A Study of Low pH-Induced Refolding of Env of Avian Sarcoma and Leukosis Virus into a Six-Helix Bundle (original) (raw)
Abstract
The fusion protein of avian sarcoma and leukosis virus is likely to fold into a six-helix bundle as part of its final configuration. A peptide, R99, inhibits fusion, probably by binding into the grooves of the triple-stranded coiled coil that becomes the central core of the six-helix bundle. The stages at which the envelope protein (Env) of avian sarcoma and leukosis virus subgroup A folds into a bundle during low pH-induced fusion were determined. Effector cells expressing Env were bound to target cells expressing the cognate receptor Tva, and intermediates of fusion were created. R99 was added and the extent of fusion inhibition was used to distinguish between a prebundle state with exposed grooves and a state in which the grooves were no longer exposed. The native conformation of Env was not sensitive to R99. But adding a soluble form of Tva to effector cells conferred sensitivity. Acidic pH applied at low temperature created an intermediate state of local hemifusion. Surprisingly, R99 caused these locally hemifused membranes to separate. This indicates that the grooves of Env were still exposed, that prebundle configurations of Env stabilized hemifused states, and that binding of R99 altered the conformation of Env. In the presence of an inhibitory lipid that blocks fusion before hemifusion, applying low pH at 37°C created an intermediate in which R99 was without effect. This suggests that the six-helix bundle can form before hemifusion and that subsequent conformational changes, such as formation of the trimeric hairpin, are responsible for pore formation and/or growth.
INTRODUCTION
Class I fusion proteins of envelope viruses, by definition, fold into six-helix bundles (6HBs) (Skehel and Wiley, 1998; Eckert and Kim, 2001; Colman and Lawrence, 2003). These proteins are assembled from three identical monomers and each monomer consists of two subunits. The surface subunit binds to receptors on target cells, and the transmembrane (TM) subunit is responsible for membrane fusion. At some point in the process of membrane fusion, the three TM subunits fold into a hairpin structure; the N-terminal segments are in the form of a trimeric helical coiled coil and the C-terminal segments are bound peripherally, in an antiparallel orientation, into each of the hydrophobic grooves within the coiled coil. This creates a rigid, rodlike structure known as a “trimeric hairpin” (Chan and Kim, 1998). The stretch of the trimeric hairpin where bound C-terminal segments are folded into _α_-helices is the portion of the hairpin known as the 6HB.
The final postfusion, trimeric hairpin structure for several class I proteins has been determined; it is likely that all proteins in this class have their fusion peptides and membrane-spanning domains (MSDs) in close proximity (Hughson, 1997; Weissenhorn et al., 1997; Chan and Kim, 1998). Recently, x-ray crystallography has determined the structure of the ectodomains of fusion proteins from dengue virus (Modis et al., 2004), Semliki Forest virus (Gibbons et al., 2004), and tick-borne encephalitis virus (Bressanelli et al., 2004) (all class II proteins which, by definition, do not form 6HBs). Although these ectodomains are missing relevant stretches of amino acids, judging from the structures it is likely that fusion peptides and MSDs are brought into proximity in the ectodomains' final, postfusion conformation. Because this motif of proximity appears to be universal, it is expected to be critical to the fusion process. We and others have speculated as to how the creation of this proximity may be central to the fusion process (Chan and Kim, 1998; Eckert and Kim, 2001; Russell et al., 2001; Borrego-Diaz et al., 2003; Markosyan et al., 2003; Park et al., 2003).
Synthetic peptides composed of the amino acid sequence of the C-terminal helical region of a 6HB bind as helices into the grooves of the coiled coil, and in so doing prevent bundle formation and inhibit fusion. The ability of these peptides to bind into the grooves of the coiled coil allows one to determine the intermediate stage of fusion at which 6HBs form: For proteins that are known to form 6HBs, the bundles are extremely thermostable (Weissenhorn et al., 1998; Lu et al., 1995; Baker et al., 1999; Malashkevich et al., 1999). Therefore, if, after arresting fusion at an intermediate stage, the addition of the peptide inhibits fusion, it follows that the bundle has not yet formed. In contrast, if addition of the peptide is without effect, then either the bundle has already formed or the grooves have become inaccessible for some other reason. Identifying the stage at which a bundle forms allows one to infer the roles that bundle formation may serve in the fusion process. This strategy has been successfully employed to show that for HIV Env and SV5 F glycoproteins, membrane merger must occur in order for bundles to form (Melikyan et al., 2000; Russell et al., 2001), and in the case of HIV, all relevant copies of Env do not fold into a bundle until after a fusion pore has formed (Markosyan et al., 2003).
Both HIV and SV5 fuse directly to the plasma membrane at neutral pH. For viruses that fuse within the acidic environment of an endosome, the stages of fusion during which the fusion proteins fold into 6HBs have not been determined. Influenza is the prototypic virus containing a class I fusion protein, hemagglutinin (HA), that requires low pH for fusion. For HA, the 6HB comprises only a small portion of the trimeric hairpin (Chen et al., 1999), and here the stage at which the bundle forms has not yet been identified. The folding of HA into a final trimeric hairpin structure is required for fusion (Borrego-Diaz et al., 2003; Park et al., 2003); this folding induces the formation and enlargement of the fusion pore (Borrego-Diaz et al., 2003).
For avian sarcoma and leukosis virus (ASLV), binding of Env to its receptor induces hydrophobic binding of Env to the target membrane, probably via membrane-insertion of fusion peptides (Hernandez et al., 1997; Damico et al., 1998; Earp et al., 2003; Melikyan et al., 2004). It is now generally agreed that a virus infects a cell only after encapsulation within the low pH environment of an endosome (Mothes et al., 2000; Diaz-Griffero et al., 2002; Narayan et al., 2003; Melikyan et al., 2004). As would be expected, low pH is needed for efficient fusion between cells expressing ASLV Env and target cells expressing the receptor (Mothes et al., 2000; Earp et al., 2003; Melikyan et al., 2004). It has also been biochemically demonstrated that ASLV Env undergoes early conformational changes upon receptor binding, and additional changes upon subsequent exposure to low pH (Mothes et al., 2000; Matsuyama et al., 2004; Smith et al., 2004).
ASLV is divided into 10 subgroups (A–J); each subgroup requires a specific receptor for infectivity (Hunter, 1997). ASLV-A utilizes a relatively small membrane protein, designated Tva (Bates et al., 1993). The ectodomain of Tva has a well-known cysteine-rich motif; this motif is repeated seven times in the low density lipoprotein receptor. A water-soluble form of Tva (sTva) that contains one copy of this motif is sufficient to initiate ASLV entry into receptor-deficient cells (Damico and Bates 2000).
The TM subunit of ASLV Env almost certainly forms a 6HB (although this has not yet been established through crystallography) because i), the hydrophobic pattern of the amino acid sequence contains two heptad repeat domains that should result in a trimeric coiled coil (Gallaher, 1996); ii), the TM subunit sequence is homologous to the fusion subunit GP2 of Ebola, shown by crystallography to form a bundle (Weissenhorn et al., 1998; Malashkevich et al., 1999); and iii), most importantly for this study, a C-terminal peptide, R99—which encompasses the predicted helical portion of the C-terminal segment of ASLV Env—inhibits fusion and infection (Earp et al., 2003). It is not known, however, when in the fusion process the 6HB forms for this low pH-requiring protein.
To study the temporal relation between membrane rearrangements during fusion and the refolding of ASLV Env into a 6HB, we utilized a strategy we originally developed for HIV Env (Melikyan et al., 2000; Markosyan et al., 2003). We arrested intermediates of fusion between effector cells expressing ASLV EnvA and target cells expressing Tva (Melikyan et al., 2004) and added the peptide R99. By optimizing conditions to release the intermediate, we determined whether R99 was able to inhibit fusion. We show that receptor binding exposes the grooves of the coiled coil and we delineate the stages at which low pH causes the R99-binding sites of Env to become sequestered.
MATERIALS AND METHODS
Reagents and cells
The cells for all experiments in this study—effector 3T3 cells constitutively expressing EnvA, as well as the target 293 cells expressing Tva950—have been described previously (Narayan et al., 2003; Melikyan et al., 2004). The inhibitory peptide, R99 derived from the C-terminal helical domain of the TM domain of ASLV Env was originally synthesized at the University of Pennsylvania (Earp et al., 2003). For the majority of the experiments reported here, the peptide was prepared by Macromolecular Resources (Fort Collins, Colorado). The sequence of R99, FNLSDHSESIQKKFQLMKEHVNKIG (N-acetylated and C-amidated), corresponds to residues 99–123 of the TM subunit of Env A. Lauroyl-lysophosphatidylcholine (L-LPC) was purchased from Avanti Polar Lipids (Alabaster, AL); CPZ (chlorpromazine) and BSA were obtained from Sigma (St. Louis, MO); and all fluorescent dyes (calcein AM, CMAC [7-amino-4-chloromethylcoumarin], and DiI) were bought from Molecular Probes (Eugene, OR). A soluble fragment of the entire 83 residue ectodomain of the Tva receptor (sTva) was produced and purified from insect cells as described in (Balliet et al., 1999).
Cell-cell fusion experiments
A two-color video fluorescence microscopy assay was used to monitor cell-cell fusion, as has been described in detail (Melikyan et al., 2000, 2004). Briefly, the effector (E) cells that express EnvA were loaded with calcein AM, and the target (T) cells expressing Tva950 (which we refer to as Tva) were loaded with CMAC according to the manufacturer's instructions. When lipid dye spread was also monitored, a three-color assay was used in which, in addition to aqueous dye labeling, the membranes of target cells were labeled with the lipophilic fluorescent dye, DiI. Effector and target cells were mixed in 1:1 ratio, resuspended in DMEM supplemented with 0.1% BSA (DMEM/BSA), and plated on 8-well slides (LabTech, Naperville, IL) coated with poly-lysine. Unless stated otherwise, to induce fusion, E/T cells were coincubated for 1 h at neutral pH at 37°C and then exposed to pH 5.4 for 10 min via exchange of the bathing solution. Fifteen minutes after reneutralization, the extent of cell-cell fusion was quantified in each well of the slide as the fraction of ∼100 E/T cell pairs for which the two aqueous dyes have mixed. For experiments in which EnvA was receptor-activated by sTva, effector cells were detached from a culture dish by incubation in divalent cation-free PBS. They were pretreated with 500 ng/ml sTva for 30 min at 37°C, and unbound sTva was removed by washing the cells three times with DMEM/BSA. These effector cells were then coincubated with receptor-deficient 293T cells at 37°C for 1 h and fusion was induced by a brief (2 min) exposure to pH 5.4.
Arresting fusion at intermediate stages that precede pore formation
The cold-arrested stage (CAS) of fusion was obtained as previously described (Melikyan et al., 2004). In essence, after coincubating E/T cells, temperature was brought to 4°C and the pH was then transiently (usually for 10 min) lowered to 5.4 and returned to pH 7.2, yielding CAS. To create the lipid-arrested stage (LAS) of fusion, E/T cells were incubated together and 0.2 mg/ml of L-LPC was then added at 23°C for 3 min. The pH was lowered to 5.4 for 3 min (in the presence of L-LPC) and after reneutralization, temperature was reduced to 4°C. The L-LPC was then removed at 4°C by washing three times with DMEM/BSA. This is defined as LAS. When L-LPC was removed at 23°C rather than at 4°C, some fusion occurred. Fusion was triggered from CAS or LAS by raising the temperature to 37°C for 15 min. The R99 peptide was added at different stages of fusion, and maintained unless indicated otherwise. The extent of fusion was then monitored.
Arresting nascent fusion pores formed by ASLV Env
Pore growth was controlled and monitored by a procedure that we previously developed and described in detail (Melikyan et al., 2000; Markosyan et al., 2003). In brief: effector cells, labeled with calcein AM, and unlabeled target cells were adhered on cover slips and coincubated in DMEM/BSA. The cover slips were placed in a chamber that was maintained at 4°C and the chamber was mounted on the stage of the microscope. Fusion of selected cell pairs was triggered by lowering the pH of the solution surrounding them to 5.4 by continuously applying a small volume of an acidic solution around the cells, via a micropipette positioned adjacent to the cells. Simultaneously with lowering of pH, the temperature of the selected cell pairs was increased to 27°C by illuminating with an IR laser diode. As soon as fusion was detected in fluorescence microscopy as the movement of calcein from effector to target cell, temperature was returned to 4°C by shutting off the laser diode and pH was reneutralized by stopping the flow of the acidic solution. The fluorescence intensity was proportional to the concentration of calcein within each of the fused cells, so the rate of change of fluorescence yielded the pore permeability (Markosyan et al., 2003).
RESULTS
R99 inhibits fusion after EnvA is receptor-activated
We coincubated effector and target cells and added R99 at a sequence of arrested intermediates. After a 1-h coincubation of effector and target cells at pH 7.2 (37°C), R99 was added and pH was then lowered. The R99 potently inhibited the cell-cell fusion that normally would have been induced by acidification to pH 5.4. The IC50 of 6.0 nM of R99 (Fig. 1) is reasonably close to its IC50 of ∼25 nM for inhibiting ASLV A infection (Earp et al., 2003).
FIGURE 1.

R99 peptide potently blocks EnvA-induced fusion to Tva-expressing target cells. E/T cells were coincubated at 37°C for 1 h and then exposed to varied concentrations of R99 for 10 min. The pH was then briefly (5 min) lowered to 5.4 and the extent of fusion was determined after an additional 15 min-incubation at neutral pH. The IC50 of R99 was determined by fitting the experimental data with a Langmuir binding isotherm, as described previously (Markosyan et al., 2002). Error bars are standard errors of the mean. Each point was obtained from at least three independent measurements, each carried out in duplicate.
ASLV A virions fuse to target cells that are devoid of receptor when sTva is added to the solution; the extent of infection varies nonlinearly with sTva concentration (Damico and Bates, 2000). We added sTva to effector cells and receptor-deficient target cells, maintaining neutral pH at 37°C for 1 h, and then lowered pH. The extent of fusion varied sigmoidally with the concentration of sTva; ∼15 ng/ml sTva yielded the half-maximal extent of fusion (Fig. 2 A). This concentration of sTva is ∼3-fold higher than the half-maximal concentration required to induce infection of receptor-deficient cells by ASLV A virus (Damico and Bates, 2000).
FIGURE 2.
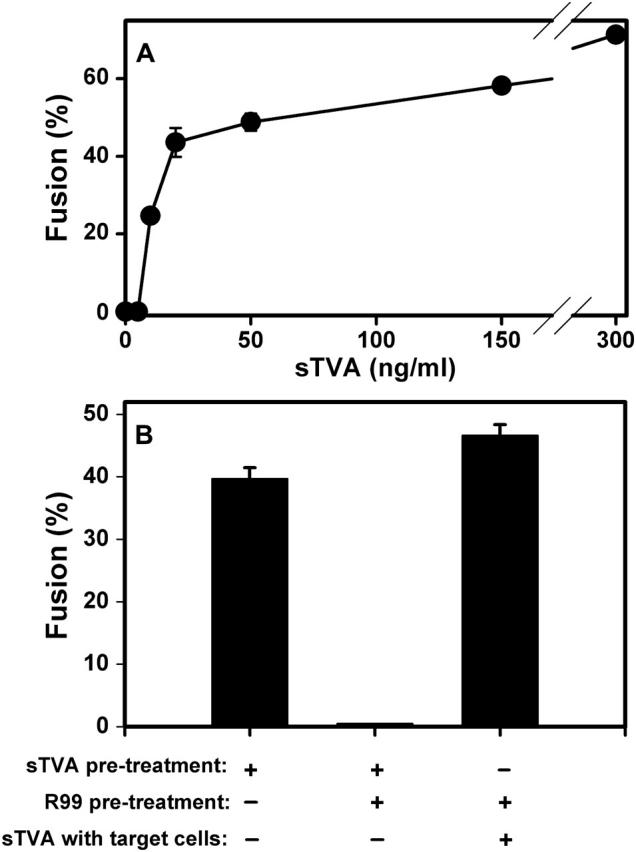
(A) Pretreatment of effector cells with sTva promotes conformational changes in EnvA that allow low pH to induce fusion to receptor-deficient cells. 3T3/EnvA cells were coincubated with 293T cells at 37°C for 1 h in the presence of the indicated concentrations of sTva. Cell-cell fusion was then triggered by lowering pH to 5.4 for 10 min. (B) Binding sTva to EnvA causes exposure of R99 binding sites. Coincubating effector and receptor-deficient 293T cells did not lead to fusion (A). Pretreating the effector cells with sTva yielded extensive fusion (first bar). When both sTva and R99 were present during the pretreatment of the effector cells, fusion to receptor-deficient cells was negligible (second bar). Pretreating effector cells with R99 alone, followed by extensive washing and then coculturing with receptor-deficient cells in the presence of sTva led to an appreciable extent of fusion (third bar). Five hundred ng/ml sTva and 3.3 _μ_M R99 were used for experiments of panel B. Error bars are standard errors on the mean for at least four measurements.
Adding sTva to ASLV A viral particles or to ectodomains of EnvA induces hydrophobic binding to liposomes, probably through insertion of Env's fusion peptides (Damico et al., 1998; Earp et al., 2003; Hernandez et al., 1997). The state of Env after binding sTva in the absence of a target membrane is known as “receptor-activated” (Mothes et al., 2000). To determine whether the grooves of the coiled coil were exposed after Env was receptor-activated, 500 ng/ml sTva, either alone or mixed with 3.3 _μ_M R99, was added to effector cells in the absence of other cells. After a 30 min incubation at 37°C, all unbound sTva (and R99, if present) was washed away. When R99 was not included in the mix, coincubating these receptor-activated effector cells with cells that did not contain receptor for 1 h at 37°C followed by low pH, led to extensive fusion (Fig. 2 B, bar 1). When R99 was included in the mix, fusion was blocked (bar 2). As a control, the effector cells were incubated with R99 and the peptide then washed away. When these washed effector cells were coincubated with receptor-deficient cells for 1 h at 37° in the presence of sTva, low pH led to extensive fusion (bar 3). Coincubating effector and receptor-deficient cells did not yield fusion if sTva was not added (Fig. 2 A). Thus, R99 bound to receptor-activated Env, but did not bind to native, nonactivated EnvA. In other words, receptor-activation of EnvA either creates or exposes the grooves of the coiled coil, but it does not induce folding of EnvA into a 6HB.
Fusion from LAS is resistant to peptide inhibition, suggesting that 6HB formation can occur before membrane merger
After effector cells expressing EnvA bind to Tva-expressing target cells, pH must be lowered for cell-cell fusion to occur (Melikyan et al., 2004). The incorporation of LPC as a lipid component into membranes has been found to inhibit fusion in all systems for which it has been tested. Incorporated LPC is thought to inhibit fusion by preventing membranes from bending in the direction necessary for hemifusion to occur (Chernomordik et al., 1995).
Preincubating E/T cells at neutral pH for 1 h at 37°C, adding LPC, and then lowering pH to 5.4 at 23°C did not lead to fusion (Fig. 3), whereas the same protocol without the addition of LPC led to efficient fusion (Fig. 3 A, bar 1). When LPC had been added, reneutralization and subsequent removal of LPC from the cell membranes by repetitive washings at a temperature below 8°C did not result in aqueous dye mixing (bar 2). At this LAS of fusion, lipid dye had also not yet spread (data not shown). But increasing temperature at this point to 37°C (at neutral pH), resulted in a significant extent of fusion (bar 3), showing that LPC's inhibitory effect was largely reversible. Because fusion could proceed at neutral pH after the removal of LPC, we conclude that low pH-dependent conformational changes of EnvA precede hemifusion. CPZ did not lead to fusion when added (at 4°C) either immediately (data not shown) or 10 min (bar 5) after creating LAS. This indicates that LAS is a prehemifusion state and did not progress to restricted hemifusion.
FIGURE 3.
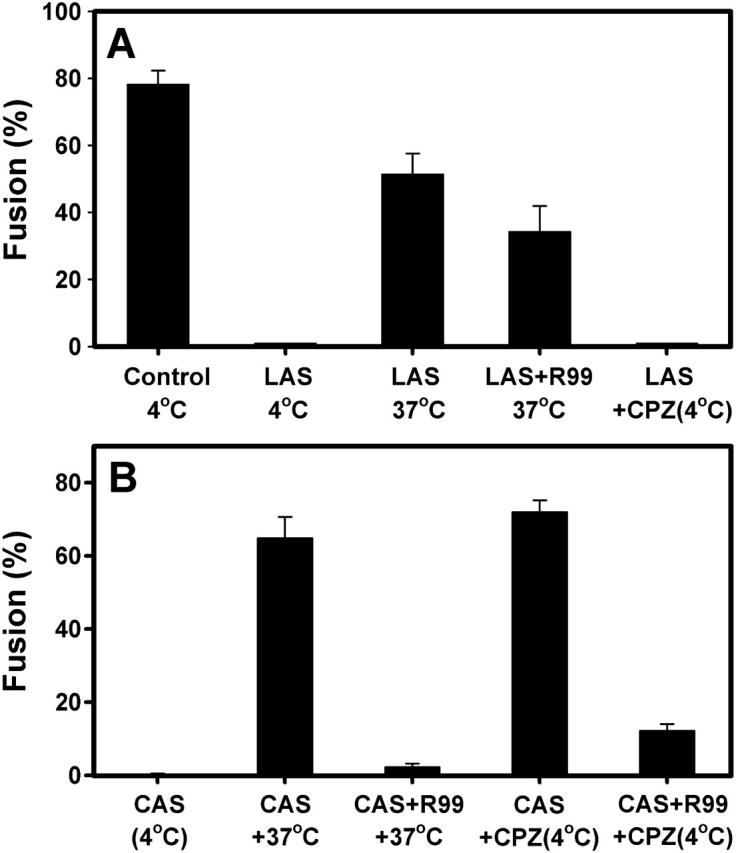
The LAS, but not the CAS, of ASLV Env-induced fusion is resistant to R99 peptide. (A) All procedures to create LAS were performed in the control (first bar), except that 0.2 mg/ml L-LPC was not included in any solutions. Fusion did not occur when LAS was created (second bar). But raising temperature to 37°C for 15 min resulted in significant fusion (third bar). Adding R99 (1.7 _μ_M) at LAS did not significantly reduce the extent of fusion upon raising temperature to 37°C (fourth bar). Adding CPZ at LAS did not lead to fusion (fifth bar). (B) Fusion did not occur when CAS was created (first bar). A subsequent incubation at 37°C (pH 7.2) for 15 min (second bar) or an exposure to 0.5 mM CPZ for 2 min at 4°C (fourth bar) resulted in extensive fusion. Incubating the cells with R99 (1.7 _μ_M) for 10 min (at 4°C) after creating CAS abolished the 37°C-induced (third bar) and suppressed the CPZ-induced (fifth bar) fusion at 4°C.
When a high concentration of R99 was added to E/T cells captured at LAS, the extent of fusion induced by raising temperature to 37°C (Fig. 3 A, bar 4) was comparable to that in the absence of peptide (bar 3). The finding that R99 does not inhibit fusion demonstrates that grooves of the coiled coil are no longer accessible, and suggests that EnvA is already folded into a 6HB at LAS. If indeed EnvA has folded into a bundle at LAS, it would mean that ASLV EnvA can fold into a 6HB before membrane merger.
Local hemifusion can occur before all copies of Env fold into 6HBs
We have previously shown (Melikyan et al., 2004) that by reducing pH to 5.4 at 4°C, an intermediate—referred to as a CAS—is captured (see Materials and Methods). Neither lipid (data not shown) nor aqueous dye (Fig. 3 B, bar 1) has spread at CAS. If lipid dye is observed to spread in the absence of fusion pores, hemifusion has certainly occurred. But many states that probably are states of hemifusion do not allow movement of dye between cells. Such states are known as “restricted hemifusion” (Chernomordik et al., 1998). It is generally agreed that when the addition of CPZ promotes aqueous dye transfer, the cells were in a state of restricted hemifusion (reviewed in Cohen et al., 2002). CAS is readily converted to full fusion by treating cells with CPZ at neutral pH, 4°C (Fig. 3 B, bar 4, also see Melikyan et al., 2004), implying that CAS is a state of restricted hemifusion. Fusion was induced from CAS by raising temperature to 37°C at neutral pH (bar 2): aqueous dye spread was observed for more than 60% of the cell pairs. CAS is relatively stable at 4°C: CPZ promotes fusion (at neutral pH) to almost the same extent when it is added 45 min after creating CAS (data not shown) as when added immediately (bar 4). This indicates that the state of restricted hemifusion is stable. But the ability of Env to induce fusion from CAS decreases with time. The extent of fusion was greater upon raising temperature to 37°C immediately after creating CAS (bar 2) than 45 min later (data not shown). Also, fusion from CAS was still completely inhibited when R99 was added 45 min after creating CAS (data not shown). Thus CAS does not evolve to a more advanced state with time at 4°C. This is in contrast to influenza HA where holding a restricted-hemifusion state, equivalent to CAS, for extended times at 4°C can promote advancement to a more downstream state (Chernomordik et al., 1998).
When R99 was added at CAS and temperature raised to 37°C, fusion did not occur (Fig. 3 B, bar 3). Because R99 inhibited fusion, not all copies of Env that participate in fusion had completed their folding into 6HB at CAS (assuming bundle formation is irreversible). Unexpectedly, the addition of R99 also strongly reduced the ability of CPZ to induce fusion at the point of CAS (bar 5). This implies that binding of R99 to prebundle conformations of the TM subunit caused the local lipid connections between the hemifused E/T cells at CAS to dissociate. In other words, upon binding R99, Env converts into a state that no longer supports hemifusion. Thus, prebundle configurations of Env and the hemifusion state are reversible, and specific prebundle configurations of the TM subunits are critical for sustaining the state of local hemifusion.
R99 does not inhibit pore growth
The growth of the initial ASLV Env-induced fusion pore can be arrested by lowering temperature (Melikyan et al., 2004). After coincubating effector and target cells at neutral pH, we acidified at 27°C and, immediately after pore formation, we lowered temperature to 4°C and reneutralized the solution (Fig. 4, A and B, downward arrow). The rate of aqueous dye transfer between the cells decreased, showing that pore growth ceased and, in fact, pore size even decreased (Fig. 4). When temperature was raised to 37°C, pore growth immediately resumed (Fig. 4 A, upward arrow). Adding the R99 peptide before raising temperature did not hinder pore growth (Fig. 4 B). Thus, Env had either already folded into a 6HB by the time a pore formed (even when formed at the relatively low temperature of 27°C), or it folded into a bundle soon after pore formation, before R99 could be added.
FIGURE 4.

Small fusion pores formed by ALSV Env are not affected by R99. Fusion was triggered by local perfusion of selected cell pairs with a solution at pH 5.4 and simultaneous raising of temperature from 4°C to 27°C. Immediately after calcein began to transfer into the target cell, the temperature was dropped back to 4°C (downward arrows) and the bathing solution was replaced by one containing 5 _μ_M R99 peptide at neutral pH (B, arrowhead) or, in control experiments, without peptide (A, arrowhead). After holding the temperature at 4°C for an additional 3–4 min, temperature was quickly raised to 37°C (upward arrows). The average fluorescence of representative target cells as a result of calcein transfer from the effector cell is shown by the noisy solid lines. The pore permeability (open circles) was calculated from the rate of calcein transfer, as previously described (Markosyan et al., 2003).
DISCUSSION
In this study, we captured ASLV EnvA-induced fusion at a receptor-activated state, at LAS, at CAS, and at a small pore. At each arrested stage, we determined the ability of R99 to inhibit aqueous dye transfer and the ability of either CPZ or temperature to effect aqueous dye transfer. As a result, we were able to make several characterizations (Fig. 5). First, since low pH is necessary to create CAS and LAS, it appears that low pH is necessary for hemifusion to occur. Second, using low temperature to arrest a state of hemifusion (i.e., CAS), we found that adding R99 causes the merged membranes (CAS) to revert back to separate membranes (Fig. 5, prebundle to extended conformation). We assume that R99 causes this reversion by binding to the grooves of the coiled coil of Env. Third, the folding of ASLV Env into a 6HB requires low pH. Fourth, grooves of the central coiled coil of Env become occluded from R99 at low pH, 37°C when membrane merger is prevented by the presence of LPC (Fig. 5, LAS). In contrast, exposing Env to acidic pH at low temperature keeps the grooves exposed while promoting membrane merger (CAS). Fifth, because different triggers are required to induce fusion from the receptor-activated state and from CAS, we can conclude that there are at least two distinct prebundle configurations. The receptor-activated conformation requires low pH to induce fusion. The prebundle configuration of CAS no longer requires low pH, but does require high temperature for fusion to occur.
FIGURE 5.

Proposed sequence for ASLV Env-refoldings and lipid rearrangements that lead to fusion pore formation. The progression of conformational changes in Env: an extended conformation after receptor-activation (left panel), prebundles (i.e., CAS, lower panel), 6HB (LAS, upper panel) and the final trimeric hairpin conformation (small fusion pore, right panel). LPC molecules that disfavor membrane hemifusion are shown by open triangles. For visual clarity, only the TM subunit of Env is shown. Shaded cylinders are C-terminal segments and open cylinders are N-terminal segments that form the coiled coil. A relatively long intervening stretch of amino acids between the C-terminal segments and MSDs allows a 6HB for form without necessitating close approach of fusion peptides and MSDs. Prebundle conformations are capable of inducing hemifusion at low temperature (CAS). 6HBs have not formed at this state. In our experiments, LAS was created after removal of L-LPC at 4°C. Although not strictly correct, we illustrate, for artistic convenience, LAS as a state at 37°C in the presence of L-LPC. At LAS, the grooves of the coiled coil are no longer exposed and thus the 6HBs may have formed. Formation of the trimeric hairpin promotes creation of the fusion pore and/or its enlargement.
R99 has the amino acid sequence of the portion of the C-terminal segment expected to pack into the grooves of the coiled coil. In the absence of a crystal structure, one cannot be certain that R99 does indeed bind to the grooves to prevent bundle formation and if it does, that this is the only site of R99 binding. To formulate mechanistic interpretations to our experimental results, we assume that the sole consequence of R99 binding is to prevent EnvA from folding into a bundle. Because bundle formation is probably irreversible, it is expected that R99 would no longer be able to bind to EnvA (and thereby inhibit fusion) once EnvA has folded into a bundle. Therefore, if the addition of R99 inhibits fusion, one can conclude that EnvA has not yet folded into a 6HB at the stage of addition. If R99 no longer inhibits fusion, either the 6HB has formed or the grooves are no longer exposed for other reasons (e.g., steric hindrance). Experimental means to distinguish between the two possibilities are not yet available.
The occurrence of conformational changes and oligomerization of ASLV Env by receptor-activation followed by low pH has been tracked by migration of Env in mildly denaturing SDS gels (Mothes et al., 2000; Matsuyama et al., 2004; Smith et al., 2004). After receptor-activating EnvA of ASLV particles and adding liposomes, the initial monomeric 37 kD band is reduced, and 70 kD and 150 kD bands appear. Lowering pH after receptor-activation results in a 90–100 kD band (as well as higher molecular weight bands, >150 kD) (Matsuyama et al., 2004; Smith et al., 2004). The presence of R99 or LPC prevented the formation of the 90–100 kD band, leading to the proposal that this band represents either a 6HB or a more complete final trimeric hairpin (Matsuyama et al., 2004; Smith et al., 2004). The fact that R99 prevented the formation of this band (Matsuyama et al., 2004) is consistent with the assumption that R99 inhibits fusion by preventing bundle formation. If the 90–100 kD band represents the complete trimeric hairpin (rather than the 6HB which comprises only a portion of the trimeric hairpin as discussed below), the biochemical studies and our functional measures would be in complete agreement.
For the same reasons we discussed for HIV Env (Markosyan et al., 2003), we consider it more likely that the 6HB of ASLV Env forms by the sequential insertion of the three C-terminal segments into the grooves of the coiled coil, rather than by simultaneous insertion (Fig. 5). Even though the 6HB is stable, unable to revert to upstream configurations, this may not be the case for prebundle configurations: the insertion of one or two (but not all three) C-terminal segments into grooves may be reversible. If this is the situation, binding of the synthetic C-peptide R99 into grooves would drive Env with one or two inserted natural C-terminal segments into earlier, more upstream prebundle configurations. We conjecture that late prebundle configurations are necessary to maintain the state of hemifusion at CAS.
For several viruses, such as HIV, SV5, human T-cell leukemia virus, and others, peptides designed against formation of 6HBs have been shown to prevent viral infection and fusion (Jiang et al., 1993; Lambert et al., 1996; Russell et al., 2001; Pinon et al., 2003; Wild et al., 1994). These viruses fuse directly to the plasma membrane at neutral pH. In contrast, in the case of viruses that utilize low pH alone (i.e., without receptor-activation) for fusion, no peptide directed against formation of a 6HB has yet been shown to inhibit fusion. It is possible that prehairpin configurations of neutral pH fusion proteins exist for appreciably longer times than low pH proteins. This would expose the grooves of the coiled coil of neutral pH fusion proteins for relatively long times. It would also account for the generally slower fusion kinetics of proteins that fuse at neutral pH (Hoekstra et al., 1984; Pal et al., 1988; Bron et al., 1993; Plonsky and Zimmerberg, 1996; Melikyan et al., 2000; Markosyan et al., 2001). For ASLV, low pH can induce fusion only after receptor-activation exposes the grooves of the coiled coil of Env (Fig. 2) and the fusion peptides have inserted into the target membrane (Melikyan et al., 2004). Because the neutral pH steps in the process are appreciably slower than the low pH steps (Melikyan et al., 2004), the grooves of ASLV Env should be exposed to R99 for long times.
For HIV Env and SV5 F, bundle formation requires membrane merger (Melikyan et al., 2000; Russell et al., 2001) and for HIV Env, bundle formation continues after pore formation (Markosyan et al., 2003). For these proteins, the 6HB comprises almost the entire trimeric hairpin and bundle formation alone should bring the MSDs and the fusion peptides into close proximity. For influenza HA, local hemifusion occurs before the formation of the trimeric hairpin has been completed (Borrego-Diaz et al., 2003), but HA must complete its folding into a trimeric hairpin for fusion to occur (Park et al., 2003) and for the pore to grow (Borrego-Diaz et al., 2003). Because there is a long intervening stretch of amino acids (termed a “leash”; Park et al., 2003) between the C-terminal helices of the 6HB and the MSDs, the formation of the 6HB, per se, is not sufficient to bring the MSDs and fusion peptides into proximity. But the creation of auxiliary structures within the final trimeric hairpin does bring the MSDs and fusion peptides into proximity (Chen et al., 1999). In short, for some proteins, such as HA, there is a subtle but important difference between the 6HB and trimeric hairpin structures, whereas for others, such as HIV Env, there is no distinction between the two.
We have suggested that the flexibility conferred by the long intervening stretch of HA allows the 6HB to form before the trimeric hairpin is complete (Borrego-Diaz et al., 2003). It is not known whether there is an intervening sequence between the 6HBs and the MSDs for ASLV Env. Based on the crystal structure of GP2 of Ebola and the amino acid sequence of ASLV Env, if the intervening sequence exists, it must be shorter (∼20 amino acid residues) than that of influenza HA (∼60 amino acid residues). But if the structure of this short intervening sequence were flexible, a 6HB could form without forcing the MSDs and fusion peptides into proximity; an auxiliary structure would be required to bring about this proximity. In view of the inability of R99 to inhibit fusion when added at LAS, the 6HB of ASLV Env may be able to form before membrane merger, even though the trimeric hairpin cannot. We propose that for all class I proteins, the formation of the trimeric hairpin is tightly coupled to creation of the fusion pore and/or its growth.
Acknowledgments
We thank Drs. Richard Barnard and John Young for providing the EnvA-expressing and Tva950-expressing cell lines, and Ms. Sofya Brener for excellent technical assistance.
This work was supported by National Institutes of Health grants AI053668, GM27367, AI43455, and CA76256.
References
- Baker, K. A., R. E. Dutch, R. A. Lamb, and T. S. Jardetzky. 1999. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 3:309–319. [DOI] [PubMed] [Google Scholar]
- Balliet, J. W., J. Berson, C. M. D'Cruz, J. Huang, J. Crane, J. M. Gilbert, and P. Bates. 1999. Production and characterization of a soluble, active form of Tva, the subgroup A avian sarcoma and leukosis virus receptor. J. Virol. 73:3054–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, P., J. A. Young, and H. E. Varmus. 1993. A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell. 74:1043–1051. [DOI] [PubMed] [Google Scholar]
- Borrego-Diaz, E., M. E. Peeples, R. M. Markosyan, G. B. Melikyan, and F. S. Cohen. 2003. Completion of trimeric hairpin formation of influenza virus hemagglutinin promotes fusion pore opening and enlargement. Virology. 316:234–244. [DOI] [PubMed] [Google Scholar]
- Bressanelli, S., K. Stiasny, S. L. Allison, E. A. Stura, S. Duquerroy, J. Lescar, F. X. Heinz, and F. A. Rey. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 23:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron, R., J. M. Wahlberg, H. Garoff, and J. Wilschut. 1993. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 12:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, D. C., and P. S. Kim. 1998. HIV entry and its inhibition. Cell. 93:681–684. [DOI] [PubMed] [Google Scholar]
- Chen, J., J. J. Skehel, and D. C. Wiley. 1999. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple- stranded coiled coil. Proc. Natl. Acad. Sci. USA. 96:8967–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik, L., M. M. Kozlov, and J. Zimmerberg. 1995. Lipids in biological membrane fusion. J. Membr. Biol. 146:1–14. [DOI] [PubMed] [Google Scholar]
- Chernomordik, L. V., V. A. Frolov, E. Leikina, P. Bronk, and J. Zimmerberg. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140:1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, F. S., R. M. Markosyan, and G. B. Melikyan. 2002. The process of membrane fusion: nipples, hemifusion, pores, and pore growth. Curr. Top. Membr. 501–529.
- Colman, P. M., and M. C. Lawrence. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4:309–319. [DOI] [PubMed] [Google Scholar]
- Damico, R., and P. Bates. 2000. Soluble receptor-induced retroviral infection of receptor-deficient cells. J. Virol. 74:6469–6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damico, R. L., J. Crane, and P. Bates. 1998. Receptor-triggered membrane association of a model retroviral glycoprotein. Proc. Natl. Acad. Sci. USA. 95:2580–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero, F., S. A. Hoschander, and J. Brojatsch. 2002. Endocytosis is a critical step in entry of subgroup B avian leukosis viruses. J. Virol. 76:12866–12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earp, L. J., S. E. Delos, R. C. Netter, P. Bates, and J. M. White. 2003. The avian retrovirus avian sarcoma/leukosis virus subtype A reaches the lipid mixing stage of fusion at neutral pH. J. Virol. 77:3058–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810. [DOI] [PubMed] [Google Scholar]
- Gallaher, W. R. 1996. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell. 85:477–478. [DOI] [PubMed] [Google Scholar]
- Gibbons, D. L., M. C. Vaney, A. Roussel, A. Vigouroux, B. Reilly, J. Lepault, M. Kielian, and F. A. Rey. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 427:320–325. [DOI] [PubMed] [Google Scholar]
- Hernandez, L. D., R. J. Peters, S. E. Delos, J. A. Young, D. A. Agard, and J. M. White. 1997. Activation of a retroviral membrane fusion protein: soluble receptor- induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol. 139:1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra, D., T. de Boer, K. Klappe, and J. Wilschut. 1984. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry. 23:5675–5681. [DOI] [PubMed] [Google Scholar]
- Hughson, F. M. 1997. Enveloped viruses: a common mode of membrane fusion? Curr. Biol. 7:R565–R569. [DOI] [PubMed] [Google Scholar]
- Hunter, E. 1997. Viral entry and receptors. In Retroviruses. J. Coffin, S. M. Hughes, and H. E. Varmus, editors. Cold Spring Harbor Press, Cold Spring Harbor, NY. 71–119. [PubMed]
- Jiang, S., K. Lin, N. Strick, and A. R. Neurath. 1993. HIV-1 inhibition by a peptide. Nature. 365:113. [DOI] [PubMed] [Google Scholar]
- Lambert, D. M., S. Barney, A. L. Lambert, K. Guthrie, R. Medinas, D. E. Davis, T. Bucy, J. Erickson, G. Merutka, and S. R. Petteway. 1996. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA. 93:2186–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, M., S. C. Blacklow, and P. S. Kim. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 2:1075–1082. [DOI] [PubMed] [Google Scholar]
- Malashkevich, V. N., B. J. Schneider, M. L. McNally, M. A. Milhollen, J. X. Pang, and P. S. Kim. 1999. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9Å resolution. Proc. Natl. Acad. Sci. USA. 96:2662–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan, R. M., F. S. Cohen, and G. B. Melikyan. 2003. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell. 14:926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan, R. M., X. Ma, M. Lu, F. S. Cohen, and G. B. Melikyan. 2002. The mechanism of inhibition of HIV-1 Env-mediated cell-cell fusion by recombinant gp41 ectodomain cores. Virology. 302:174–184. [DOI] [PubMed] [Google Scholar]
- Markosyan, R. M., G. B. Melikyan, and F. S. Cohen. 2001. Evolution of intermediates of influenza virus hemagglutinin-mediated fusion revealed by kinetic measurements of pore formation. Biophys. J. 80:812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama, S., S. E. Delos, and J. M. White. 2004. Sequential roles of receptor binding and low pH in forming prehairpin and hairpin conformations of a retroviral envelope glycoprotein. J. Virol. 78:8201–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G. B., R. J. Barnard, R. M. Markosyan, J. A. Young, and F. S. Cohen. 2004. Low pH is required for Avian Sarcoma and Leukosis Virus Env-induced hemifusion and fusion pore formation but not for pore growth. J. Virol. 78:3753–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G. B., R. M. Markosyan, H. Hemmati, M. K. Delmedico, D. M. Lambert, and F. S. Cohen. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151:413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis, Y., S. Ogata, D. Clements, and S. C. Harrison. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature. 427:313–319. [DOI] [PubMed] [Google Scholar]
- Mothes, W., A. L. Boerger, S. Narayan, J. M. Cunningham, and J. A. Young. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell. 103:679–689. [DOI] [PubMed] [Google Scholar]
- Narayan, S., R. J. Barnard, and J. A. Young. 2003. Two retroviral entry pathways distinguished by lipid raft association of the viral receptor and differences in viral infectivity. J. Virol. 77:1977–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal, R., Y. Barenholz, and R. R. Wagner. 1988. Pyrene phospholipid as a biological fluorescent probe for studying fusion of virus membrane with liposomes. Biochemistry. 27:30–36. [DOI] [PubMed] [Google Scholar]
- Park, H. E., J. A. Gruenke, and J. M. White. 2003. Leash in the groove mechanism of membrane fusion. Nat. Struct. Biol. 10:1048–1053. [DOI] [PubMed] [Google Scholar]
- Pinon, J. D., S. M. Kelly, N. C. Price, J. U. Flanagan, and D. W. Brighty. 2003. An antiviral peptide targets a coiled-coil domain of the human T-cell leukemia virus envelope glycoprotein. J. Virol. 77:3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plonsky, I., and J. Zimmerberg. 1996. The initial fusion pore induced by baculovirus GP64 is large and forms quickly. J. Cell Biol. 135:1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, C. J., T. S. Jardetzky, and R. A. Lamb. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel, J. J., and D. C. Wiley. 1998. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 95:871–874. [DOI] [PubMed] [Google Scholar]
- Smith, J. G., W. Mothes, S. C. Blacklow, and J. M. Cunningham. 2004. The mature avian leukosis virus subgroup A envelope glycoprotein is metastable, and refolding induced by the synergistic effects of receptor binding and low pH is coupled to infection. J. Virol. 78:1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn, W., A. Carfi, K. H. Lee, J. J. Skehel, and D. C. Wiley. 1998. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell. 2:605–616. [DOI] [PubMed] [Google Scholar]
- Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 387:426–430. [DOI] [PubMed] [Google Scholar]
- Wild, C. T., D. C. Shugars, T. K. Greenwell, C. B. McDanal, and T. J. Matthews. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA. 91:9770–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]