p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis (original) (raw)
Abstract
The activity of the cyclin-dependent kinase inhibitor p27 is controlled by its concentration and subcellular localization. However, the mechanisms that regulate its intracellular transport are poorly understood. Here we show that p27 is phosphorylated on Ser10 in vivo and that mutation of Ser10 to Ala inhibits p27 cytoplasmic relocalization in response to mitogenic stimulation. In contrast, a fraction of wild-type p27 and a p27(S10D)-phospho-mimetic mutant translocates to the cytoplasm in the presence of mitogens. G1 nuclear export of p27 and its Ser10 phosphorylation precede cyclin-dependent kinase 2 (Cdk2) activation and degradation of the bulk of p27. Interestingly, leptomycin B-mediated nuclear accumulation accelerates the turnover of endogenous p27; the p27(S10A) mutant, which is trapped in the nucleus, has a shorter half-life than wild-type p27 and the p27(S10D) mutant. In summary, p27 is efficiently degraded in the nucleus and phosphorylation of Ser10 is necessary for the nuclear to cytoplasmic redistribution of a fraction of p27 in response to mitogenic stimulation. This cytoplasmic localization may serve to decrease the abundance of p27 in the nucleus below a certain threshold required for activation of cyclin–Cdk2 complexes.
Keywords: cell cycle/p27/phosphorylation/subcellular localization/ubiquitin-dependent proteolysis
Introduction
The cyclin-dependent kinase (Cdk) inhibitor p27_Kip1_ (p27) is an important regulator of the mammalian cell cycle (reviewed in Sherr and Roberts, 1995, 1999; Hengst and Reed, 1998). p27 negatively regulates G1 progression by binding to cyclin–Cdk2 complexes and preventing their activity. Accordingly, the levels of p27 are high in quiescent cells and decline upon mitogenic stimulation. In addition, both p27 and its homolog p21_Cip1_ positively regulate cell cycle progression by promoting the assembly and activity of cyclin D–Cdk4/6 complexes (Soos et al., 1996; LaBaer et al., 1997; Cheng et al., 1999).
The activity of p27 is controlled by its concentration, its distribution among different cellular complexes and its subcellular localization (Ekholm and Reed, 2000; Slingerland and Pagano, 2000). One of the key mechanisms involved in the regulation of p27 abundance is proteolysis by the ubiquitin–proteasome pathway (Pagano et al., 1995). p27 is phosphorylated on Thr187 by cyclin E–Cdk2 (Muller et al., 1997; Sheaff et al., 1997; Vlach et al., 1997; Montagnoli et al., 1999), and is then recognized and targeted for ubiquitylation by the SCF_Skp2_ ubiquitin–protein ligase and its cofactor, the Cdk subunit 1 (Cks1; Carrano et al., 1999; Sutterluty et al., 1999; Tsvetkov et al., 1999; Nakayama et al., 2000; Ganoth et al., 2001; Spruck et al., 2001). Thus, the timing of p27 degradation is dependent on both the accumulation of cyclin E and concomitant activation of Cdk2, and the mitogen-stimulated induction of Skp2 and Cks1 expression. The biosynthesis of p27 is also subject to regulation by transcriptional (Servant et al., 2000) and translational mechanisms (Agrawal et al., 1996; Hengst and Reed, 1996; Millard et al., 1997; Miskimins et al., 2001). In addition to changes in expression levels, inactivation of p27 also occurs through sequestration by cyclin D–Cdk4/6 complexes (Sherr and Roberts, 1999).
To exert its inhibitory action, p27 needs to be transported into the nucleus (Reynisdottir and Massague, 1997; Orend et al., 1998; Soucek et al., 1998; Tomoda et al., 1999). The nuclear import of p27 is dependent on the presence of a nuclear localization signal (NLS) localized near the C-terminus of the protein (Zeng et al., 2000). Moreover, association of p27 with nuclear pore-associated protein 60 (NPAP60) also contributes to nuclear import (Muller et al., 2000). p27 was shown to physically interact with Jab1, a component of the COP9–signalosome complex, whose overexpression induces p27 translocation to the cytoplasm and promotes p27 degradation (Tomoda et al., 1999). Thus, it is generally believed that degradation of p27 requires its cytoplasmic relocalization. Despite these observations, the regulation of p27 cellular localization remains poorly understood. Here, we show that phosphorylation of Ser10 is a necessary signal for nuclear export of p27 upon cell cycle re-entry. In addition, we demonstrate that p27 is efficiently degraded in the nucleus and that cytoplasmic localization is not a prerequisite for p27 degradation.
Results
p27 is phosphorylated on Ser10 in vivo
We have analyzed the phosphorylation state of endogenous p27 in Rat1 fibroblasts. Results of in vivo labeling studies indicated that p27 is phosphorylated in quiescent cells (Figure 1A). The level of p27 phosphorylation transiently increases upon cell cycle re-entry. After 6–12 h of mitogenic stimulation, p27 phosphorylation declines with no apparent change in the amount of protein. At the G1/S transition, the amount of p27 protein and phosphorylation fall concomitantly (Figure 1A). Analysis of tryptic phosphopeptide maps revealed that p27 is phosphorylated on two major peptides (Figure 1B, spots 2 and 3) and one minor peptide of variable intensity (Figure 1B, spot 1). The two major labeled peptides were found to contain exclusively phosphoserine (data not shown).

Fig. 1. p27 is phosphorylated on Ser10 in vivo. (A) Quiescent Rat1 cells were labeled with 0.5 mCi/ml [32P]phosphoric acid for 4 h and then stimulated with 10% serum for the times indicated. The cells were lysed and the endogenous p27 was immunoprecipitated with anti-p27 antibody. The precipitated proteins were resolved by SDS–gel electrophoresis, transferred to PVDF membrane and analyzed by autoradiography (top). The abundance of p27 protein was monitored by immunoblotting (bottom). (B) The 32P-labeled p27 protein band (from cells stimulated for 12 h with serum) was excised from the membrane and digested with trypsin. The resulting phosphopeptides were separated by thin layer electrophoresis followed by ascending chromatography. The arrow denotes the position of sample application. (C) Rat1 cells were transfected with HA-tagged wild-type p27 or p27(S10A) mutant. After 48 h, the cells were labeled with [32P]phosphoric acid and ectopic p27 was immunoprecipitated from cellular lysates with an anti-HA antibody. The extent of phosphorylation and abundance of ectopic p27 were analyzed as in (A). (D) Phosphopeptide mapping analysis of ectopically expressed HA-tagged p27 and p27(S10A) mutant. (E) Specificity of p27 phospho-Ser10 antibody. Recombinant purified wild-type p27 and p27(S10A) mutant were phosphorylated in vitro by either cyclin E–Cdk2 (lanes 1 and 2) or ERK2 (lanes 3 and 4). Phosphorylation products were analyzed by immunoblotting with anti-phospho-Ser10-specific antibody (top), anti-phospho-Thr187 specific antibody (middle) or with a mouse anti-p27 antibody (bottom). (F) NIH 3T3 cells were transfected with human wild-type p27 (lane 1), p27(S10A; lane 2), p27(S10D; lane 3) or p27(S10E; lane 4) mutants. Protein extracts were analyzed by immunoblotting with either anti-phospho-Ser10 antibody or anti-p27 antibody. The human p27 can be distinguished from the endogenous mouse p27 because it migrates slower on SDS gels. (G) Lysates from NIH 3T3 cells transfected with wild-type p27 (wt) or p27(S10A) mutant were subjected to immunoprecipitation with either anti-p27 antibody (lanes 1 and 2) or anti-phospho-Ser10 antibody (lanes 3 and 4). The precipitated proteins were analyzed by immunoblotting with anti-p27 antibody.
A site-directed mutagenesis approach was used to identify the phosphorylation sites in p27. Because of the important role of proline-directed kinases in cell cycle regulation, we mutated the three serine and threonine residues (Ser10, Ser178 and Thr187) that are immediately followed by a Pro residue. Rat1 cells were transfected with expression vectors encoding wild-type or alanine mutants of hemagglutinin (HA)-tagged p27, metabolically labeled with [32P]orthophosphate, and the ectopically expressed p27 was immunoprecipitated with anti-HA antibody. The extent of phosphorylation of p27-HA was analyzed by autoradiography and normalized to the amount of immunoprecipitated protein. As shown in Figure 1C, replacement of Ser10 by Ala resulted in a strong reduction in 32P incorporation, indicating that Ser10 is a major site of p27 phosphorylation. No significant change in the total level of p27-HA phosphorylation was observed for the p27(S178A) mutant, while the p27(T187A) mutant showed a decrease in phosphorylation only when cyclin E and Cdk2 were co-transfected to phosphorylate this site (data not shown). The phosphopeptide map of HA-tagged p27 was comparable with that of the endogenous protein, except for the presence of four additional minor spots labeled 4–7 (Figure 1D). The physiological significance of these four minor phosphopeptides remains to be established, as they were reproducibly detected only upon overexpression of the protein. The two major phosphopeptides (spots 2 and 3) observed in wild-type p27 were absent in the p27(S10A) mutant, confirming that p27 is phosphorylated on Ser10 in vivo (Figure 1D). Further analysis revealed that spots 2 and 3 are derived from the same peptide containing phosphorylated Ser10 exclusively (data not shown). During the course of this work, Ishida et al. (2000) also reported that Ser10 is a major phosphorylation site of p27.
To explore the regulation of p27 phosphorylation on Ser10, we raised a phospho-specific antibody against a synthetic peptide that spans the phosphorylated Ser10 residue of p27. The specificity of the purified antibody is illustrated in Figure 1E. The purified antibody recognized p27 recombinant protein only after phosphorylation in vitro by the MAP kinase ERK2 (Figure 1E, lane 3, top), and substitution of Ser10 by Ala completely abolished the immunodetection of the protein (Figure 1E, lane 4). In addition, p27 was not recognized by the anti-phospho-Ser10 antibody when phosphorylated in vitro by cyclin E–Cdk2 (Figure 1E, lanes 1 and 2, top), which phosphorylated both wild-type and p27(S10A) protein on Thr187 efficiently instead, as detected with a phospho-Thr187-specific antibody (Carrano et al., 1999; Montagnoli et al., 1999; Figure 1E, lanes 1 and 2, middle). We further tested the specificity of the antibody on cellular p27. Asynchronous NIH 3T3 cells were transfected with expression plasmids encoding wild-type p27, p27(S10A), p27(S10D) or p27(S10E) mutants. Immunoblot analysis of total cell lysates revealed that the anti-phospho-Ser10 antibody recognizes endogenous murine p27 as well as ectopically expressed human wild-type p27 (which migrates slower than mouse p27), but fails to detect the p27(S10A) mutant, thereby confirming the specificity of the antibody for phosphorylated p27 (Figure 1F, lanes 1 and 2). Notably, this antibody recognized p27(S10D) and p27(S10E) mutants (Figure 1F, lanes 3 and 4), indicating that these substitutions effectively mimic the negative charge of the phosphate in position 10. The specificity of the antibody was also demonstrated in immunoprecipitation (Figure 1G) and in immunofluorescence experiments (not shown).
Phosphorylation of p27 on Ser10 is predominant in G0/G1 cells
To determine whether p27 is phosphorylated on Ser10 in a cell cycle-dependent manner, quiescent primary T lymphocytes were stimulated with mitogens (Figure 2A). Progression of T lymphocytes into the cell cycle was associated with an increase in the levels of cyclin E and cyclin A, and a gradual decrease in the abundance of p27. The decrease in p27 expression coincided with a marked reduction in phosphorylation of the protein on Ser10. Similar results were obtained in synchronized NIH 3T3 and HeLa cells (data not shown). To demonstrate that changes in p27 phospho-Ser10 immunoreactivity reflect changes in the stoichiometry of Ser10 phosphorylation, not just variations in the amount of protein, extracts of NIH 3T3 cells, synchronized in G0/G1 by serum starvation and released into the cell cycle for the times indicated, were assayed for their ability to phosphorylate a constant amount of recombinant p27 protein in vitro. Immunoblot analysis with anti-phospho-Ser10- and anti-phospho-Thr187-specific antibodies revealed that the kinase activity responsible for Ser10 phosphorylation was elevated in G0/G1 and declined as cells advanced to S phase (16 h after serum re-addition; Figure 2B).

Fig. 2. Phosphorylation of p27 on Ser10 is cell cycle regulated. (A) Mouse T lymphocytes were stimulated with concanavalin A and interleukin-2 for the times indicated. Cellular extracts were analyzed by immunoblotting with specific antibodies to the indicated proteins. (B) NIH 3T3 fibroblasts were synchronized in G0/G1 by serum starvation and restimulated with 10% serum for the times indicated. Cell extracts were prepared and incubated with recombinant purified p27 in kinase assay buffer as described in Materials and methods. The reaction products were analyzed by immunoblotting using anti-phospho-Ser10- (upper) and Thr187 (lower)-specific antibodies. (C) MCF-7 breast cancer cells were depleted of estradiol and treated with 1 µM tamoxifen for the times indicated (lanes 1–4). After 48 h, arrested cells were stimulated to re-enter the cell cycle by addition of 500 nM estradiol for the intervals of time indicated (lanes 5–9). Cell extracts were analyzed by immunoblotting with specific antibodies to the indicated proteins. (D) Cell extracts (50 µg protein) from proliferating (AS) or tamoxifen-treated (TMX) MCF-7 cells were sequentially immunoprecipitated four times with anti-phospho-Ser10 antibody. The resultant pellets (lanes 1–4 and 6–9) and the final supernatant (lanes 5 and 10), as well as the input lysate (lanes 11 and 12), were analyzed by immunoblotting with anti-p27 antibody. (E) MCF-7 cell extracts described in (C) (lanes 1–4) were subjected to immunoprecipitation with anti-p27 antibody and analyzed by immunoblotting with specific antibodies to the indicated proteins. (F) The same MCF-7 cell extracts described in (E) were subjected to immunoprecipitation with anti-phospho-Ser10 antibody and analyzed by immunoblotting as indicated.
Additional evidence for the cell cycle regulation of p27 phosphorylation on Ser10 was obtained in MCF-7 breast cancer cells. Asynchronous MCF-7 cells were arrested in G1 by treatment with tamoxifen combined with estrogen deprivation, as monitored by cyclin A down-regulation (Figure 2C, lanes 1–4). In this cell line, the total abundance of p27 did not change significantly during cell cycle exit, but the level of its phosphorylation on Ser10 increased significantly (Figure 2C, lanes 1–4). When the cells were re-stimulated to enter the cell cycle by addition of estradiol, p27 protein abundance decreased in parallel with a decrease in Ser10 phosphorylation (Figure 2C, lanes 5–9). To quantify more accurately the extent of p27 Ser10 phosphorylation in exponentially proliferating versus G1-arrested MCF-7 cells, cellular lysates were subjected to successive rounds of immunoprecipitation with the anti-phospho-Ser10-specific antibody to ensure that all the phosphorylated p27 was precipitated effectively. The pellets and the final supernatant were analyzed by immunoblotting with anti-p27 antibody to estimate the percentage of p27 phosphorylated on Ser10 (Figure 2D). From four different experiments using both MCF-7 and NIH 3T3 cells and densitometry analysis of the bands, we calculated that ∼33% of total p27 was phosphorylated on Ser10 in arrested cells, while only 5% of the protein was phosphorylated at this site in proliferating cells (Figure 2D and data not shown). Importantly, we determined that the fraction of p27 phosphorylated on Ser10 is still able to bind to cyclin D1 and cyclin E, and that the anti-phospho-Ser10 antibody effectively precipitates cyclin–Cdk complexes together with phosphorylated p27 (Figure 2E and F).
Phosphorylation on Ser10 stabilizes p27 in vivo but does not affect p27 ubiquitylation in vitro
Phosphorylation of p27 on Thr187 by cyclin E–Cdk2 promotes p27 degradation by the ubiquitin–proteasome pathway (Carrano et al., 1999; Montagnoli et al., 1999; Sutterluty et al., 1999). While this work was in progress, Nakayama and co-workers suggested that phosphorylation on Ser10 stabilizes p27 (Ishida et al., 2000). We confirmed that the phospho-mimetic p27(S10D) mutant was more stable than the non-phosphorylatable p27(S10A) mutant; the estimated half-lives of p27(S10D) and p27(S10A) were 9.5 and 5.9 h, respectively, whereas wild-type p27 was degraded at a rate intermediate between that of p27(S10D) and p27(S10A) mutants (Figure 3).

Fig. 3. Phosphorylation of p27 on Ser10 increases its half-life. (A) Rat1 cells were transfected with HA-tagged p27 wild-type or p27(Ser10) mutants and serum deprived for 24 h. The cells were then pulse-labeled for 2 h with [35S]methionine or [35S]cysteine and chased for the times indicated in fresh medium containing 10% serum. Cell lysates were subjected to immunoprecipitation with anti-HA antibody and the labeled p27 protein was analyzed by SDS–gel electrophoresis and fluorography. (B) Densitometric analysis of p27 degradation rate. Data points correspond to the experiment shown in (A). The half-life values of wild-type and mutant p27 represent the mean ± SEM of four independent pulse–chase experiments.
We next explored the molecular basis for the stabilization effect of Ser10 phosphorylation on p27 turnover in vivo. We first examined the impact of the S10D mutation on the phosphorylation of p27 on Thr187 and its association with the F-box protein Skp2. For these experiments, cellular extracts prepared from Rat1 cells transiently transfected with the different p27-HA mutants were incubated with in vitro translated Skp2. The p27-HA complexes were isolated by anti-HA immunoprecipitation and analyzed by autoradiography and immunoblotting. Mutation of Ser10 to Ala or Asp did not affect the ability of p27-HA to associate with Skp2, while under the same conditions, the T187A mutant failed to co-precipitate labeled Skp2 (Figure 4). These experiments also showed that p27 Ser10 mutants are phosphorylated on Thr187 to the same extent as the wild-type protein, consistent with the results of in vitro phosphorylation (Figure 1E). These data indicate that phosphorylation of Ser10 does not impact on the targeting of p27 by the SCFSkp2 ubiquitin–protein ligase complex.
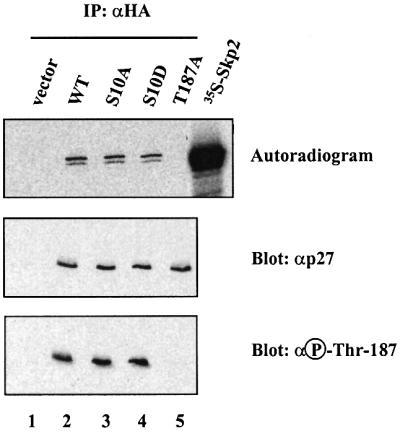
Fig. 4. Phosphorylation of p27 on Ser10 does not affect its interaction with the F-box protein Skp2. Rat1 cells were transfected with HA-tagged p27 wild-type or p27 mutants and treated for 5 h with 5 µM MG132 (to increase the amount of p27 phosphorylated on Thr187). Lysates were prepared, incubated with in vitro translated [35S]Myc6Skp2 for 3 h and subjected to immunoprecipitation with anti-HA antibody. The precipitated proteins were resolved by SDS–gel electrophoresis, transferred to nitrocellulose membrane and visualized by autoradiography (upper). The membrane was further subjected to immunoblot analysis with antibodies to p27 (middle) and phospho-Thr187 (lower).
To confirm that p27 Ser10 mutants are still ubiquitylated, we used an established purified recombinant system (Ganoth et al., 2001), which includes the four components of the SCF ubiquitin ligase (Skp1, Cul1, Skp2 and Roc1/Rbx1), Cks1, the ubiquitin-conjugating enzyme Ubc3, the ubiquitin-activating enzyme E1 and cyclin E–Cdk2 complex. We performed in vitro ubiquitylation assays using this purified system and in vitro translated 35S-labeled wild-type p27 and Ser10 mutants as substrates (Figure 5). Both p27(S10A) and p27(S10D) mutants were efficiently ubiquitylated in vitro to the same extent as the wild-type protein (∼75% of ubiquitin conjugation after 1 h reaction). Accordingly, both Ser10 mutants were degraded in vitro with the same kinetics as the wild-type protein (data not shown). Thus, phosphorylation on Ser10 does not affect p27 ubiquitylation or stability in vitro.

Fig. 5. Phosphorylation on Ser10 does not affect p27 in vitro ubiquitylation. 35S-labeled in vitro translated p27 wild-type (lanes 1–5), p27(S10A) mutant (lanes 6–10) or p27(S10D) mutant (lanes 11–15) were subjected to a ubiquitylation reaction for the times indicated using purified components as described in Materials and methods. The reaction products were analyzed by SDS–gel electrophoresis followed by autoradiography. The bracket on the left side marks a ladder of bands corresponding to polyubiquitylated p27.
Phosphorylation on Ser10 is necessary for the nuclear export of p27
Since Ser10 mutants of p27 are ubiquitylated and degraded efficiently in vitro while expression of p27(S10D) is stabilized in vivo, we reasoned that in the cell-free system a regulatory step was missing. One such regulatory mechanism could be p27 transport, since the subcellular localization of p27 appears to play an important role in controlling its stability (Tomoda et al., 1999). Therefore, we evaluated the role of Ser10 phosphorylation on p27 cellular localization. We first examined the subcellular distribution of endogenous p27 during cell cycle re- entry of Rat1 cells. In quiescent cells, p27 was localized almost exclusively in the nucleus (Figure 6A, t = 0 h). Stimulation with serum promoted the redistribution of a fraction of p27 to the cytoplasm in all cells (Figure 6A, t = 4 and 8 h, top). After 12 h of stimulation, the staining of p27 decreased in parallel with the abundance of the protein (data not shown). Detailed kinetic studies revealed that the cytoplasmic relocalization of p27 precedes activation of Cdk2, induction of Skp2 and degradation of the bulk of p27 protein (Figure 6B).

Fig. 6. Mitogenic stimulation promotes the nuclear export of a fraction of p27. (A) Rat1 cells were made quiescent by serum starvation and restimulated to enter the cell cycle by addition of 10% serum for the times indicated. The subcellular localization of endogenous p27 was determined by fluorescence microscopy. When present, LMB (2 ng/ml) was added simultaneously with serum. (B) Rat1 cells were made quiescent and restimulated with serum for the times indicated. The expression of p27 and Skp2 was monitored by immunoblotting using specific antibodies. The activity of Cdk2 was assayed using histone H1 as substrate.
To confirm these findings, p27-HA was ectopically expressed in Rat1 cells and its localization was visualized by immunofluorescence with an antibody against the HA epitope. Under the transfection conditions used for these studies, ∼50% of cells expressing p27 enter S phase after 20 h of serum stimulation (data not shown). In serum-starved cells, p27-HA was predominantly nuclear (Figure 7A and B). Addition of serum caused a time-dependent translocation of p27-HA to the cytoplasm, with ∼35% of cells displaying cytoplasmic staining after 24 h (Figure 7A and B). A higher proportion of cells exhibited cytoplasmic redistribution of endogenous p27 as compared with ectopic p27-HA, which possibly reflects the inhibitory effect of ectopic p27 expression on cell cycle progression. Treatment of Rat1 cells with leptomycin B (LMB), a specific inhibitor of CRM1-dependent nuclear export (Nishi et al., 1994), completely prevented the cytoplasmic relocalization of endogenous and ectopic p27 (Figures 6A, bottom and 7C).

Fig. 7. Phosphorylation of Ser10 is necessary for the nuclear to cytoplasmic translocation of p27 upon cell cycle re-entry. (A) Rat1 cells were transfected with HA-tagged p27 wild-type or Ser10 mutants and serum starved for 24 h. The cells were then stimulated with 10% serum for the times indicated. The subcellular localization of ectopic p27 was determined by fluorescence microscopy after staining with anti-HA antibody. (B) Quantitative evaluation of the cellular localization of p27 wild-type and Ser10 mutants. At least 300 cells were scored for each coverslip. The results are expressed as the percentage of cells showing both nuclear and cytoplasmic staining. The graph represents the mean ± SEM of three separate experiments. (C) Effect of LMB. Rat1 cells were transfected with HA-tagged p27 wild-type or Ser10 mutants. After 24 h, the cells were made quiescent and restimulated with 10% serum for 24 h in the absence or presence of 2 ng/ml LMB. The results are expressed as the percentage of cells showing both nuclear and cytoplasmic staining. The graph represents the mean ± SEM of five separate experiments.
We next evaluated the effect of Ser10 mutations on the subcellular localization of p27. The phospho-mimetic p27(S10D) mutant behaved as the wild-type protein and was relocalized to the cytoplasm upon serum stimulation (Figure 7A and B). In contrast, the Ser10 to Ala mutation almost completely abolished the cytoplasmic redistribution of the protein, indicating that phosphorylation of Ser10 is necessary for nuclear export of p27. We also tested the effect of LMB on the localization of Ser10 mutants. Treatment with LMB completely inhibited the serum-dependent relocalization of the p27(S10D) mutant to the cytoplasm (Figure 7C).
Finally, we examined the localization of endogenous p27 phosphorylated on Ser10 in MCF-7 cells. In quiescent tamoxifen-treated cells, ∼90% of total p27 was detected in the nuclear compartment (Figure 8). Treatment of cells with estradiol, which allows cell cycle re-entry, caused the redistribution of a fraction of p27 to the cytoplasm, similar to what was observed in Rat1 cells. We also analyzed the distribution of the Ser10-phosphorylated pool of p27 in the same conditions using the anti-phospho-Ser10-specific antibody (Figure 8). We observed that in arrested MCF-7 cells, the Ser10-phosphorylated form of p27 (∼33% of total p27 as quantified in Figure 2D) was localized mainly in the nucleus. However, when the cells were stimulated with estradiol, Ser10-phosphorylated p27 (∼5% of total p27) was found exclusively in the cytoplasm. Altogether, these data are consistent with the hypothesis that phosphorylation of Ser10 provides a necessary but not sufficient signal for CRM1-dependent nuclear to cytoplasmic transport of p27 during cell cycle progression.

Fig. 8. p27 phosphorylated on Ser10 is localized into the cytoplasm of proliferating cells. MCF-7 cells were growth arrested by depletion of estradiol and treatment with tamoxifen for 48 h. The cells were then restimulated to enter the cell cycle by addition of 500 nM estradiol for a period of 8 h. The subcellular localizations of endogenous p27 and Ser10 phosphorylated p27 were determined by immunofluorescence using anti-p27 and anti-phospho-Ser10, respectively. Exposure time in proliferating cells was enhanced to better visualize cytoplasmic p27.
Degradation of p27 occurs efficiently in the nucleus
It has been suggested that translocation of p27 to the cytoplasm is associated with its subsequent degradation (Tomoda et al., 1999). However, we observed that the p27(S10A) mutant, which remains in the nucleus, is degraded efficiently upon cell cycle re-entry. These results suggest that degradation of p27 can occur in the nucleus. To confirm this result, we compared the turnover rate of endogenous p27 in Rat1 cells treated with or without LMB (Figure 9). Inhibition of nuclear export with LMB did not prevent the degradation of p27 but rather decreased the half-life of the protein, consistent with the fact that p27(S10A), which is found exclusively in the nucleus, is degraded efficiently (Figure 3). These results clearly demonstrate that p27 is degraded efficiently in the nucleus.

Fig. 9. Effect of LMB on the degradation rate of endogenous p27. (A) Rat1 cells were made quiescent by serum starvation for 24 h. Pulse–chase experiments were performed as described in the legend to Figure 3 except that LMB was added during the chase period. The endogenous p27 protein was immunoprecipitated with anti-p27 and analyzed by SDS–gel electrophoresis and fluorography. (B) Densitometric analysis of p27 degradation rate.
Discussion
We report here that cellular p27 is phosphorylated on Ser10, a putative target site for proline-directed kinases, during the G1 phase of the cell cycle. This site is conserved in all mammalian p27 homologs, but not in the related proteins p21Cip1 and p57Kip2. Interestingly, a similar Ser-Pro motif is also found at position 16 in the Xenopus Cip/Kip homolog p27Xic1 (Su et al., 1995). We show that phosphorylation of Ser10 has a significant impact on the subcellular localization and consequent turnover of the p27 protein. Moreover, we provide strong evidence that p27 is degraded in the nuclear compartment.
The activity of p27 is regulated by its subcellular compartmentalization. To interact with its targets (Cdk2 complexes) and consequently inhibit G1 progression, p27 needs to be transported into the nucleus (Reynisdottir and Massague, 1997; Orend et al., 1998; Soucek et al., 1998; Tomoda et al., 1999). In this study, we have shown that endogenous p27 protein is found almost exclusively in the nucleus in G0/G1 cells and that a fraction of p27 is translocated to the cytoplasm in response to mitogenic signaling (Figures 6 and 7). Furthermore, using a phospho-Ser10-specific antibody, we found that the fraction of p27 phosphorylated on Ser10 is localized exclusively in the cytoplasm of proliferating cells (Figure 8). This cytoplasmic redistribution of p27 is critically dependent on the availability and phosphorylation of the Ser10 residue, since the p27(S10D) mutant, which mimics phosphorylation, is efficiently exported to the cytoplasm whereas the p27(S10A) mutant is not (Figure 7). However, although necessary, phosphorylation of Ser10 is not sufficient to induce nuclear export of p27 as the p27(S10D) mutant or the wild-type protein (phosphorylated on Ser10) are not exported efficiently in G0/G1 cells. Therefore, another signal provided by serum growth factors appears necessary to direct p27 to the cytoplasm. These findings are reminiscent of the regulation of p21_Cip1_, which is relocalized to the cytoplasm and inactivated through phosphorylation of Thr145 by Akt (Zhou et al., 2001).
The observation that LMB treatment inhibits the nucleo-cytoplasmic redistribution of wild-type p27 and the p27(S10D) mutant (Figures 6 and 7) strongly suggests that phosphorylation of Ser10 modulates p27 localization by promoting its active nuclear export. Examination of the p27 sequence does not reveal any motif with obvious homology to leucine-rich nuclear export signals (Fornerod et al., 1997), suggesting that an additional bridging factor may be required for binding to and export by CRM1. One putative candidate for such a factor is Jab1, which interacts with p27 and induces its cytoplasmic accumulation upon overexpression (Tomoda et al., 1999). As yet, we have been unable to detect any physical interaction between Jab1 and p27 (either wild type or mutants; data not shown). Future studies are warranted to clarify the role of Jab1 in p27 nuclear export. In their study, Tomoda et al. (1999) also reported that overexpression of Jab1 accelerates p27 degradation, but failed to identify the mechanism by which it induces this effect. The authors proposed that Jab1 could stimulate p27 phosphorylation on Thr187, which, in turn, induces p27 proteolysis. In light of our results, we propose that Jab1-induced degradation of p27 is an indirect consequence of its ability to stimulate the cell cycle and might not have any relationship with the possible role of Jab1 in p27 nuclear export. An alternative explanation has recently been proposed by Boussiotis et al. (2000), who suggested that it is indeed p27 that inhibits Jab1 by sequestering it in the cytoplasm. Owing to the well established role of Jab1 as a co-activator of Jun transcription factors, this results in defective _trans_-activation of AP-1.
What is the function of the nuclear to cytoplasmic transport of p27 in the G1 phase? The G1 phosphorylation of p27 on Ser10 and the export of a fraction of p27 precede Cdk2 activation, phosphorylation of p27 on Thr187 and expression of Skp2 (Figure 6). These observations are consistent with the idea that p27 export serves to remove this inhibitor from its nuclear targets (such as cyclin E– Cdk2), rather than to promote its degradation. Thus, the Ser10 phosphorylation- and mitogen-dependent translocation of p27 (possibly in its free, non-Cdk2-bound form) to the cytoplasm may serve to lower its effective nuclear concentration under a critical threshold. This, in turn, would allow the activation of the increasing subpopulation of free cyclin E–Cdk2, and the subsequent phosphorylation and degradation of the remaining nuclear p27 by the ubiquitin–proteasome pathway via SCF_Skp2_. The cytoplasmic localization of p27 may also impact on the activity of cyclin D–Cdk4/6 complexes, since previous studies have shown that Cip/Kip family members promote the assembly and activation of cyclin D–Cdk complexes (LaBaer et al., 1997; Cheng et al., 1999). Alternatively, cytoplasmic p27 might regulate cellular functions that are apparently Cdk independent, such as cell migration (Nagahara et al., 1998).
While this work was in progress, Nakayama and colleagues have shown that upon overexpression in cells, p27 is phosphorylated on Ser10 and that this phosphorylation stabilizes its expression (Ishida et al., 2000). Our data indicate that phosphorylation of Ser10 does not directly control p27 stability, since p27(S10D) and p27(S10A) mutants are both recognized by Skp2 in vitro and in vivo (Figure 4 and data not shown) and are ubiquitylated and degraded in vitro with identical kinetics (Figure 5 and data not shown). Instead, we suggest that the shorter half-life of the p27(S10A) mutant is due to its prevalent nuclear localization, which results in a more efficient degradation than that observed for a p27 protein able to translocate to the cytoplasm. Indeed, our results clearly demonstrate that p27 is degraded efficiently within the nucleus and that cytoplasmic localization is not a prerequisite for p27 degradation. This conclusion is supported by the following observations. (i) Treatment with LMB does not suppress, but accelerates, the turnover rate of endogenous p27 protein (Figure 9). (ii) The p27(S10A) mutant, which is not exported efficiently from the nucleus, has a shorter half-life than the wild-type protein (Figure 3). (iii) Recent studies have demonstrated that the Xenopus homolog of p27, p27Xic1, is ubiquitylated and degraded exclusively in the nucleus (Swanson et al., 2000; Chuang and Yew, 2001). (iv) p27 fused to a nuclear export signal that sequesters it into the cytoplasm is not less stable than wild-type p27 (Tomoda et al., 1999). (v) The 26S proteasome is localized both in the cytoplasmic and nuclear compartments (Rivett, 1998). (vi) While the generic components of the SCF ligase (Cul1, Skp1 and Roc1/Rbx1) are present both in the nucleus and cytoplasm, the p27-specific SCF subunit Skp2, as well as its co-factor Cks1, is found exclusively in the nucleus (Lisztwan et al., 1998; T.Bashir and M.Pagano, unpublished observations). The relative contributions of the nuclear and cytoplasmic compartments in the degradation of different subpopulations of p27 remain to be determined.
Materials and methods
Cell culture, cell synchronization and transfections
Rat1 cells were cultured and synchronized by serum starvation as described previously (Meloche, 1995). Rat1 cells were transiently transfected with p27 constructs using FuGENE 6 transfection reagent (Roche Molecular Biochemicals). Asynchronous human MCF-7 breast cancer cells were arrested by transferring them to phenol red-free medium supplemented with 5% charcoal-stripped calf serum in the presence of 1 µM tamoxifen (Sigma) for 48 h. The cells were then released from quiescence by addition of 500 nM estradiol (Sigma). NIH 3T3 mouse fibroblasts were synchronized in G0/G1 by serum starvation for 48 h and then stimulated to enter the cell cycle by re-addition of serum. T lymphocytes in suspension were prepared from mouse lymph nodes as described (Latres et al., 2001) and stimulated with a mitogenic combination of concanavalin A (5 µg/ml) and interleukin-2 (50 ng/ml).
Reagents, antibodies and DNA constructs
LMB was a gift from M.Yoshida (University of Tokyo). MG132 was obtained from Biomol. Monoclonal antibody (mAb) 12CA5 to influenza HA protein was a gift from M.Dennis (SignalGene). The p27 phospho-Ser10-specific antibody was generated in collaboration with Zymed by immunizing rabbits with the synthetic peptide RVSNG*SPSLERMDC, corresponding to amino acids 5–18 of human p27 sequence with a phosphoserine at position 10 (*S). The antibody was purified from serum by two rounds of affinity chromatography on a phospho-Ser10 peptide column followed by a non-phosphopeptide column. The polyclonal antibody specific to phosphorylated Thr187 of p27, as well as antibodies to cyclin A, cyclin E and cyclin D1, have been described previously (Montagnoli et al., 1999; Carrano and Pagano, 2001). Commercial antibodies were from the following suppliers: polyclonal anti-p27 antibody (sc-528), Santa-Cruz Biotechnology; anti-p27 mAb, Zymed and Transduction Laboratories; anti-HA mAb for immunofluorescence applications (F-7), Santa-Cruz Biotechnology; anti-Skp2 antibody, Zymed; biotinylated anti-rabbit IgG, fluorescein–avidin D and fluorescein–streptavidin, Vector Laboratories and fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG, Jackson Laboratories.
All recombinant p27 DNA constructs were derived from the human cDNA (Polyak et al., 1994). To generate a p27 cDNA tagged with an HA epitope (p27-HA), a synthetic oligonucleotide encoding the amino acid sequence YDVPDYASL was inserted at the C-terminus of p27 cDNA using the altered sites in an in vitro mutagenesis system (Promega). The p27 and p27-HA cDNAs were used as templates for in vitro mutagenesis to replace Ser10 with Ala (S10A) or Asp (S10D) and Thr187 with Ala (T187A). The cDNAs encoding the various p27 proteins were subcloned into the _Eco_RI site of the expression vector pcDNA3 (Invitrogen). The human Skp2 cDNA was ligated into the _Eco_RI site of pcDNA3-Myc6 (P.Coulombe and S.Meloche, unpublished data).
In vitro phosphorylation of p27
Recombinant purified His-p27 (1 µg) was incubated for 1 h at 30°C in the presence of 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM ATP and 200 ng of recombinant purified cyclin E–Cdk2 or 20 U of activated ERK2 (New England Biolabs). For in vitro kinase assays, NIH 3T3 cells were lysed using the nitrogen disruption bomb as described (Montagnoli et al., 1999) and 100 µg of extract were used for each time point in the kinase reaction. Recombinant proteins were produced and purified as described (Montagnoli et al., 1999).
In vivo phosphorylation analysis and phosphopeptide mapping
Quiescent Rat1 cells in 100 mm Petri dishes were stimulated with serum for the times indicated and labeled for the last 4 h in bicarbonate and phosphate-free HEPES-buffered MEM containing 0.5 mCi/ml [32P]phosphoric acid and 10% dialyzed calf serum. For analysis of ectopically expressed p27, exponentially growing Rat1 cells were labeled 48 h after the transfection. The cells were then washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in Triton X-100 lysis buffer (Servant et al., 2000). After clarification, the lysates were incubated for 4 h at 4°C with either 10 µl of anti-p27 antibody (endogenous protein) or 3 µl of anti-HA antibody 12CA5 pre-adsorbed on protein A–Sepharose beads (Pharmacia). The immune complexes were washed with Triton X-100 lysis buffer and fractionated by SDS–gel electrophoresis. The proteins were then transferred to PVDF membranes and visualized by autoradiography. Phosphopeptide mapping was performed as described previously (Fleurent et al., 1997).
Immunoblot analysis and Cdk2 kinase assay
Cell lysis, immunoprecipitation and immunoblot analysis were performed as described previously (Servant et al., 2000; Carrano and Pagano, 2001). For association studies, Skp2 was produced by in vitro transcription– translation of pcDNA3-Myc6 Skp2 with T7 RNA polymerase (Promega) in the presence of [35S]methionine and mixed with lysates of exponentially growing Rat1 cells transfected with the various p27-HA constructs. After 3 h of incubation at 4°C, the mixed extracts were incubated with 12CA5 antibody and the immunoprecipitated proteins were analyzed by autoradiography. Immunoblot analysis of p27 Thr187 phosphorylation was carried out according to the manufacturer’s specifications. The phosphotransferase activity of Cdk2 was measured by immune complex kinase assay using histone H1 as substrate as described (Meloche, 1995).
In vitro ubiquitylation and degradation assay
The in vitro ubiquitylation assay was performed in a 10 µl reaction volume containing 40 mM Tris–HCl pH 7.6, 5 mM NaCl, 1 mM DTT, 10% glycerol (v/v), 10 mM phosphocreatine, 100 µg/ml creatine phosphokinase, 0.5 mM ATP, 1 mg/ml soybean trypsin inhibitor, 1 µM ubiquitin aldehyde, 1 mg/ml methylated ubiquitin, 1 pmol of E1, 50 pmol of Ubc3, 0.25 µl of Skp2–Skp1, 0.25 µl of Roc1-Cul-1, 1 ng of Cks1, 0.1 µl of cyclin E–Cdk2 and 0.5 µl of in vitro translated 35S-p27 (Ganoth et al., 2001). After incubation at 30°C, samples were analyzed by SDS–gel electrophoresis and autoradiography. The degradation reaction was performed in the same buffer with the exception that methyl ubiquitin was substituted by ubiquitin and 30 µg of HeLa cell extract (prepared by hypotonic lysis and supplemented with 0.25 µl of Skp2–Skp1 and 0.1 µl of cyclin E–Cdk2) were used instead of the purified system.
Pulse–chase experiments
To examine the turnover of p27 mutant proteins, Rat1 cells were serum starved for 24 h and pulse labeled for 2 h with 120 µCi/ml [35S]methionine and [35S]cysteine (ICN). The cells were then chased for the times indicated in MEM medium containing 10% calf serum and excess methionine and cysteine. After washing with ice-cold PBS, cellular lysates were prepared and subjected to immunoprecipitation with anti-p27 or anti-HA antibody 12CA5 as described above. Immune complexes were washed five times with Triton X-100 lysis buffer and the eluted proteins were analyzed by SDS–gel electrophoresis on 12% acrylamide gels. The p27 protein was detected by fluorography and quantified using a PhosphorImager.
Immunofluorescence studies
For localization of endogenous p27, Rat1 cells were plated onto glass coverslips and serum starved for 24 h. The cells were then stimulated with 10% calf serum and fixed at the times indicated with 3.7% paraformaldehyde/PBS for 20 min at 37°C. After quenching in 0.1 M glycine for 5 min, the cells were permeabilized by incubation in 0.1% Triton X-100 for 5 min at room temperature. Staining was performed by incubating cells for 1 h at 37°C with anti-p27 antibody, followed by incubation with biotinylated anti-rabbit IgG for 1 h and then with fluorescein–avidin D for 30 min. For localization of endogenous p27 phosphorylated on Ser10, MCF-7 cells were synchronized as indicated and staining was performed by incubation with anti-phospho-Ser10 antibody, followed by incubation with biotinylated anti-rabbit IgG and fluorescein–streptavidin. For localization of ectopically expressed p27, the cells were growth arrested 24 h after transfection, stimulated with serum and processed as described. Staining was performed using anti-HA as primary antibody and FITC-conjugated anti-mouse IgG as secondary reagent. Cell samples were viewed by fluorescence microscopy (Leica DM RB). In some experiments, exposures were enhanced to help visualize the pool of cytoplasmic p27. A minimum of 300 cells were scored for each coverslip. The results are expressed as the percentage of cells showing both nuclear and cytoplasmic staining. For experiments with LMB, the drug was added at a final concentration of 2 ng/ml.
Acknowledgments
Acknowledgements
We thank E.Latres, J.Massagué, D.Morgan, M.Yoshida and H.Zhang for reagents, N.V.Dorrello, R.Piva and M.Rialland for their contribution to this work, C.Charbonneau and A.Marcil for help with fluorescence microscopy, and M.Saba-El-Leil and A.Hershko for suggestions. We also thank J.Slingerland and M.Boehm for communicating their results before publication. M.P. is grateful to L.Yamasaki for continuous support. G.R. and P.C. are recipients of fellowships from the National Cancer Institute of Canada and the Canadian Institutes for Health Research, respectively. L.D.M. is a recipient of a fellowship from the American Italian Cancer Foundation. M.P. is recipient of the Irma T.Hirschl Scholarship. S.M. is an Investigator of the Canadian Institutes for Health Research. This work was supported by grants from the Canadian Institutes for Health Research (MOP-14168) to S.M., the Human Frontier Science Program Organization (RG0229) and the NIH (R01-CA76584 and R01-GM57587) to M.P.
References
- Agrawal D., Hauser,P., McPherson,F., Dong,F., Garcia,A. and Pledger,W.J. (1996) Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol. Cell. Biol., 16, 4327–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussiotis V.A., Freeman,G.J., Taylor,P.A., Berezovskaya,A., Grass,I., Blazar,B.R. and Nadler,L.M. (2000) p27kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nature Med., 6, 290–297. [DOI] [PubMed] [Google Scholar]
- Carrano A.C. and Pagano,M. (2001) Role of the F-box protein Skp2 in adhesion-dependent cell cycle progression. J. Cell Biol., 153, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A.C., Eytan,E., Hershko,A. and Pagano,M. (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature Cell Biol., 1, 193–199. [DOI] [PubMed] [Google Scholar]
- Cheng M., Olivier,P., Diehl,J.A., Fero,M., Roussel,M.F., Roberts,J.M. and Sherr,C.J. (1999) The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J., 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang L.C. and Yew,P.R. (2001) Regulation of nuclear transport and degradation of the Xenopus cyclin-dependent kinase inhibitor, p27Xic1. J. Biol. Chem., 276, 1610–1617. [DOI] [PubMed] [Google Scholar]
- Ekholm S.V. and Reed,S.I. (2000) Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol., 12, 676–684. [DOI] [PubMed] [Google Scholar]
- Fleurent M., Gingras,A.C., Sonenberg,N. and Meloche,S. (1997) Angiotensin II stimulates phosphorylation of the translational repressor 4E-binding protein 1 by a mitogen-activated protein kinase-independent mechanism. J. Biol. Chem., 272, 4006–4012. [DOI] [PubMed] [Google Scholar]
- Fornerod M., Ohno,M., Yoshida,M. and Mattaj,I.W. (1997) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell, 90, 1051–1060. [DOI] [PubMed] [Google Scholar]
- Ganoth D., Bornstein,G., Ko,T.K., Larsen,B., Tyers,M., Pagano,M. and Hershko,A. (2001) The cell-cycle regulatory protein Cks1 is required for SCFSkp2-mediated ubiquitinylation of p27. Nature Cell Biol., 3, 321–324. [DOI] [PubMed] [Google Scholar]
- Hengst L. and Reed,S.I. (1996) Translational control of p27Kip1 accumulation during the cell cycle. Science, 271, 1861–1864. [DOI] [PubMed] [Google Scholar]
- Hengst L. and Reed,S.I. (1998) Inhibitors of the Cip/Kip family. Curr. Top. Microbiol. Immunol., 227, 25–41. [DOI] [PubMed] [Google Scholar]
- Ishida N., Kitagawa,M., Hatakeyama,S. and Nakayama,K. (2000) Phosphorylation at serine 10, a major phosphorylation site of p27Kip1, increases its protein stability. J. Biol. Chem., 275, 25146–25154. [DOI] [PubMed] [Google Scholar]
- LaBaer J., Garrett,M.D., Stevenson,L.F., Slingerland,J.M., Sandhu,C., Chou,H.S., Fattaey,A. and Harlow,E. (1997) New functional activities for the p21 family of CDK inhibitors. Genes Dev., 11, 847–862. [DOI] [PubMed] [Google Scholar]
- Latres E., Chiarle,R., Schulman,B.A., Pavletich,N.P., Pellicer,A., Inghirami,G. and Pagano,M. (2001) Role of the F-box protein Skp2 in lymphomagenesis. Proc. Natl Acad. Sci. USA, 98, 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisztwan J., Marti,A., Sutterluty,H., Gstaiger,M., Wirbelauer,C. and Krek,W. (1998) Association of human CUL-1 and ubiquitin-conjugating enzyme CDC34 with the F-box protein p45SKP2: evidence for evolutionary conservation in the subunit composition of the CDC34-SCF pathway. EMBO J., 17, 368–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloche S. (1995) Cell cycle reentry of mammalian fibroblasts is accompanied by the sustained activation of p44mapk and p42mapk isoforms in the G1 phase and their inactivation at the G1/S transition. J. Cell Physiol., 163, 577–588. [DOI] [PubMed] [Google Scholar]
- Millard S.S., Yan,J.S., Nguyen,H., Pagano,M., Kiyokawa,H. and Koff,A. (1997) Enhanced ribosomal association of p27Kip1 mRNA is a mechanism contributing to accumulation during growth arrest. J. Biol. Chem., 272, 7093–7098. [DOI] [PubMed] [Google Scholar]
- Miskimins W.K., Wang,G., Hawkinson,M. and Miskimins,R. (2001) Control of cyclin-dependent kinase inhibitor p27 expression by cap-independent translation. Mol. Cell. Biol., 21, 4960–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A., Fiore,F., Eytan,E., Carrano,A.C., Draetta,G.F., Hershko,A. and Pagano,M. (1999) Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev., 13, 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D., Bouchard,C., Rudolph,B., Steiner,P., Stuckmann,I., Saffrich,R., Ansorge,W., Huttner,W. and Eilers,M. (1997) Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E–cdk2 complexes. Oncogene, 15, 2561–2576. [DOI] [PubMed] [Google Scholar]
- Muller D., Thieke,K., Burgin,A., Dickmanns,A. and Eilers,M. (2000) Cyclin E-mediated elimination of p27 requires its interaction with the nuclear pore-associated protein mNPAP60. EMBO J., 19, 2168–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara H., Vocero-Akbani,A.M., Snyder,E.L., Ho,A., Latham,D.G., Lissy,N.A., Becker-Hapak,M., Ezhevsky,S.A. and Dowdy,S.F. (1998) Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nature Med., 4, 1449–1452. [DOI] [PubMed] [Google Scholar]
- Nakayama K. et al. (2000) Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO J., 19, 2069–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi K., Yoshida,M., Fujiwara,D., Nishikawa,M., Horinouchi,S. and Beppu,T. (1994) Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J. Biol. Chem., 269, 6320–6324. [PubMed] [Google Scholar]
- Orend G., Hunter,T. and Ruoslahti,E. (1998) Cytoplasmic displacement of cyclin E–cdk2 inhibitors p21Cip1 and p27Kip1 in anchorage-independent cells. Oncogene, 16, 2575–2583. [DOI] [PubMed] [Google Scholar]
- Pagano M., Tam,S.W., Theodoras,A.M., Beer-Romero,P., Del Sal,G., Chau,V., Yew,P.R., Draetta,G.F. and Rolfe,M. (1995) Role of the ubiquitin–proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science, 269, 682–685. [DOI] [PubMed] [Google Scholar]
- Polyak K., Lee,M.H., Erdjument-Bromage,H., Koff,A., Roberts,J.M., Tempst,P. and Massague,J. (1994) Cloning of p27Kip1 a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell, 78, 59–66. [DOI] [PubMed] [Google Scholar]
- Reynisdottir I. and Massague,J. (1997) The subcellular locations of p15Ink4b and p27Kip1 coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev., 11, 492–503. [DOI] [PubMed] [Google Scholar]
- Rivett A.J. (1998) Intracellular distribution of proteasomes. Curr. Opin. Immunol., 10, 110–114. [DOI] [PubMed] [Google Scholar]
- Servant M.J., Coulombe,P., Turgeon,B. and Meloche,S. (2000) Differential regulation of p27Kip1 expression by mitogenic and hypertrophic factors: involvement of transcriptional and posttranscriptional mechanisms. J. Cell Biol., 148, 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaff R.J., Groudine,M., Gordon,M., Roberts,J.M. and Clurman,B.E. (1997) Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev., 11, 1464–1478. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev., 9, 1149–1163. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Slingerland J. and Pagano,M. (2000) Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell Physiol., 183, 10–17. [DOI] [PubMed] [Google Scholar]
- Soos T.J. et al. (1996) Formation of p27–CDK complexes during the human mitotic cell cycle. Cell Growth Differ., 7, 135–146. [PubMed] [Google Scholar]
- Soucek T., Yeung,R.S. and Hengstschlager,M. (1998) Inactivation of the cyclin-dependent kinase inhibitor p27 upon loss of the tuberous sclerosis complex gene-2. Proc. Natl Acad. Sci. USA, 95, 15653–15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruck C., Strohmaier,H., Watson,M., Smith,A.P., Ryan,A., Krek,W. and Reed,S.I. (2001) A CDK-independent function of mammalian Cks1. Targeting of SCFSkp2 to the CDK inhibitor p27Kip1. Mol. Cell, 7, 639–650. [DOI] [PubMed] [Google Scholar]
- Su J.Y., Rempel,R.E., Erikson,E. and Maller,J.L. (1995) Cloning and characterization of the Xenopus cyclin-dependent kinase inhibitor p27XIC1. Proc. Natl Acad. Sci. USA, 92, 10187–10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterluty H., Chatelain,E., Marti,A., Wirbelauer,C., Senften,M., Muller,U. and Krek,W. (1999) p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nature Cell Biol., 1, 207–214. [DOI] [PubMed] [Google Scholar]
- Swanson C., Ross,J. and Jackson,P.K. (2000) Nuclear accumulation of cyclin E–Cdk2 triggers a concentration-dependent switch for the destruction of p27Xic1. Proc. Natl Acad. Sci. USA, 97, 7796–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda K., Kubota,Y. and Kato,J. (1999) Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature, 398, 160–165. [DOI] [PubMed] [Google Scholar]
- Tsvetkov L.M., Yeh,K.H., Lee,S.J., Sun,H. and Zhang,H. (1999) p27Kip1 ubiquitination and degradation is regulated by the SCFSkp2 complex through phosphorylated Thr187 in p27. Curr. Biol., 9, 661–664. [DOI] [PubMed] [Google Scholar]
- Vlach J., Hennecke,S. and Amati,B. (1997) Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J., 16, 5334–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y., Hirano,K., Hirano,M., Nishimura,J. and Kanaide,H. (2000) Minimal requirements for the nuclear localization of p27Kip1, a cyclin-dependent kinase inhibitor. Biochem. Biophys. Res. Commun., 274, 37–42. [DOI] [PubMed] [Google Scholar]
- Zhou B.P., Liao,Y., Xia,W., Spohn,B., Lee,M.H. and Hung,M.C. (2001) Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature Cell Biol., 3, 245–252. [DOI] [PubMed] [Google Scholar]