Electrochemical imaging of fusion pore openings by electrochemical detector arrays (original) (raw)
Abstract
Opening of individual exocytotic fusion pores in chromaffin cells was imaged electrochemically with high time resolution. Electrochemical detector arrays that consist of four platinum microelectrodes were microfabricated on a glass coverslip. Exocytosis of single vesicles containing catecholamines from a cell positioned on top of the array is detected by the individual electrodes as a time-resolved oxidation current, reflecting the time course of arrival of catecholamine molecules at the electrode surfaces. The signals exhibit low noise and reveal foot signals indicating fusion pore formation and expansion. The position of individual release events is determined from the fraction of catecholamines recorded by the individual electrodes. Simultaneous fluorescence imaging of release of acridine orange from individual vesicles confirmed the electrochemical position assignments. This electrochemical camera provides very high time resolution, spatiotemporal localization of individual fusion pore openings and quantitative data on the flux of transmitter from individual vesicles. Analysis of the amperometric currents employing random walk simulations indicates that the time course of amperometric spikes measured near the cell surface is due to a low apparent diffusion coefficient of cat-echolamines near the cell surface and not due to slow dissociation from the granular matrix.
Keywords: amperometry, exocytosis, transmitter release
Exocytosis (1) is responsible for release of neurotransmitters, peptides, hormones, and various mediators of the immune system. During exocytosis the molecules stored within membrane-bound secretory vesicles inside the cell are released by fusion of the vesicle membrane with the cell's plasma membrane. A critical event that attracted much attention is the formation and expansion of the fusion pore (reviewed in ref. 2) through which the vesicular cargo molecules are released.
Chromaffin cells from the adrenal gland are a widely used model system to study neuronal exocytosis, and the fusion pore properties in these cells have been studied in great detail (3–6). Release of single vesicles (quanta) of epinephrine or norepinephrine can be detected electrochemically with a carbon fiber electrode (CFE) as amperometric spikes (7, 8). These spikes are often preceded by a so-called foot signal (8), which is due to slow discharge of catecholamines through a narrow fusion pore (3) and indicates the time of fusion pore formation and expansion.
To obtain not only temporal but also spatial resolution of opening and expansion of single fusion pores in chromaffin cells, we fabricated electrochemical detector (ECD) arrays of four Pt electrodes patterned on glass coverslips (9). For quantal events, the oxidation currents recorded from the four electrodes allow the desired determination of fusion pore dynamics with millisecond time resolution as well as localization of the fusion event. Simultaneous observation of the cell by fluorescence microscopy can be performed. Here we show that the spatiotemporal assignments of single exocytotic events obtained by electrochemical imaging of fusion events correlate with release of a fluorescent vesicle marker during exocytosis in chromaffin cells.
Materials and Methods
ECD Array. Pt conductors insulated by photoresist were fabricated photolithographically on standard microscope coverslips No. 1.5 (160–190 μm thick) as described in ref. 9, forming a fourelectrode ECD array. Each ECD array consists of a square window ≈10 × 10 μm2 in size in the insulating 0.5-μm-thick photoresist. The active electrodes are formed by the tips of the four Pt conductors, ≈3 μm wide and 150 nm thick, that protrude into the corners of the square. The area and shape of the electrode arrays were imaged and quantified by using atomic force microscopy and optical microscopy.
Cell Preparation and Recording Conditions. Bovine adrenal glands were obtained from a local slaughterhouse and prepared as described in ref. 10. Cells were cultured on 12-mm coverslips that were coated with 0.02% poly(d)-lysine and used on days 1–10. Before the experiment, a small coverslip with cells attached was placed in a 35-mm dish with 1 ml of bath solution containing 150 mM NaCl, 10 mM Hepes, 5 mM CaCl2, 5 mM KCl, 2 mM MgCl2 (pH 7.25) (318 mosM) and supplemented with 3 μM acridine orange (Molecular Probes) for 15 min in the dark. An ECD array coverslip was mounted on a custom-built stage on a Zeiss 135TV microscope, and the coverslip with the cells was placed at some distance from the ECD array. Bath solution (150–200 μl) was then added to cover the cells and the active electrode array. By using a patch pipette, a cell was lifted off the coverslip and manipulated over the center of an ECD array. Some cells were stimulated by including ionomycin in the pipette or adding ionomycin to the bath. Experiments were performed at room temperature.
Electrochemical Recording and Analysis. The ECD electrodes were connected via their contact pads to four current amplifiers operating on a single power supply with a four-channel head stage (VA-10, NPI Electronic, Tamm, Germany) and the built-in filters set to 500 Hz. The amperometric currents were simultaneously recorded by using a holding potential of +700 mV relative to a common Ag/AgCl electrode immersed into the bath and sampled at 1 kHz. Amperometric current spikes were subjected to a rolling integration to obtain the fraction of charge detected at each electrode. Position assignments from amperometric data were obtained as described in Results. For graphical display only, current traces were further smoothed by using a binomial algorithm in igor pro (WaveMetrics, Lake Oswego, OR).
Random Walk Simulations and Deconvolution Fits. Random walk simulations were programmed by using the Java programming language and forte ce 3.0 for Java (Sun Microsystems, Santa Clara, CA) as described in Results, and the position of measured events was determined by least squares fitting to give best agreement between measured and simulated fractional charges for each of the four electrodes. For time course analysis of a given event, random walk simulations were performed for its location by using 2,000,000 molecules to obtain a low-noise time course for the simulated signals of the four electrodes. The experimental ECD signals and simulated signals were integrated by using igor pro. Because the data were sampled at 1 ms per point, the release time course was approximated by a series of instantaneous release events of different amplitudes, each occurring every 1 ms. At each time point ti, the release amplitude r(ti) was fit to the experimental time course D(ti), using the random walk diffusion signals [_D_RW(t)]. These amplitudes were fit by using the equation
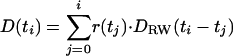
employing procedures written in igor pro. The release amplitudes were fit globally such that convolution with the four integrated random walk diffusion functions gave best agreement with the four integrated experimental signals. Fits were performed for various diffusion constants until the best fit was obtained for each secretory event.
Fluorescence Imaging. During the experiment the bottom of the cell was imaged by using a 100 × 1.30-numerical aperture Fluar oil-immersion objective and low-fluorescence immersion oil (Immersol 518 F, Zeiss) with a cooled, 12-bit charge-coupled device camera (Quantix 57, Photometrics, Tucson, AZ) mounted to the bottom port of the microscope. Images were 2 × 2 binned, and the exposure for each image was 100 ms with a 9-ms interexposure period. At these settings the image pixel size was 260 nm. For synchronization of image exposure times the exposing output of the Quantix 57 was recorded in a separate channel on a 16-bit A/D converter together with the four amperometric currents. Epi-illumination was from a 75-W Xenon arc lamp attenuated by using a neutral density filter of optical density 3.0 and a 480/40-nm excitation filter, a 505-nm LP dichroic filter, and a 535/50-nm emission filter (Omega Optical and Chroma Technology, both of Brattleboro, VT).
Image Analysis. Images were acquired and processed by using v++ software and its built-in routines (Digital Optics, Auckland). Single frames (n) from acquired sequences were extracted as signed 32-bit images. To recognize dequenching “flashes” occurring upon exocytosis of a vesicle, a rolling subtraction procedure was implemented providing difference images according to the following scheme: For a sequence of six images (IMAGEn,..., IMAGEn+5), difference images (DIFFERENCEi = IMAGEi – AVERAGE, i = n,..., n + 5) were generated by subtracting the average of 10 images preceding the first image in the sequence (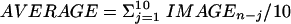 ). The 32-bit difference images contained both negative and positive values. The difference images of the sequence (n,..., n + 5), were scaled and converted to 8-bit gray scale images such that the minimum and maximum pixel values found in the sequence were converted to values of 0 and 255, respectively. Difference sequences that contained clear indications of a sudden intensity increase (flashes) were manually selected and automatically logged.
). The 32-bit difference images contained both negative and positive values. The difference images of the sequence (n,..., n + 5), were scaled and converted to 8-bit gray scale images such that the minimum and maximum pixel values found in the sequence were converted to values of 0 and 255, respectively. Difference sequences that contained clear indications of a sudden intensity increase (flashes) were manually selected and automatically logged.
Results
ECD Recordings. For a typical experiment a chromaffin cell was picked up by a patch pipette sealed onto its surface by using standard methods (11). Using the pipette, the cell was manipulated over an ECD array of four amperometric Pt electrodes and mechanically pushed gently onto the surface as indicated by the bottom of the cell being in the same focal plane as the electrodes (Fig. 1_A_). In the presence of calcium, amperometric spikes indicating individual exocytotic events were recorded from all four electrodes in response to the mechanical stimulation (Fig. 1_B_). One of these events is shown on an expanded time scale in Fig. 2_A_. For this event, the signal from electrode C is largest, followed by that of electrode B and electrode D. The signal from electrode A is just detectable. In this cell, 57 of 211 events showed a detectable amperometric spike in three or four electrodes. The remaining events showed amperometric spikes in only one or two electrodes. In a second mechanically stimulated cell, 14 of 27 events showed amperometric spikes in three or four electrodes. In ionomycin-stimulated cells, <5% of events showed amperometric spikes in three or four electrodes.
Fig. 1.

ECD array recording from a chromaffin cell. (A) Light microscope image of a chromaffin cell placed on top of an ECD array, with four electrodes labeled A through D. (B) Amperometric recordings from the four electrodes (channel A, red trace; channel B, green trace; channel C, black trace; channel D, blue trace) recorded from a single bovine chromaffin cell mechanically stimulated by a patch pipette in calcium containing external solution.
Fig. 2.

Analysis of an exocytotic event. (A) One exocytotic event from the experiment of Fig. 1 shown on an expanded time scale. Arrows mark start and end of the foot signal (black trace) indicating fusion pore opening (O) and expansion (E). (Inset) Foot signal on expanded current scale. (B) Running integrals of the amperometric currents provide the total charge detected by the individual electrodes. (C) Currents from the four electrodes after normalization of the amplitudes reveal differences in time course for the different signals. (D) Spatiotemporal correlation of secretion of catecholamines and release of the fluorescent vesicle marker acridine orange. (Left) Original sequence of fluorescence images. (Right) Difference images obtained by subtracting the average of 10 images preceding the sequence. The exposure times for each frame are indicated on the horizontal axes of A_–_C. The fluorescence “flash” in the difference images indicates the position of the exocytotic event. The release position calculated from the electrochemical signals (B) is indicated in D as a red cross. (Scale bar, 5 μm.)
As with CFEs, a foot signal preceding the spike of catecholamine release is clearly visible in the large signal recorded by electrode C, indicating formation and expansion of the fusion pore (arrows in Fig. 2 A). The duration of this foot signal was 48 ms. In this cell, 87 of 211 events showed a detectable foot signal. The mean duration of these foot signals was 18 ms. In a second cell, 22 of 27 events showed a detectable foot signal with a mean duration of 22 ms. The average integrated charge of the foot signals was 51 fC and 70 fC in the two cells, respectively. In two cells stimulated with the calcium ionophore ionomycin, 40% of the events showed foot signals with mean durations of 12 ms and 15 ms, and mean foot charges of 34 fC and 73 fC. The properties of foot signals observed with ECD arrays are thus similar to those reported for CFE amperometry of chromaffin cells (8, 12).
Fig. 2_B_ shows the running integrals of the four currents indicating the charge measured by each electrode. They provide a low-noise measure of catecholamine release and again clearly indicate the foot signal. The total amperometric charge for this event (sum of the four partial charges) was 5.3 pC corresponding to 1.7 × 107 catecholamine molecules. The mean total charge of all events from this cell was 2.0 ± 1.5 pC (SD, n = 211). The mean charge of single events in a second mechanically stimulated cell was 1.5 ± 1.4 pC (SD, n = 27). The mean charge in two ionomycin-stimulated cells was 1.3 ± 1.9 pC (SD, n = 160) and 0.63 ± 0.36 pC (SD, n = 143). The quantal size of events recorded by ECD arrays is thus in the same range as that of events recorded with CFEs (3, 7, 12, 13).
ECD Imaging. Assuming that free diffusion of released molecules in the space between the cell surface and the surface of the coverslip between the ECD electrodes is equally possible in all directions, and that each electrode effectively oxidizes all molecules arriving at its surface, we expect the individual release event to be located closest to the electrode detecting the largest fraction of charge. Based on this assumption we would locate the event depicted in Fig. 2 to be closest to electrode C and far from electrode A. Fig. 2_C_ shows the four amperometric signals superimposed after normalization to the same amplitude. The signals are slower the smaller they are, consistent with longer diffusion times to the more distant electrodes.
To determine quantitatively the position of release from the fractional charges measured by the four electrodes, random walk simulations were performed to estimate the fraction of molecules arriving at the different electrodes, depending on the position where vesicle contents were released. The positions and shapes of the detectors used in a particular experiment were determined by atomic force microscopy and optical microscopy from which a matrix was created in the computer reflecting the actual electrode geometries (Fig. 3_A_). The cell was modeled as a semiinfinite cylinder with a flat 10-μm-diameter bottom surface suspended 100 nm above the Pt electrode surfaces. Release sites were set up in a 0.5-μm grid on this surface. Release from individual sites was assumed to be instantaneous, and diffusion was modeled by random walk simulations for 10,000 molecules per vesicle. These fractional charges depend only on the locations of the particular release sites and on the geometries of the different electrodes. They depend neither on the particular diffusion coefficient assumed in the simulation nor on the particular time course of release. The position of the measured events was fit to give best agreement between measured and simulated fractional charges for each of the four electrodes, using interpolation between the release sites chosen in the random walk simulations.
Fig. 3.

Correlation between electrochemical and fluorescence imaging. (A) Outline of the detector map (red lines) used for the random walk simulations overlayed on image of the actual electrodes. The dashed blue circle indicates the central area with a 2-μm radius. (B) Difference images (a_–_v) of all 22 fluorescence flashes from this cell for which position assignments were obtained. The red crosses (at end of arrows) indicate the position assignments from electrochemical imaging. Difference image i shows the event of Fig. 2. In all cases the fluorescence flash appeared in the image frame immediately following the spike of catecholamine release measured electrochemically. (C) Comparison of the measured x coordinates obtained from electrochemical and fluorescence imaging. (D) Comparison of y coordinates. The dashed lines with slope 1 would be expected for perfect agreement between the two independent measurements. The red data points refer to events located outside the blue circle of Fig. 3_A_.
We have chosen to model the cell by a semiinfinite cylinder rather than a disk to approximate the cell geometry encountered by molecules diffusing out of the 10-μm disk area. Although some molecules released from the bottom surface escape detection in the simulation, the estimated fraction of “lost” molecules from simulated exocytotic events was always very small (<1%). As expected, many release events are recorded in only one or two channels. For events that appeared in traces from two electrodes, these electrodes were always adjacent to each other as expected for events at the ECD periphery. However, neither can we assign a position to events that appear in only one or two electrodes, nor can the fluorescence dequenching flash be observed. Thus, these events were not included in the analysis performed here. Release events that do not occur near the bottom surface produce amperometric signals with very small amplitude and very slow time course. Such signals were never observed in more than two electrodes.
Comparison with Fluorescence Imaging. To independently confirm the positions obtained with electrochemical imaging, we simultaneously performed fluorescence imaging on the same cell. The secretory vesicles were loaded with acridine orange, a dye that accumulates in acidic compartments at high concentrations such that its green fluorescence is partially quenched and red-shifted (14). Upon exocytosis the dye is diluted, which manifests itself by a dequenching flash of green fluorescence (15, 16). Fig. 2_D_ Left shows a sequence of six fluorescence images. The average of 10 images preceding this sequence was subtracted from each individual image in the sequence providing the difference images (Fig. 2_D_ Left). The dequenching flash in the difference images is correlated with the time course of catecholamine secretion (Fig. 2_B_). The position of this event obtained from electrochemical imaging (Fig. 2_D_, red cross) matches the position of the dequenching flash.
The 22 fluorescence dequenching flashes (Fig. 3_B_) from difference images obtained as in Fig. 2_D_ agree well with the corresponding ECD position assignments (red crosses at arrow tips). For a quantitative comparison, we assigned a position to each fluorescence dequenching flash (Fig. 3_B_) as the center of the brightest four pixels in the difference image. For most events, determining center of mass gave essentially the same result. However, for events that had a flash that was partly covered by one of the electrodes, the center of mass approach was less suitable. To analyze all events in a consistent way, we have thus chosen to use the four brightest pixels to determine the position. A coordinate system was chosen according to the symmetry of the ECD array as shown in Fig. 3_A_. Fig. 3 C and D show the comparison between the x and y positions obtained by electrochemical array imaging and the positions of the corresponding fluorescence dequenching flashes. The dashed lines indicate a slope of 1 as expected for perfect agreement between electrochemical imaging and fluorescence imaging. The data points are close to these lines.
The mean absolute deviation between the coordinates obtained from electrochemical and fluorescence imaging in this cell was 410 nm for the x direction and 280 nm for the y direction (n = 22). Because the precision of electrochemical imaging is better in the central area between the electrodes, analysis using only data points lying within 2 μm from the center (Fig. 3_A_, circle) gave smaller mean deviations (330 nm for the x direction and 190 nm for the y direction; n = 18). It should be noted that the fluorescence dequenching flashes are very diffuse and the assignment of its position is not very precise. The values obtained for the accuracy of the ECD should thus be considered upper limits of the error of electrochemical imaging.
Kinetic Analysis. For comparison with CFE recordings, we analyzed the width of the amperometric spikes at half maximum (_t_1/2) for the largest signal of each event, which is obtained from the electrode closest to the release site. The mean _t_1/2 values were 19.5 ± 14.4 ms (SD, n = 211) and 9.2 ± 2.7 ms (SD, n = 27) for the mechanically stimulated cells and 15.1 ± 22.6 ms (SD, n = 160) and 12.1 ± 8.3 ms (SD, n = 143) for the ionomycin-stimulated cells. These values agree well with those obtained with conventional CFE recordings with cell-electrode distances of 1 μm or less (13).
The random walk simulations for various release positions provide not only the fractional charges but also time courses of the amperometric signals. Assuming that quantal release is instantaneous, and using the standard diffusion constant D = 6 × 10–6 cm2/s for epinephrine (17), the simulated random walk signals were always faster than the measured signals (data not shown). This is not unexpected (13) because the measured signals are in fact a convolution of the release kinetics with the appropriate diffusion function. Using the diffusion functions determined by the random walk simulations, we thus performed a deconvolution to determine the underlying release kinetics (Fig. 4_A_) by least squares fitting of the release function to the running integrals of the measured amperometric signals (see Materials and Methods). Whereas the largest signal is rather well reproduced by the fit, the smaller signals are not fitted well. For the small signals the measured time course is significantly slower than the simulated time course (Fig. 4_A_). Because the electrodes measuring smaller signals are further away from the release site, this result suggested that diffusion is in fact slower than assumed in the simulation. We thus varied the diffusion coefficient assumed for determining the diffusion function and repeated the deconvolution fits. Diffusion constants up to 30 times lower than the value for free diffusion were tested. The best agreement was obtained by using a diffusion coefficient D = 8 × 10–7 cm2/s (Fig. 4_B_). For seven events from two different cells the best fits were obtained with diffusion coefficients in a narrow range between 8 × 10–7 cm2/s and 13 × 10–7 cm2/s. Thus, the apparent diffusion coefficient near the cell surface is ≈10 times slower than that for free diffusion.
Fig. 4.

Integrated experimental (solid lines) and fitted (dashed lines) amperometric signals using the standard diffusion constant D = 6 × 10–6 cm2/s (A) and a diffusion constant of D = 8 × 10–7 cm2/s(B). (Insets) Time course of release resulting from the fits. All four traces were fitted simultaneously.
Discussion
Pt ECD arrays appropriate for cellular dimensions can be fabricated directly on glass coverslips (9). Patterned Pt microelectrodes are thus suitable to perform amperometric measurements of single exocytotic events by oxidation of the catecholamines released from individual vesicles. Mechanical stimulation of chromaffin cells manipulated over the ECD array evokes exocytotic events that generate low-noise amperometric signals at the different electrodes. Mechanically stimulated release has been shown to be calcium-dependent (3, 5, 7) and thus presumably involves the same neuronal SNARE-mediated fusion mechanism as release that is stimulated by receptor agonists, K+ depolarization, or calcium influx into permeabilized cells. The properties of single events recorded with the Pt ECD arrays are indistinguishable from those reported for previous CFE amperometry with respect to quantal size, spike half width, and foot duration. It can thus be concluded that the recorded events resemble those induced by physiological stimulation.
Electrochemical Imaging. Events that produce amperometric spikes in three or four electrodes occur in the central area between the electrodes. About 30% of events from mechanically stimulated cells occurred in this area, and analysis of the fractions of molecules detected by the different electrodes allows for spatiotemporal localization of individual fusion pore openings on the cell surface. The ECD array thus acts as an electrochemical imaging device with millisecond time resolution.
By simultaneous fluorescence imaging, we showed directly that secretion of the fluorescent vesicle marker acridine orange occurs in concert with the secretion of catecholamine from the same vesicle. The observed fluorescence cloud decays much more slowly than the amperometric current. This is expected because the catecholamine molecules are rapidly consumed during the oxidation process, whereas the fluorescent molecules are diluted slowly in the diffusion process. A similarly slow decay is observed when catecholamines are measured with cyclic voltammetry, a method that reversibly oxidizes and reduces the molecules without the consumption of molecules that occurs in constant-voltage amperometry (13).
The spatial precision was estimated by comparing positions obtained by electrochemical imaging with positions obtained by fluorescence imaging. The mean distance between positions obtained with the two methods was 510 nm. However, the fluorescence dequenching flashes are very diffuse; the assigned position is thus not very precise, and the distance of 510 nm is presumably an overestimate. For events within the 4-μm-diameter central circle, the mean absolute deviation between the coordinates from fluorescence imaging and from electrochemical imaging was 330 nm for the x direction and 190 nm for the y direction. The deviation is somewhat larger for events outside of that area. Whereas the agreement is very good for the y direction (Fig. 3_D_), fitting the points for the x direction (Fig. 3_C_) gave a slope of 1.37 ± 0.08, suggesting that a small systematic error exists. Such errors may result from small errors in the boundaries of the electrodes or from some debris near the boundaries of the electrodes. However, the mean deviations are rather small and comparable to the point-spread-function of the microscope.
Proper localization of fusion pore openings requires that all electrodes of the array are equally sensitive catecholamine detectors. The good correspondence between electrochemical and fluorescence imaging indicates that this is the case; but how can this be ensured independent of fluorescence imaging? An important criterion for a “good” sensitive amperometric electrode is that it rapidly oxidizes catecholamine molecules, indicated by brief amperometric spikes with short half-width. Release events from chromaffin cells typically have a half-width of 10–20 ms when recorded with CFEs very close to the cell surface (7, 8, 13). The amperometric signals for a given exocytotic event measured with an ECD array differ among different electrodes in their time course depending on the fusion pore–electrode distance. However, the largest amperometric signal of a given event is recorded by the closest electrode and thus has a similarly short half-width. As a criterion that all electrodes of an ECD array are equally sensitive, we thus require that each electrode gave at least one amperometric spike with a half-width <20 ms. If one of the electrodes measured no or only small and slow signals during an experiment, the experiment was discarded.
Electrochemical imaging has several advantages over fluorescence imaging. It selectively images exocytotic events at the cell surface. Although this is also possible using total internal reflection fluorescence (TIRF) microscopy (18), the electrochemical imaging has much higher time resolution. Our recordings filtered at 500 Hz were ≈10 times faster than video rate, and at higher bandwidth, submillisecond time resolution is possible. The high time resolution and signal-to-noise ratio allows one to detect amperometric foot signals (8), which reflect the opening and expansion of the exocytotic fusion pore (3). Thus, using ECD arrays, electrochemical imaging of fusion pore opening and expansion can be performed with high temporal and spatial resolution. In contrast, it has yet been impossible to detect the opening of the initial narrow fusion pore by fluorescence techniques.
Exocytosis in neuronal cell types critically depends on so-called SNARE proteins in the vesicle and the plasma membrane (19). Interactions of these proteins may be directly involved in the fusion event (20, 21), but direct evidence is yet to be obtained and alternative hypotheses have been put forward (22). Conformational changes in the SNARE complex can be observed by using fluorescence microscopy (23). It will thus be of great interest to image simultaneously the opening and expansion of individual fusion pores and to correlate the fusion pore dynamics with fluorescence of tagged proteins.
Kinetics of Quantal Release. Quantal release of catecholamines is usually measured by using a CFE positioned very close to the cell surface (7, 8). It was found that for cell-CFE distances of 5 μm, the individual amperometric spikes were well reproduced by the diffusion kinetics. However, for small distance, <1 μm, spikes were broader than expected for diffusion (13). This finding was interpreted to reflect release from the vesicle being rate-limiting, reflecting dissociation of catecholamines from the vesicular matrix (13). However, spikes measured very close to the cell surface were also well reproduced assuming a reduced diffusion coefficient of 3 × 10–7 cm2/s near the cell surface (8).
The experiments with the ECD array described here allow one to distinguish directly between these two possibilities. The diffusion to the electrodes occurs over relatively large distances along the cell surface. If diffusion were normal, as in free solution, then the deconvolution fit of Fig. 4_A_ would have provided the release function reflecting dissociation of catecholamines form the granular matrix. However, this approach did not provide a satisfactory fit of the measured signals. In contrast, good fits were obtained when a significantly lower diffusion coefficient was used (Fig. 4_B_). The half-width of the release function for this event was only 8 ms, which is an overestimate due to the 500-Hz low pass filter. The time course of amperometric spikes measured with a CFE near the cell surface is thus largely determined by a low diffusion coefficient of catecholamines near the cell surface rather than by slow dissociation form the granular matrix.
A low apparent diffusion coefficient is produced if the diffusing molecules bind reversibly to the cell membrane (24). The binding mechanisms that reduce the apparent diffusion coefficient near the cell surface are currently unknown. ECD array recordings as described here may be performed on cells pretreated with proteases to obtain information on the involvement of proteins in catecholamine binding to the cell surface. In addition, reversible binding of catecholamine to the phospholipid bilayer or the glycocalyx that surrounds the cell may be involved. Pretreatment of the cells with a series of endoglycosidases may thus also provide interesting information.
Future ECD Array Developments and Applications. In the experiments described here, cells were placed on the ECD array by using a patch pipette. A difficulty with this procedure is that cells must be detached from the surface, a step that often disrupts the cell. To overcome this limitation, cells may be targeted to ECD arrays by other means. Cells may be directed to microelectrode arrays by dielectrophoresis (25), and preliminary experiments in our laboratory indicate that the ECD array itself can be used to trap cells in the ECD center. Using this method, we anticipate a significant increase in throughput. Another limitation is that typically only one out of three arrays worked well, i.e., all electrodes were sensitive and exhibited low noise. We recently succeeded in replacing the photoresist insulation by fused silica (SiO2) insulation creating more robust devices that show low noise more consistently. The fused silica surface also opens the possibility to guiding neuronal and other cell growth on surfaces by using surface chemical patterning as previously described for directing growth to electrodes recording electrical activity at multiple sites (26).
Acknowledgments
We thank Lori Kwan and Joan Lenz for the cell preparations and excellent technical assistance. This work was supported by the Nanobiotechnology Center (an STC program of National Science Foundation Agreement ECS-9876771) and National Institutes of Health Grant R01 NS38200, and was performed in part at the Cornell Nanofabrication Facility (a member of the National Nanofabrication Users Network), which is supported by the National Science Foundation under Grant ECS-9731293, its users, Cornell University, and Industrial Affiliates.
Author contributions: G.D. and M.L. designed research; I.H., K.K., and G.D. performed research; K.K., K.B., V.V., and H.G.C. contributed new reagents/analytic tools; I.H., K.K., K.B., G.D., M.G.Y., and M.L. analyzed data; I.H., K.K., K.B., and M.L. wrote the paper; and H.G.C. provided advice and resources for nanofabrication of the devices used here.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CFE, carbon fiber electrode; ECD, electrochemical detector.
References
- 1.Almers, W. (1990) Ann. Rev. Physiol. 52**,** 607–624. [DOI] [PubMed] [Google Scholar]
- 2.Lindau, M. & Alvarez de Toledo, G. (2003) Biochim. Biophys. Acta 1641**,** 167–173. [DOI] [PubMed] [Google Scholar]
- 3.Albillos, A., Dernick, G., Horstmann, H., Almers, W., Alvarez de Toledo, G. & Lindau, M. (1997) Nature 389**,** 509–512. [DOI] [PubMed] [Google Scholar]
- 4.Alés, E., Tabares, L., Poyato, J. M., Valero, V., Lindau, M. & Alvarez de Toledo, G. (1999) Nat. Cell Biol. 1**,** 40–44. [DOI] [PubMed] [Google Scholar]
- 5.Dernick, G., Alvarez De Toledo, G. & Lindau, M. (2003) Nat. Cell Biol. 5**,** 358–362. [DOI] [PubMed] [Google Scholar]
- 6.Tabares, L., Ales, E., Lindau, M. & Alvarez De Toledo, G. (2001) J. Biol. Chem. 276**,** 39974–39979. [DOI] [PubMed] [Google Scholar]
- 7.Wightman, R. M., Jankowski, J. A., Kennedy, R. T., Kawagoe, D. T., Schroeder, T. J., Leszczyszyn, D. J., Near, J. A., Diliberto, E. J., Jr., & Viveros, O. H. (1991) Proc. Natl. Acad. Sci. USA 88**,** 10754–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow, R. H., von Rüden, L. & Neher, E. (1992) Nature 356**,** 60–63. [DOI] [PubMed] [Google Scholar]
- 9.Dias, A. F., Dernick, G., Valero, V., Yong, M. G., James, C. D., Craighead, H. G. & Lindau, M. (2002) Nanotechnology 13**,** 285–289. [Google Scholar]
- 10.Parsons, T. D., Coorssen, J. R., Horstmann, H. & Almers, W. (1995) Neuron 15**,** 1085–1096. [DOI] [PubMed] [Google Scholar]
- 11.Hamill, O. P., Marty, A., Neher, E., Sakmann, B. & Sigworth, F. J. (1981) Pflügers Arch. Eur. J. Physiol. 391**,** 85–100. [DOI] [PubMed] [Google Scholar]
- 12.Zhou, Z., Misler, S. & Chow, R. H. (1996) Biophys. J. 70**,** 1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wightman, R. M., Schroeder, T. J., Finnegan, J. M., Ciolkowski, E. L. & Pihel, K. (1995) Biophys. J. 68**,** 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams, R. M. & Webb, W. W. (2000) J. Cell Sci. 113**,** 3839–3850. [DOI] [PubMed] [Google Scholar]
- 15.Steyer, J. A., Horstmann, H. & Almers, W. (1997) Nature 388**,** 474–478. [DOI] [PubMed] [Google Scholar]
- 16.Avery, J., Ellis, D. J., Lang, T., Holroyd, P., Riedel, D., Henderson, R. M., Edwardson, J. M. & Jahn, R. (2000) J. Cell Biol. 148**,** 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerhardt, G. A. & Adams, R. N. (1982) Anal. Chem. 54**,** 2618–2620. [Google Scholar]
- 18.Steyer, A. & Almers, W. (1999) Biophys. J. 76**,** 2262–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Söllner, T., Whiteheart, S., Brunner, M., Erdjument-Bromage, H., Geromanos, M., Tempst, P. & Rothman, J. E. (1993) Nature 362**,** 318–323. [DOI] [PubMed] [Google Scholar]
- 20.Weber, T., Parlati, F., McNew, J. A., Johnston, R. J., Westermann, B., Söllner, T. H. & Rothman, J. E. (2000) J. Cell Biol. 149**,** 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen, Y. A., Scales, S. J., Patel, S. M., Doung, Y.-C. & Scheller, R. H. (1999) Cell 97**,** 165–174. [DOI] [PubMed] [Google Scholar]
- 22.Morel, N. (2003) Biol. Cell 95**,** 453–457. [DOI] [PubMed] [Google Scholar]
- 23.An, S. J. & Almers, W. (2004) Science 306**,** 1042–1046. [DOI] [PubMed] [Google Scholar]
- 24.Hochstrate, P. & Ruppel, H. (1980) Biophys. Struct. Mech. 6**,** 125–138. [DOI] [PubMed] [Google Scholar]
- 25.Gray, D. S., Tan, J. L., Voldman, J. & Chen, C. S. (2004) Biosens. Bioelectron 19**,** 1765–1774. [DOI] [PubMed] [Google Scholar]
- 26.James, C. D., Spence, A. J., Dowell-Mesfin, N. M., Hussain, R. J., Smith, K. L., Craighead, H. G., Isaacson, M. S., Shain, W. & Turner, J. N. (2004) IEEE Trans. Biomed. Eng. 51**,** 1640–1648. [DOI] [PubMed] [Google Scholar]