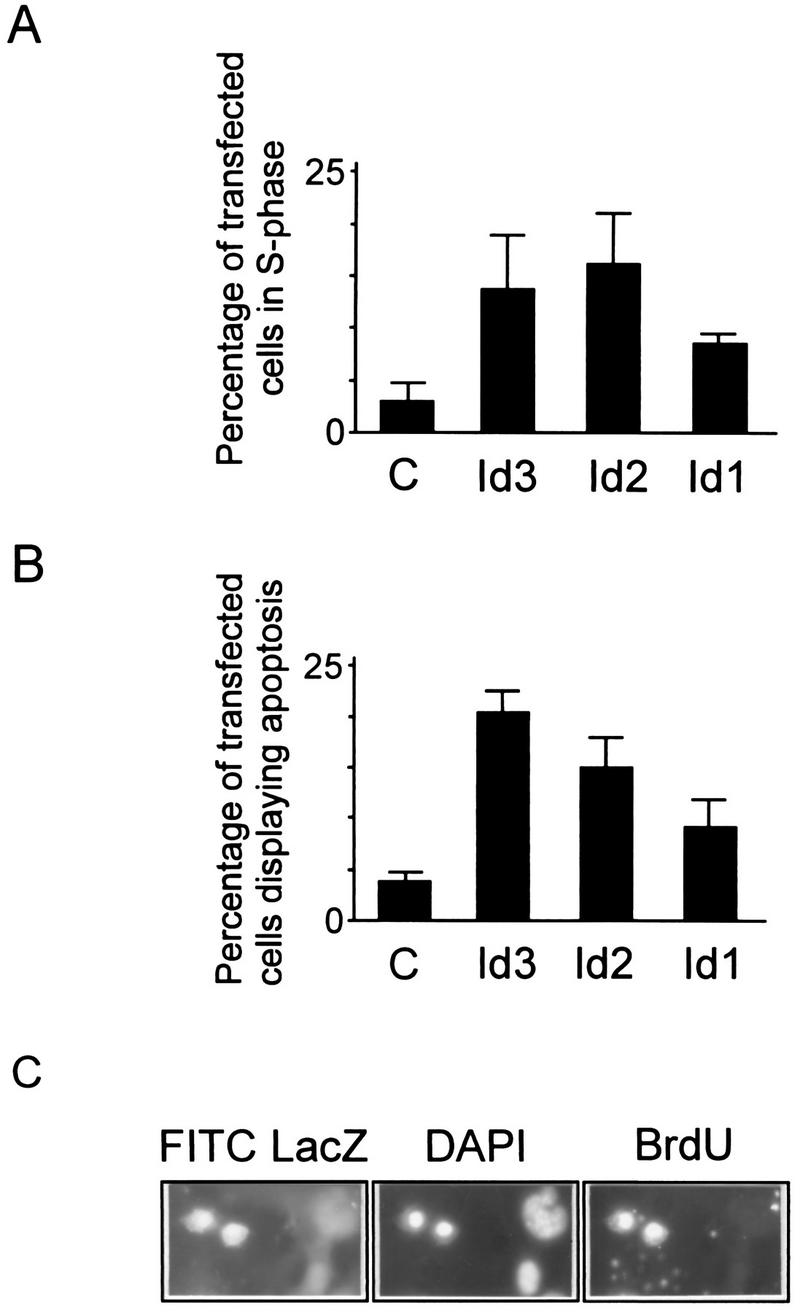
Induction of apoptosis by transient transfection of Id genes. Subconfluent replicate monolayers of primary REF cells in 3.5-cm-diameter dishes were transfected with the LacZ-expressing marker gene pEQ176 in combination with carrier DNA alone (bars C) or with each of the Id vectors as indicated. At 36 h posttransfection, cells were deprived of serum for 24 h and then pulse-labelled with BrdU, after which plates were fixed and immunostained for LacZ expression and for BrdU incorporation and the DAPI stained for evaluation of nuclear morphology. At least 200 LacZ-positive cells were scored for each transfection. Panel C illustrates the appearance of typical fragmented nuclei revealed by DAPI staining in LacZ-positive (Id3-expressing) cells that were also BrdU positive.
To obtain more conclusive evidence for this Id-mediated apoptosis, we derived stable transfectants in which the Id3 gene was conditionally expressed under control of the tTA protein, facilitating negative regulation by the drug TET (20, 49). By maintaining cells in TET-containing medium throughout selection and subsequent propagation, we could obviate the potential toxicity caused by exogenous Id3 expression. Initially, we examined polyclonal populations of such transfectants. Because these cells were generated by cotransfection with three different plasmid vectors (pUHId3, under control of the tTA protein; pJEF33, encoding the tTA protein itself; and pSV2Neo, to facilitate selection), only a fraction of the resulting G418-resistant population (10 to 20% of cells) expressed detectable exogenous Id3 protein when assessed by immunofluorescence (Fig. 6A). This expression was almost totally suppressed when cells were cultured in the presence of the drug TET (Fig. 6A and B). When the Id-expressing transfectants were cultured in the presence of serum, the nuclear morphology of Id3-expressing cells as assessed by DAPI staining appeared normal, whereas on removal of serum for 24 h, the Id3-expressing cells, but not nonexpressing cells in the same population (or cells in control transfectants), acquired a highly condensed nuclear morphology, characteristic of apoptotic cells (Fig. 6A). These experiments with cells stably transfected with inducible Id3 vector essentially confirm the results obtained with transient transfections. As additional criteria of apoptosis, the cell population stably transfected with Id3 stained positive following ISEL analysis (59) and annexin labelling (38) when cultured in the absence of TET and serum (Fig. 6C). As with the DAPI analysis, no significant staining with these markers was observed in Id3 transfectants cultured in the presence of TET or in control, empty vector transfectants in the absence of TET (Fig. 6C).
FIG. 6.
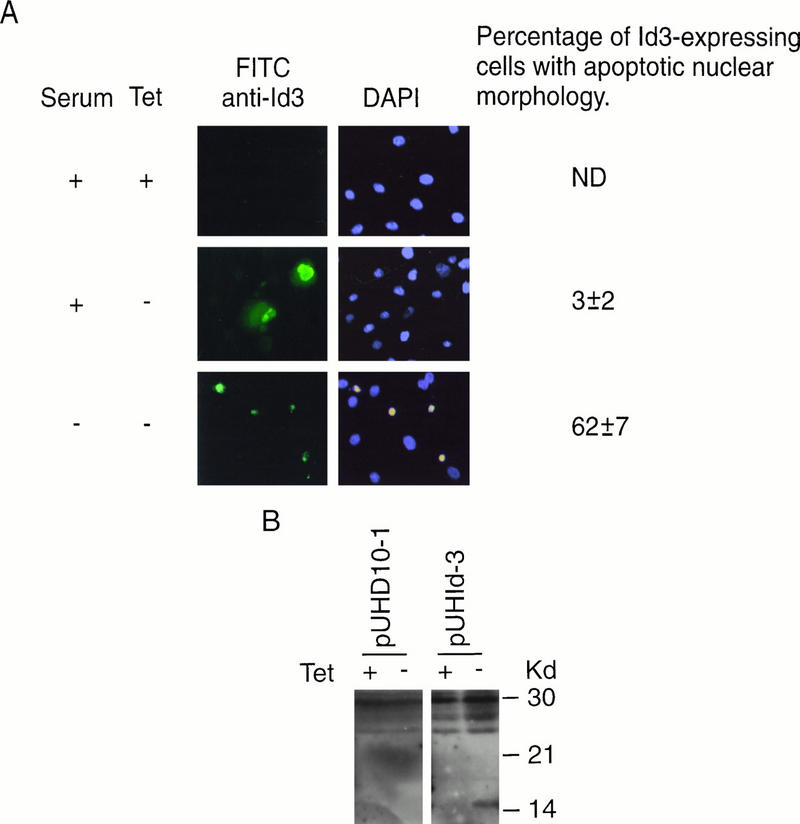

Id3 overexpression induces apoptosis in stable transfectants. (A) Primary REF cells stably transfected with the Id3 vector pUHD-Id3 and then selected as a polyclonal population were seeded onto slides and cultured in either the presence or absence of TET for 2 days. An additional parallel culture was deprived of serum during the second day of the experiment. Cells were immunostained by using anti-Id3 antibody (FITC) and counterstained with DAPI for evaluation of nuclear morphology. The photograph shows representative fields of view for each of the culture conditions. The percentage of Id3-positive cells displaying an apoptotic nuclear morphology was assessed from evaluation of at least 200 Id3-positive cells under each set of culture conditions. Control, empty vector (pUHD10-1)-transfected cell populations assessed in parallel displayed only low (endogenous) levels of Id3 protein expression and did not exhibit significant apoptosis under the different growth conditions used in the experiment (data not shown). From the number of G418-resistant colonies obtained following drug selection, the transfection efficiency of these cells was comparable to that obtained with pUHDId3. ND, not determined since there were too few cells with levels of Id3 protein significantly higher than the endogenous level to permit evaluation. (B) Lysates from cells transfected with either empty vector control (pUHD10-1) or pUHId3 and cultured in the presence or absence of TET for 48 h were analyzed by Western blotting for Id3 protein, migrating as a 15-kDa species. (C) Cells stably transfected with control empty vector (pUHD10-1) or with the Id3 vector pUHDId3 were seeded onto microscope slides and were cultured in low-serum medium for up to 24 h. Where indicated, TET was removed from cultures 24 h prior to the shift to serum deprivation conditions. Cells were stained for ISEL at 24 h and for annexin (in parallel with PI) after 2 h of serum deprivation. As a marker of late apoptosis and as a positive control, pUHDId3-transfected cells were further stained with PI after 24 h of serum deprivation. For pUHDId3-transfected cells cultured in the absence of TET (permissive for inducible Id3 expression [see panel A]), 5 to 10% of the total population of serum-deprived cells was apoptotic by ISEL and annexin analysis in several experiments (400 to 500 cells were evaluated). By contrast, fewer than 0.5% of pUHDId3-transfected cells grown in the presence of TET or pUHD10-1 (control) cells grown in the absence of drug were apoptotic.
To confirm and extend these findings, we also isolated a number of single clones of Id3 transfectants from these polyclonal populations. As shown in Fig. 7, three clones which expressed exogenous Id3 were identified in this way. In two cell clones, Trld3-4 and Trld3-12, expression of Id3 protein was TET repressible, whereas in the third (Trld3-13), the expression of exogenous protein was essentially constitutive, with only a marginal induction on removal of TET. The immunofluorescence data in Fig. 7 also illustrate the predominantly perinuclear intracellular distribution of exogenous Id3 protein, as reported in previous studies (11).
FIG. 7.
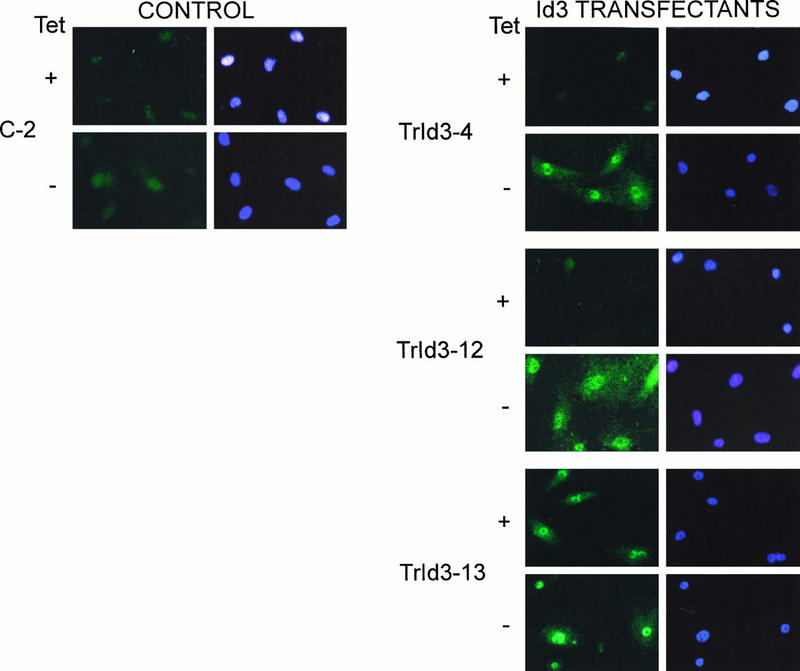
Detection of exogenous Id3 protein in stable transfectant clones. Cells from a control transfectant (transduced with empty vector [pUHD10-1]) and from the three Id3 transfectants indicated were seeded on microscope slides and cultured in the presence or absence of TET for 48 h prior to fixation and immunostaining with RD1 anti-Id3 antibody and counterstaining with DAPI as a cell/nuclear marker.
Cultures of all three transfectant clones died after 1 to 2 days following removal of serum in the absence of TET, with virtually all cells rounding up and forming cytoplasmic protrusions prior to detachment, in a manner typical of apoptosis (data not shown). In the two clones Trld3-4 and Trld3-12, this cell death was TET suppressible. To more empirically evaluate the apoptotic responses of these Id3 transfectants, we analyzed the three clones in parallel with two control clones (transduced with empty vector, pUHD10-1) by DAPI staining and ISEL analysis, as shown in Fig. 8. The apoptotic responses in Id3 clones Trld3-4 and Trld3-12 as assessed by both criteria were strongly regulatable by TET, whereas in Trld3-13, expressing constitutive levels of Id3, the apoptotic response was largely independent of the presence of the drug.
FIG. 8.

TET-regulated apoptotic response in Id3-transfected cell clones. Replicate cultures of two control cell lines and three Id3 transfectants on slides were incubated in the presence or absence of serum, having been removed from TET-containing medium 48 h prior to this where indicated. Cells were assessed by ISEL analysis at 24 h or by DAPI staining at 24 h, and the results were evaluated for at least 100 cells in each culture. Data for apoptotic response in the presence of serum are not shown, since in all cases this was minimal (less than 5%). Error bars indicate standard deviations.
Mechanisms of Id-induced apoptosis.
Recent studies have shown that cyclin A- and cyclin E-dependent Cdk2 phosphorylation of the Id2 and Id3 proteins at a conserved serine residue at position 5 modulates the ability of these proteins to antagonize bHLH-dependent gene expression (21) and abrogates their function in promoting cell cycle S phase in serum-deprived fibroblasts (13). To determine whether this cell cycle-regulated phosphorylation of Id proteins affects their apoptotic functions, we transfected phospho-ablation (Ala5) and phospho-mimicking (Asp5) mutants of the Id3 expression construct into REF cells and then subjected the cells to serum deprivation. In addition to empty vector and wild-type Id3 controls, we also included another Id3 mutant, the Pro49 mutant, in which substitution of a proline residue in the H1 domain severely impairs its ability to heterodimerize with bHLH protein targets (11, 58). The expression levels of wild-type and mutant proteins were comparable in transfected cells (data not shown). As shown by the experiment in Fig. 9A, whereas the Asp5 mutant had a much-reduced function in promoting cell cycle S phase (comparable to that of the Pro49 mutant), the phospho-ablation Ala5 mutant exhibited a gain-of-function effect compared with wild-type Id3, a finding consistent with our previous studies (13). When the cells in these transfection experiments were DAPI stained for nuclear morphology (Fig. 9B), the apoptotic responses exactly mirrored those of the cell cycle S phase. This implies that the Id determinants and cell cycle-linked mechanisms responsible for promoting the G1-to-S phase transition are tightly coupled to those driving apoptosis.
FIG. 9.

Effect of phospho-ablation and phospho-mimicking mutants of Id3 on cell cycle S phase and apoptotic responses. Replicate subconfluent cultures of primary REF cells were transfected with pEQ176 LacZ vector together with either the control, empty vector (pcDNA3) or expression constructs encoding wild-type Id3 (WT) or Id3 mutants (Ala5, Asp5, and Pro49). At 24 h posttransfection, cells were shifted to low-serum medium, and after a further 24 h they were pulsed with BrdU and then fixed and immunostained for LacZ expression and BrdU incorporation and counterstained with DAPI. At least 200 LacZ-positive cells were evaluated in each transfection for BrdU positivity (A) and for apoptotic nuclear morphology (B). C, control. Error bars indicate standard deviations.
To further investigate the mechanisms of Id-induced apoptosis, we evaluated how the presence of other gene products affected the ability of Id3 to induce apoptosis in serum-deprived fibroblasts. Consistent with the data obtained in CFE and cell transformation assays (Fig. 3 and Table 1), cotransfection of Id3 with constructs carrying the Bcl2 and BclXL oncogenes efficiently rescued cells from Id3-induced apoptosis (Fig. 10A). The bHLH protein E47 is known to arrest the G1-to-S phase transition of cell cycle progression (43) and, as a principle bHLH target for Id proteins, effectively opposes their functions (2, 5, 26, 39, 43, 53, 54, 61). Previously, we showed that in cells cotransfected with Id3 and E47 vectors, the Id3 protein is associated with E47 as a stable heterodimer in coimmune precipitates and that the E47 protein is in functional excess (11). Consistent with this, when cells were cotransfected with a combination of E47 and Id3 vectors, only 5% of the cells were found to be in S phase, compared with 15% for cells transfected with Id3 alone (Fig. 10B). As shown in Fig. 10A, cotransfection of an E47 construct was almost as efficient as Bcl2 or BclXL in rescuing cells from Id3-driven apoptosis. Finally, we investigated whether Id3-induced apoptosis is mediated through p53-dependent or p53-independent mechanisms by comparing the responses of wild-type and p53−/− primary MEF to enforced expression of Id3. These experiments were complicated by the greater propensity of MEF cells than of REF cells to detach from the culture dish and/or to undergo spontaneous apoptosis in serum-depleted medium (34). However, as shown in Fig. 10A, wild-type and p53−/− MEF cells displayed comparable susceptibilities to Id3-induced apoptosis, a finding which implicates mechanisms that are at least in part independent of p53 function.
FIG. 10.

Effect of apoptosis-regulatory gene products on Id3-induced apoptosis and cell cycle S phase. Replicate subconfluent cultures of either primary REF cells or primary MEF cells (from wild-type [WT] or p53−/− mice) were cotransfected with pEQ176, encoding the LacZ marker, either alone or in combination with Id3, Bcl2, BclXL, or E47 expression constructs as indicated. At 24 h posttransfection, REF cells were shifted to low-serum medium for 24 h, while MEF cells were serum shifted for only 18 h to minimize cell loss through detachment from the monolayer. Both sets of cells were pulsed with BrdU for 2 h prior to fixation and immunostaining for LacZ expression, BrdU incorporation, and DAPI counterstaining for evaluation of nuclei with apoptotic morphology. Error bars indicate standard deviations.
Since the experiments in Fig. 9 showed that Cdk2-dependent phosphorylation determinants could not dissociate the apoptotic from the G1-to-S cell cycle functional effects of Id3, we also examined how the transfection experiments in Fig. 10, affected cell cycle S phase. As can be seen from Fig. 10B, the profile of S phase responses elicited by overexpression of Id3 closely mirrored the apoptotic responses (Fig. 10A). This observation further highlights the close association between the mechanisms of Id function driving cell cycle progression into S phase and those driving apoptosis.
DISCUSSION
A number of positive regulators of G1 cell cycle progression, such as E2F (27, 28, 50) and cyclin D1 (24, 37), have recently been reported to display oncogenic potential in transformation assays of primary embryo fibroblasts. Indeed, several of the classical, mitogen-responsive proto-oncogenes exemplified by c-myc, c-fos, and c-jun are themselves known to be intimately involved in the regulation of G1 cell cycle progression (40). Since enforced ectopic expression of Id genes has been reported to promote growth in at least some cell types (2, 16, 25, 35), we reasoned that they might also be capable of functioning as dominant oncogenes. However, our previous attempts to demonstrate oncogenic potential of Id genes by transfection of established fibroblast cell lines revealed a capacity to induce a transformed cell morphology but none of the other features characteristic of malignant transformation (12). Moreover, this response could not be further augmented by cotransfection with other oncogenes (1b). By contrast, using a more sensitive primary embryo fibroblast transformation assay (32), we have now found that transfection of Id genes alone readily induced foci in monolayer cultures but that these failed to grow out as immortalized lines unless supplied with cooperating functions provided by the antiapoptotic Bcl2 or BclXL gene. Neither of the two dominant oncogenes, c-myc and ras, significantly affected the response seen with Id3 alone. Interestingly, Hara et al. (22) previously reported that the Id1 gene is capable of inducing a small but significant DNA synthesis response in senescent human fibroblasts when cotransfected with an attenuated mutant of the simian virus 40 T antigen. Such an observation is consistent with the ability of enforced overexpression of Id genes to impart immortalization functions under appropriate conditions. In this regard, it will be of interest to determine if Id genes can cooperate with a spectrum of other classes of viral and cellular oncogenes in immortalization of primary rodent fibroblasts and how this compares with the responses seen for other G1 cell cycle regulators, since this might provide important clues about mechanisms of function.
The observations that Id3-overexpressing REF cell lines could be readily established by cotransfection with the antiapoptotic Bcl2 and BclXL oncogenes and that the suppression in CFE induced either by Id3 alone or by the Bcl2 and BclXL genes themselves could be reversed by cotransfection of the two genes in combination suggested that Id3 in isolation might induce cell death. This would account for our inability to propagate Id3-transfected REF cell lines and would also explain the reported difficulties encountered by several laboratories in obtaining cell lines stably expressing high levels of Id genes (2, 26). Evidently, this is a cell-type-specific phenomenon, since a number of established cell lines besides NIH 3T3 cells tolerate high levels of Id gene expression without discernible effects on cell growth or survival characteristics (1a, 12, 43). In addition, several of the other aforementioned positive regulators of early G1 cell cycle progression which, like Id proteins, function as cooperating oncoproteins, are also capable of promoting apoptosis under appropriate growth conditions (30, 31, 44, 51). In evaluating this functional property of Id proteins, we found that in both transient- and stable-transfection assays, Id3 promotes apoptosis in serum-deprived REF cells. In transient assays we also demonstrated a similar effect of the Id1 and Id2 genes. Besides assessing apoptotic cells from DAPI-stained nuclear morphology, we also established the apoptotic nature of Id3-induced cell death in stable transformants by the independent criteria of cell morphology, ISEL analysis of DNA fragmentation (59), and detection of membrane phosphatidylserine by staining with annexin V (38).
Induction of apoptosis by several other positive regulators of G1 cell cycle progression and by viral and cellular oncogenes is mediated through either p53-dependent or p53-independent pathways (57). For example, the prototype c-myc oncogene induces apoptosis (18), at least in part through a p53-dependent pathway coupled to induction of the Cdc25A phosphatase gene (19). Apoptosis in this case is evidently independent of the ability of this oncogene to activate Cdk activity involved in cell cycle progression, which is apparently dispensable for Myc-induced apoptosis (46). Our observation that the extent of Id3-induced apoptosis in p53−/− MEF cells was comparable to that seen in wild-type MEF cells argues for a p53-independent mechanism, perhaps analogous to that used by cyclin D1 (31, 51), which shares many of the functional biological properties of the Id proteins. However, the cyclin D1 and Id proteins do differ in one important functional attribute. Whereas cyclin D1 cooperates with Ras to immortalize primary REF cells (37), Id proteins do not.
Induction of apoptosis by ectopic overexpression of some ERGs, such as c-myc (46) and c-jun (8), is dissociable from the ability of these genes to promote DNA synthesis. However, two observations in our study suggest a close coupling between the pathways through which Id proteins promote the G1-to-S phase cell cycle transition and those which mediate apoptosis. First, the extent of Id-induced apoptosis in different experiments was invariably correlated with the relative magnitude of cell cycle S phase promotion. Second, the Id-expressing cell populations displaying apoptosis in low-serum medium and those in S phase were largely coincident, a phenomenon which has been previously reported for E2F-1- and cyclin D1-induced apoptosis (30, 44, 51). This close association between the cell cycle and apoptosis function of Id proteins was further highlighted by the functional properties of the Id3 serine 5 phospho-ablation and phospo-mimickry mutants. Cyclin A- and cyclin E-dependent phosphorylation of the conserved serine 5 residue in the Id3 and Id2 proteins during the G1-to-S phase transition of the cell cycle alters their bHLH target protein specificities, and phospho-mimickry mutants of these Id proteins have a severely impaired ability to promote cell cycling in serum-deprived fibroblasts (13, 20). In the present study, this mutant of Id3 was correspondingly attenuated in its ability to promote apoptosis. This implies that the unphosphorylated form of Id3 (normally present throughout early G1) but not the phosphorylated form (persisting from late G1 throughout S phase of the cell cycle) drives apoptosis. Consistent with this, prevention of Cdk2-dependent phosphorylation of transfected Id3 by using the Id3 Ala5 mutant significantly enhanced both the cell cycling and apoptotic responses. A lowering of functional Cdk2 activity and consequent hypophosphorylation of Id protein in serum-deprived fibroblasts might therefore act to trigger apoptosis induced by overexpression of Id genes. Further experiments will be required to address this question.
The precise mechanisms through which the different Id proteins function in G1 cell cycle progression are currently enigmatic. Only the Id2 protein is reportedly capable of direct functional antagonism of the Rb, p107, and p130 checkpoint control pocket proteins (22, 25, 33), yet all three of the well-characterized Id family members are capable of promoting cell growth (2, 13, 16, 25, 35) and are demonstrably involved in G1 progression (3, 23, 43). Several lines of evidence do, however, implicate bHLH-dependent mechanisms in Id cell cycle function (43). It is noteworthy, therefore, that cotransfection with the E47 bHLH gene was almost as efficient as that with the Bcl2 and BclXL genes in rescuing Id3-induced apoptosis and that the Pro49 Id3 mutant, which is severely impaired in its ability to heterodimerize with bHLH partners, was also attenuated in apoptotic and growth-promoting functions. We speculate that, in addition to inhibiting bHLH-dependent expression of genes associated with terminal cell differentiation, Id functions are integrated in the cell cycle machinery through a common pathway involving bHLH-dependent mechanisms that regulate G1 progression, apoptosis, and oncogenesis.
ACKNOWLEDGMENTS
We thank S. Cory for the Bcl2 expression construct, C. Thompson for the BclXL vector, X.-H. Sun for the E47 vector, H. Land for the ras and c-myc oncogene expression vectors, and M. R. Schleiss for the LacZ expression vector. We are also indebted to L. Peterson for assistance with Western analysis, to G. Craggs and S. Harran for preparation of embryo fibroblasts, and to R. Deed, G. Peters, and M. Dexter for helpful discussions and for critical reading of the manuscript.
This work was supported by the UK Cancer Research Campaign.
REFERENCES
- 1.Andres-Barquin P J, Hernandez M-C, Hayes T E, McKay R D G, Israel M A. Id genes encoding inhibitors of transcription are expressed during in vitro astrocyte differentiation and in cell lines derived from astrocytic tumours. Cancer Res. 1997;57:215–220. [PubMed] [Google Scholar]
- 1a.Atherton, G., and J. Norton. Unpublished observations.
- 1b.Atherton, G., H. Travers, and J. Norton. Unpublished observations.
- 2.Atherton G T, Travers H, Deed R W, Norton J D. Regulation of cell differentiation in C2C12 myoblasts by the Id3 helix-loop-helix protein. Cell Growth Differ. 1996;7:1059–1066. [PubMed] [Google Scholar]
- 3.Barone M V, Pepperkok R, Peverali F A, Philipson L. Id proteins control growth induction in mammalian cells. Proc Natl Acad Sci USA. 1994;91:4985–4988. doi: 10.1073/pnas.91.11.4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol. 1996;8:805–814. doi: 10.1016/s0955-0674(96)80081-0. [DOI] [PubMed] [Google Scholar]
- 5.Benezra R, Davis R L, Lockshon D, Turner D L, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 6.Biggs J R, Murphy E V, Israel M A. A human Id-like helix-loop-helix protein expressed during early development. Proc Natl Acad Sci USA. 1992;89:1512–1516. doi: 10.1073/pnas.89.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boise L H, Gonzalez-Garcia M, Postema C E, Ding L, Lindsten T, Turka L A, Mao X, Nunez G, Thompson C B. bcl-x, a Bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 8.Bossy-Wetzel E, Bakiri L, Yaniv M. Induction of apoptosis by the transcription factor, c-jun. EMBO J. 1997;16:1695–1709. doi: 10.1093/emboj/16.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christy B A, Sanders L K, Lau L F, Copeland N G, Jenkins N A, Nathans D. An Id-related helix-loop-helix protein encoded by a growth factor-inducible gene. Proc Natl Acad Sci USA. 1991;88:1815–1819. doi: 10.1073/pnas.88.5.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crescenzi M, Fleming T P, Lassar A B, Weintraub H. MyoD induces growth arrest independent of differentiation in normal and transformed cells. Proc Natl Acad Sci USA. 1990;87:8442–8446. doi: 10.1073/pnas.87.21.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deed R W, Armitage S, Norton J D. Nuclear localization and regulation of Id protein through an E protein-mediated chaperone mechanism. J Biol Chem. 1996;271:23603–23606. doi: 10.1074/jbc.271.39.23603. [DOI] [PubMed] [Google Scholar]
- 12.Deed R W, Bianchi S M, Atherton G T, Johnston D, Santibanez-Koref M, Murphy J J, Norton J D. An immediate early human gene encodes an Id-like helix-loop-helix protein and is regulated by protein kinase C activation in diverse cell types. Oncogene. 1993;8:599–607. [PubMed] [Google Scholar]
- 13.Deed R W, Hara E, Atherton G T, Peters G, Norton J D. Regulation of Id3 cell cycle function by Cdk2-dependent phosphorylation. Mol Cell Biol. 1997;17:6815–6821. doi: 10.1128/mcb.17.12.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deed R W, Hirose T, Mitchell E L D, Santibanez-Koref M F, Norton J D. Structural organisation and chromosomal mapping of the human Id3 gene. Gene. 1994;151:309–314. doi: 10.1016/0378-1119(94)90676-9. [DOI] [PubMed] [Google Scholar]
- 15.Deed R W, Jasiok M, Norton J D. Nucleotide sequence of the cDNA encoding human helix-loop-helix Id-1 protein: identification of functionally conserved residues common to Id proteins. Biochim Biophys Acta. 1994;1219:160–162. doi: 10.1016/0167-4781(94)90261-5. [DOI] [PubMed] [Google Scholar]
- 16.Desprez P-Y, Hara E, Bissel M J, Campisi J. Suppression of mammary epithelial cell differentiation by the helix-loop-helix protein Id1. Mol Cell Biol. 1995;15:3398–3404. doi: 10.1128/mcb.15.6.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evan G I, Brown L, Whyte M, Harrington E. Apoptosis and the cell cycle. Curr Opin Cell Biol. 1995;7:825–834. doi: 10.1016/0955-0674(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 18.Evan G I, Wyllie A H, Gilbert C S, Littlewood T D, Land H, Brooks M, Waters C M, Penn L Z, Hancock D C. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 19.Galaktionou K, Chen X, Beach D. Cdc25 cell cycle phosphatase as a target for c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- 20.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hara E, Hall M, Peters G. Cdk2-dependent phosphorylation of Id2 modulates activity of E2A-related transcription factors. EMBO J. 1997;16:332–342. doi: 10.1093/emboj/16.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hara E, Uzman A, Dimri P, Nehlin J O, Testori A, Campisi J. The helix-loop-helix protein Id-1 and a retinoblastoma protein binding mutant of SV40 T antigen synergize to reactivate DNA synthesis in senescent human fibroblasts. Dev Genet. 1996;18:161–172. doi: 10.1002/(SICI)1520-6408(1996)18:2<161::AID-DVG9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Hara E, Yamaguchi T, Nojima H, Ide T, Campisi J, Okayama H, Oda K. Id-related genes encoding helix-loop-helix proteins are required for G1 progression and are repressed in senescent human fibroblasts. J Biol Chem. 1994;269:2139–2145. [PubMed] [Google Scholar]
- 24.Hinds P W, Dowdy S F, Eaton E N, Arnold A, Weinberg R A. Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci USA. 1994;91:709–713. doi: 10.1073/pnas.91.2.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iavarone A, Garg P, Lasorella A, Hsu J, Israel M A. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994;8:1270–1284. doi: 10.1101/gad.8.11.1270. [DOI] [PubMed] [Google Scholar]
- 26.Jen Y, Weintraub H, Benezra R. Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev. 1992;6:1466–1479. doi: 10.1101/gad.6.8.1466. [DOI] [PubMed] [Google Scholar]
- 27.Johnson D G, Cress W D, Jakoi L, Nevins J R. Oncogenic capacity of the E2F-1 gene. Proc Natl Acad Sci USA. 1994;91:12823–12827. doi: 10.1073/pnas.91.26.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joos K, Lam E W-F, Bybee A, Girling R, Muller R, La Thangue N B. Proto-oncogenic properties of the DP family of proteins. Oncogene. 1995;10:1529–1536. [PubMed] [Google Scholar]
- 29.Korsmeyer S J. Regulators of cell death. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- 30.Kowalik T F, De Gregori J, Schwarz J K, Nevins J R. E2F-1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995;69:2491–2500. doi: 10.1128/jvi.69.4.2491-2500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kranenberg O, Van der Eb A J, Zantima A. Cyclin D1 as an essential mediator of apoptotic neuronal cell death. EMBO J. 1996;15:46–54. [PMC free article] [PubMed] [Google Scholar]
- 32.Land H, Parada L F, Weinberg R A. Tunorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 33.Lasorella A, Iavarone A, Israel M A. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol Cell Biol. 1996;16:2570–2578. doi: 10.1128/mcb.16.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lassus P, Ferlin M, Piette J, Hibner U. Anti-apoptotic activity of low levels of wild type p53. EMBO J. 1996;15:4566–4573. [PMC free article] [PubMed] [Google Scholar]
- 35.Lister J, Forrester W C, Baron M H. Inhibition of an erythroid differentiation switch by the helix-loop-helix protein Id1. J Biol Chem. 1995;270:17939–17946. doi: 10.1074/jbc.270.30.17939. [DOI] [PubMed] [Google Scholar]
- 36.Littlewood T D, Evan G I. Transcription factors 2: helix-loop-helix. Prot Profile. 1995;2:621–702. [PubMed] [Google Scholar]
- 37.Lovec H, Sewing A, Lucibello F C, Muller R, Moroy T. Oncogenic activity of cyclin D1 revealed through cooperation with Hras: link between cell cycle control and oncogenic transformation. Oncogene. 1994;9:323–326. [PubMed] [Google Scholar]
- 38.Martin S J, Reutelingsperger C P M, McGahon A J, Rader J A, van Schie R C A A, La Face D M. Early redistribution of plasmamembrane phosphatidylserine as a general feature of apoptosis regardless of initiating stimulus: inhibition by overexpression of Bcl2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer K B, Skogberg M, Margenfeld C, Ireland J, Pettersson S. Repression of the immunoglobulin heavy chain 3′ enhancer by helix-loop-helix protein Id3 via a functionally important E47/E12 binding site: implications for development control of enhancer function. Eur J Immunol. 1995;25:1770–1777. doi: 10.1002/eji.1830250643. [DOI] [PubMed] [Google Scholar]
- 40.Murphy J J, Newton J S, Norton J D. Early response genes and B cell activation. In: Harnett M M, Rigley K P, editors. Lymphocyte signalling: mechanisms, subversion and manipulation. New York, N.Y: John Wiley and Sons; 1997. pp. 183–202. [Google Scholar]
- 41.O’Reilly L, Huang D C S, Strasser A. The cell death inhibitor, Bcl-2, and its homologues influence control of cell cycle entry. EMBO J. 1996;15:6979–6990. [PMC free article] [PubMed] [Google Scholar]
- 42.Penn L J Z, Brooks M W, Laufer E M, Land H. Negative autoregulation of c-myc transcription. EMBO J. 1990;9:1113–1121. doi: 10.1002/j.1460-2075.1990.tb08217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peverali F A, Ramqvist T, Saffrich R, Pepperkok R, Barone M V, Philipson L. Regulation of G1 progression by E2A and Id helix-loop-helix proteins. EMBO J. 1994;13:4291–4301. doi: 10.1002/j.1460-2075.1994.tb06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin X-Q, Livingston D M, Kaelin W G, Adams P D. Deregulated transcription factor E2F-1 expression leads to S phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riechmann V, Cruchten I, Sablitzky F. The expression pattern of Id4, a novel dominant negative helix-loop-helix protein, is distinct from Id1, Id2 and Id3. Nucleic Acids Res. 1994;22:749–755. doi: 10.1093/nar/22.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rudolph B, Saffrich R, Zwicker J, Henglein B, Muller R, Ansorge W, Eilers M. Activation of cyclin-dependent kinases by c-myc mediates induction of cyclin A but not apoptosis. EMBO J. 1996;15:3065–3076. [PMC free article] [PubMed] [Google Scholar]
- 47.Schleiss M R, Degnin C R, Geballe A P. Translational control of human cytomegalovirus gp48 expression. J Virol. 1991;65:6782–6789. doi: 10.1128/jvi.65.12.6782-6789.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shih C, Weinberg R A. Isolation of transforming sequences from a human bladder carcinoma line. Cell. 1982;29:161–169. doi: 10.1016/0092-8674(82)90100-3. [DOI] [PubMed] [Google Scholar]
- 49.Shockett P E, Schatz D G. Diverse strategies for tetracycline-regulated inducible gene expression. Proc Natl Acad Sci USA. 1996;93:5173–5176. doi: 10.1073/pnas.93.11.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh P, Wong S H, Hong W. Overexpression of E2F-1 in rat embryo fibroblasts leads to neoplastic transformation. EMBO J. 1994;13:3329–3338. doi: 10.1002/j.1460-2075.1994.tb06635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sofer-Levi Y, Resnitzky D. Apoptosis induced by ectopic overexpression of cyclin D1 but not cyclin E. Oncogene. 1996;13:2431–2437. [PubMed] [Google Scholar]
- 52.Sorrentino V, Pepperkok R, Davis R L, Ansorge W, Philipson L. Cell proliferation inhibited by MyoD1 independently of myogenic differentiation. Nature. 1990;345:813–815. doi: 10.1038/345813a0. [DOI] [PubMed] [Google Scholar]
- 53.Sun X H. Constitutive expression of the Id1 gene impairs mouse B cell development. Cell. 1994;79:893–900. doi: 10.1016/0092-8674(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 54.Sun X H, Copeland N G, Jenkins N A, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaux D L, Cory S, Adams J M. Bcl-2 gene promotes haemopoietic cell survival and cooperares with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 56.Voronova A, Baltimore D. Mutations that disrupt DNA binding and dimer formation in the E47 HLH protein map to distinct domains. Proc Natl Acad Sci USA. 1990;87:4722–4726. doi: 10.1073/pnas.87.12.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.White E. Life, death and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 58.Wibley J, Deed R W, Jasiok M, Douglas K, Norton J D. A homology model of the Id2 helix-loop-helix domain as a basis for structure-function predictions. Biochim Biophys Acta. 1996;1294:138–146. doi: 10.1016/0167-4838(96)00008-8. [DOI] [PubMed] [Google Scholar]
- 59.Wijsman J H, Jonker R R, Keijer R, Vandevelde C J H, Cornelisse C J, Vandierendonck J H. A new method to detect apoptosis in paraffin sections. J Histochem Cytochem. 1993;41:7–12. doi: 10.1177/41.1.7678025. [DOI] [PubMed] [Google Scholar]
- 60.Williams K J, Boyle J M, Birch J M, Norton J D, Scott D. Cell cycle arrest defect in Li Fraumeni syndrome: a mechanism of cancer predisposition? Oncogene. 1997;14:277–282. doi: 10.1038/sj.onc.1200838. [DOI] [PubMed] [Google Scholar]
- 61.Wilson R B, Kiledjian M, Shen C, Benezra R, Zwollo P, Dymecki S M, Desiderio S V, Kadesch T. Repression of immunoglobulin enhancers by the helix-loop-helix protein Id: implications for B-lymphoid cell development. Mol Cell Biol. 1991;11:6185–6191. doi: 10.1128/mcb.11.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu W, Dahmen J, Bulfone A, Rigolet M, Hernandez M-C, Kuo W L, Puelles L, Rubenstein J L R, Israel M A. Id gene expression during development and molecular cloning of the Id1 gene. Mol Brain Res. 1995;30:312–326. doi: 10.1016/0169-328x(95)00017-m. [DOI] [PubMed] [Google Scholar]