The yeast counterparts of human 'MELAS' mutations cause mitochondrial dysfunction that can be rescued by overexpression of the mitochondrial translation factor EF-Tu (original) (raw)
Abstract
We have taken advantage of the similarity between human and yeast (Saccharomyces cerevisiae) mitochondrial tRNALeu(UUR), and of the possibility of transforming yeast mitochondria, to construct yeast mitochondrial mutations in the gene encoding tRNALeu(UUR) equivalent to the human A3243G, C3256T and T3291C mutations that have been found in patients with the neurodegenerative disease MELAS (for mitochondrial 'myopathy, _e_ncephalopathy, lactic acidosis and stroke-like episodes'). The resulting yeast cells (bearing the equivalent mutations A14G, C26T and T69C) were defective for growth on respiratory substrates, exhibited an abnormal mitochondrial morphology, and accumulated mitochondrial DNA deletions at a very high rate, a trait characteristic of severe mitochondrial defects in protein synthesis. This effect was specific at least in the pathogenic mutation T69C, because when we introduced A or G instead of C, the respiratory defect was absent or very mild. All defective phenotypes returned to normal when the mutant cells were transformed by multicopy plasmids carrying the gene encoding the mitochondrial elongation factor EF-Tu. The ability to create and analyse such mutated strains and to select correcting genes should make yeast a good model for the study of tRNAs and their interacting partners and a practical tool for the study of pathological mutations and of tRNA sequence polymorphisms.
Introduction
Practically undiagnosed 30 years ago, patients with mitochondrial diseases are now a steadily increasing population (Wallace, 1999; Lestienne et al., 1999). Mendelian and non-mendelian forms of inheritance of the diseases are observed, reflecting the fact that both mitochondrial and nuclear genomes contribute to mitochondrial function (Costanzo & Fox, 1990).
Among maternally inherited mitochondrial diseases, a significant proportion is due to nucleotide changes in the mitochondrial (mt) tRNA genes and especially in the tRNALeu(UUR) gene (Moares et al., 1993; Goto et al., 1994; Hao & Moares, 1996; see also MITOMAP (www.mitomap.org)). However, because sequence polymorphisms that do not cause any apparent pathologies are also frequent, the pathological effects of individual base substitutions are often uncertain. Varying levels of heteroplasmy also contribute to these difficulties (Wallace, 1993). These problems have been widely approached by the use of cybrids, namely cultured cells in which mt DNA has been eliminated and replaced by pathological mitochondria (King & Attardi, 1989). However, to rapidly discriminate between the candidates and identify the true pathogenic mutations resulting in oxidative phosphorylation defects, yeast cells—which are homoplasmic—could be a very useful tool. Moreover, yeast can be used to screen nuclear genes that, when overexpressed, can rescue the defective phenotypes.
The effects of mutations in mt tRNA genes have been widely studied in yeast by the use of randomly occurring mutations (Najarian et al., 1986, and references therein; Francisci et al., 1998, and references therein). The study of targeted mutations has recently become possible in yeast by the establishment of a successful biolistic transformation system (reviewed in Bonnefoy & Fox, 2000). This now makes it possible to introduce mutations at any desired position in yeast mt tRNA genes (see, for example, Rohou et al., 2001), including mutations equivalent to those that are pathogenic in humans. On the basis of the fact that tRNALeu(UUR) is very similar in humans and in Saccharomyces cerevisiae, as shown in Fig. 1, we describe here the defective phenotypes obtained by introducing mutations equivalent to the known human pathogenic mutations A3243G, C3256T and T3291C at positions 14, 26 and 69, respectively, of yeast mitochondrial tRNALeu(UUR). The mutant phenotypes can be corrected by overexpression of mitochondrial elongation factor EF-Tu encoded by the nuclear gene TUF1.
Figure 1.
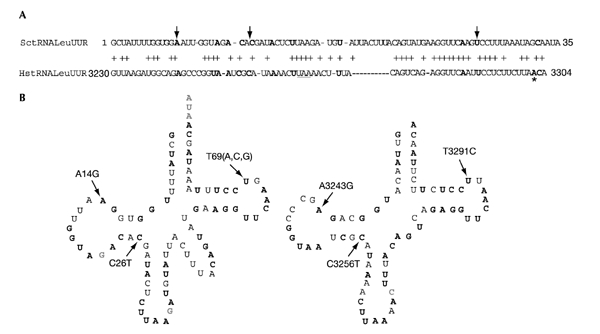
Comparison of yeast and human tRNALeu(UUR) sequences and structures. (A) Alignment of yeast and human mitochondrial tRNA. Numbers refer to positions in the tRNALeu(UUR) gene for yeast and to positions in the mitochondrial DNA sequence for humans. Plus signs indicate nucleotides that are conserved between the two tRNAs. The anticodon is underlined. Bold nucleotides represent bases that were found mutated in patients (see MITOMAP, www.mitomap.org); the asterisk indicates the only pathological position not conserved in yeast. The arrows indicate the mutations studied in this work. (B) Cloverleaf structure of yeast (left) and human (right) mitochondrial tRNALeu(UUR). Nucleotides in grey are non-conserved bases. Arrows indicate the positions of the mutations studied and specify the substitutions studied.
Results
Base substitutions into S. cerevisiae mt tRNALeu(UUR)
We have used the biolistic procedure, originally established by Fox et al. (1991) and adapted by Rohou et al. (2001), to introduce into yeast mitochondrial tRNALeu(UUR) mutations equivalent to those involved in the neurological disease MELAS (for mitochondrial '_m_yopathy, _e_ncephalopathy, _l_actic _a_cidosis and _s_troke-like episodes').
We first introduced into yeast tRNALeu(UUR) the base substitution A14G, equivalent to the most frequent human MELAS mutation (A3243G), and obtained a strain unable to grow on respiratory (glycerol) medium (Fig. 2). Cells could grow on fermentative (glucose or galactose) medium, but very rapidly lost their mt DNA (data not shown), a feature indicating a very severe mitochondrial protein synthesis defect (Myers et al., 1985). We then generated a mutation at position 26, equivalent to C3256T in humans, and obtained the same respiratory defect (Fig. 2).
Figure 2.
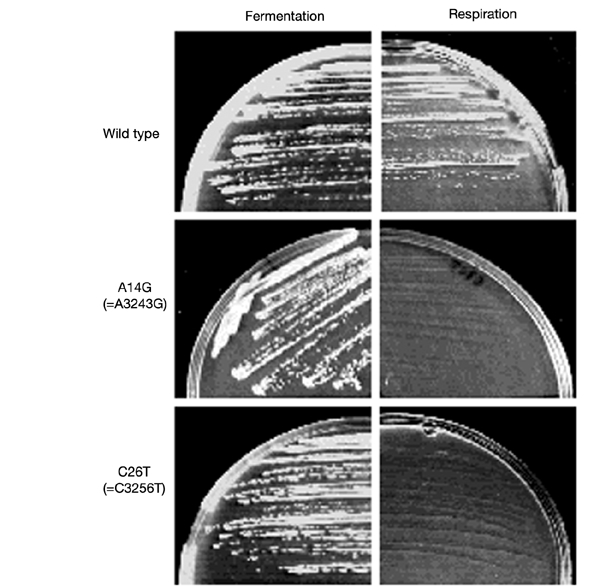
Phenotype of yeast mutant strains harbouring mutations equivalent to A3243G and C3256T. Left panels show the growth of isogenic wild-type and mutant strains after 4 d at 28 °C on complete glucose medium (fermentative medium); right panels show the same strains and conditions on complete glycerol medium (respiratory medium).
These results indicate that base substitutions that confer on humans a severe MELAS syndrome also produce a severe phenotype when introduced into yeast at equivalent positions. This conclusion was not evident a priori because the metabolism and mode of life of S. cerevisiae and animal cells are quite different (see Discussion).
Correlation with nucleotide changes in humans
To establish the described system as an effective tool it was essential to establish specificity of effects. We therefore introduced at position 69 of the mt tRNALeu(UUR) (equivalent to position 3291 in humans) the three possible base substitutions. One of them, the T→C nucleotide change corresponding to the human pathogenic mutation T3291C, conferred on the cells the same drastic phenotype as did the two other pathogenic equivalent mutations (A3243G and C3256T). In contrast, the two other nucleotide changes at position 69 (T→A and T→G) conferred on yeast cells a quasi-wild-type phenotype (Fig. 3) and only accumulated _rho_° cells in a higher proportion than the wild type (data not shown). Mutations T3291A and T3291G have never been detected in patients despite extensive screening, and although this is not formal proof that they are not pathogenic, the present results strongly support—at least for position 69 of yeast tRNALeu(UUR)—the validity of the yeast model.
Figure 3.
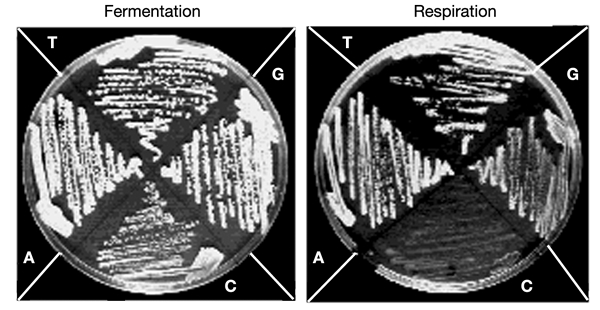
Phenotypic effect of the three different mutations at position T69 of yeast (equivalent to T3291 in humans). Growth comparison of the four strains (isogenic wild-type and T→G/T→C/T→A mutants) on fermentative and respiratory media. Conditions are the same as in Fig 2. Bases at position 69 are indicated on the corresponding sectors of the plates.
Phenotype correction by overexpression of mt EF-Tu
In previous work (Rinaldi et al., 1997; Francisci et al., 1998) we have shown that overexpression on a multicopy plasmid of the nuclear gene (TUF1) encoding the mitochondrial elongation factor EF-Tu permitted the correction of the thermosensitive respiratory defect of several mt tRNA mutations. We then examined the effect of TUF1 overexpression in cells bearing the base substitution C26T (equivalent to C3256T in humans) and found that growth on respiratory substrates was restored to normal levels (Fig. 4), whereas no effects on growth or respiration of this overexpression were observed in the wild type (data not shown).
Figure 4.
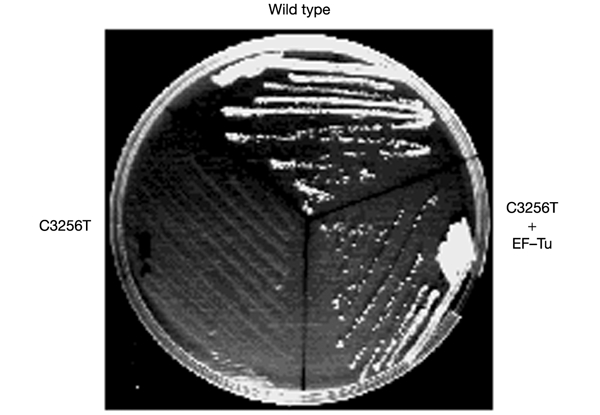
Effect of overexpression of mt EF-Tu on the defective phenotype arising from the yeast mutation equivalent to C3256T in humans. Growth comparison on respiratory medium of the wild-type strain, the strain carrying the C3265T equivalent mutation and the same mutant strain transformed with a multicopy plasmid carrying the TUF1 gene. The corrected phenotype is correlated with the presence of the plasmid (data not shown).
To verify that mitochondria were now functioning normally, we also examined the structure of the mitochondrial network. This network, which was absent in the mutant (compare Fig. 5A and B) returned to a wild-type tubular structure in the cells harbouring mt EF-Tu (compare Fig. 5A and C). We also examined, by Northern blot analysis, the effect of TUF1 overexpression on the presence of tRNALeu(UUR) in cells bearing the C3256T equivalent mutation. Results (Fig. 6A, lane 2) showed that tRNALeu(UUR) was undetectable in the mutant. This was probably due to the high percentage of _rho_° cells, because another mitochondrial tRNA such as tRNAVal was also not detected (Fig. 6B, lane 2). The tRNALeu hybridization signal was restored to a normal level in the same mutant transformed by the _TUF1_-containing plasmid (Fig. 6, lanes 3). However, the mutated tRNALeu migrated more slowly under electrophoresis on a partly denaturing gel. Similar behaviour was observed previously for various tRNA mutations (Rinaldi et al., 1994) and is also observed in the above-mentioned non-pathogenic substitutions (Fig. 6, lanes 4), probably indicating a conformational change in the three-dimensional structure of the mutated tRNA. The difference in electrophoretic mobility disappeared in completely denaturing gels.
Figure 5.
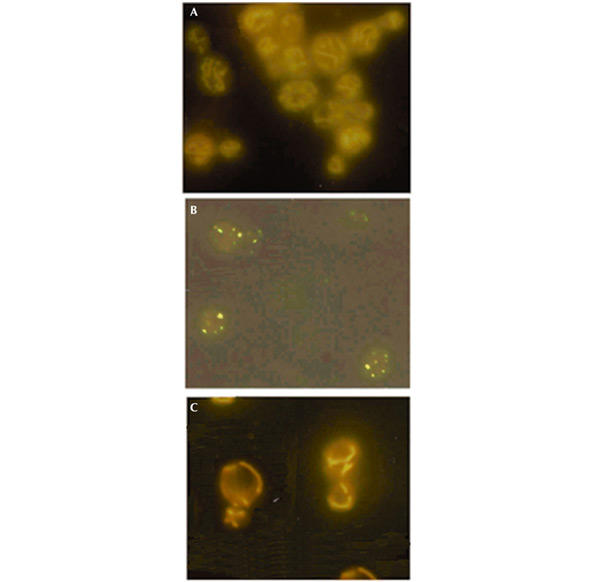
Mitochondrial morphology observed by DASPMI staining. Cells from the wild type (A), the mutant carrying the C3256T equivalent (B) and the same mutant transformed by plasmids carrying the TUF1 gene (C) were observed by fluorescence microscopy after staining with DASPMI (the magnification is the same in all pictures, 100×). The vital dye DASPMI is supposed to reveal only functional mitochondrial membranes.
Figure 6.
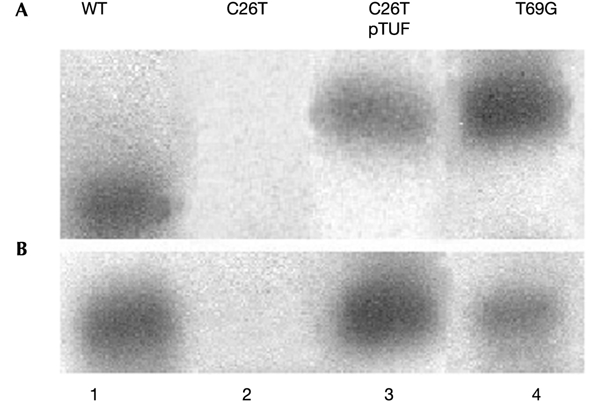
Northern blot analysis of mitochondrial transcripts in the wild type and in mutant strains transformed or not by plasmids containing the TUF1 gene. Total mtRNA (8 μg) purified from the wild-type cells (lane 1), from cells bearing the pathogenic C3256T equivalent mutation C26T (lane 2), from the same cells transformed with pTUF (lane 3) and from non-transformed cells bearing the non-pathogenic T3291G equivalent mutation T69G (lane 4) were loaded on partly denaturing 6% polyacrylamide–8 M urea gels. Procedure and probes were as described by Francisci et al. (1998). Hybridization was with the Leu probe (A) or with the Val probe (B).
We then performed a similar experiment with the multicopy plasmid pCM262 in which the TUF1 gene had been placed under the control of the regulatable Tet promoter, repressed by tetracycline (Gari et al., 1997). The engineered version of TUF1 was introduced into the two mutants A14G (equivalent to human A3243G) and T69C (equivalent to human T3291C) and growth on respiratory substrates was monitored. Figure 7 shows that, although the mutants failed to grow on glycerol medium, they both acquired the ability to use this exclusively respiratory substrate after transformation with the TUF1 gene placed under high expression conditions. The effect was lost in the presence of the repressor doxycycline, a tetracycline analogue.
Figure 7.
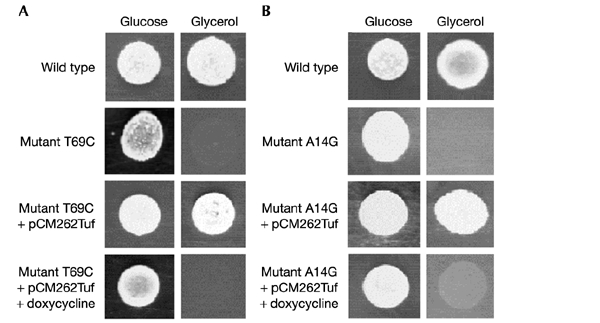
Correction of mutant phenotypes by the regulated expression of TUF1. Mutant yeast strains harbouring the mutations T69C (A) and A14G (B) were transformed by a multicopy plasmid bearing the TUF1 gene under the control of the Tet promoter. This promoter is fully active in the absence of doxycycline and is inactivated in its presence. Growth of the transformed strains was compared with mutant and wild-type strains on respiratory (glycerol) and fermentative (glucose) media, with and without doxycycline.
Discussion
The results presented here show that base substitutions known to be pathogenic in humans generate a highly defective mitochondrial phenotype in yeast, with an inability to grow on respiratory substrates and a rapid loss of mt DNA. As a control for effect specificity, we observed that two different non-pathogenic base substitutions introduced at position 69 (position 3291 in human mt DNA) had no (or a very limited) effect. This result strongly suggests a correlation between pathological mutations in humans and defective phenotypes in yeast.
In evaluating this correlation and the possibility of using yeast as a tool to investigate base substitutions in human mt tRNA, we should first take into account the fact that human cells are heteroplasmic (and heteroplasmy is important in the onset of pathologies), whereas yeast is homoplasmic. However, yeast offers a unique possibility in that mutants exhibiting complete or very severe respiratory deficiencies can grow by fermentative metabolism on glucose and are therefore amenable to molecular functional studies. Moreover, we show here that conditions can be created under which the drastic effects of homoplasmy can be alleviated (see below).
In other words, the homoplasmy of yeast might be a handicap in approaching questions such as threshold effect or heteroplasmic molecules segregation, but it is a clear advantage in investigating malfunctions of human equivalent mutations in tRNAs. For example, there has been some controversy about whether pathogenic mutations in humans such as A3243G actually affect translation or cause problems with RNA processing. The yeast data seem to argue in favour of a translational defect. Along the same line, an interesting application of this model is to check in a simple system the cellular effects of any given 'human' polymorphism leading to a relatively rapid classification of candidate mutations or to the clarification of complex cases.
However, we should stress that the success of the transposition from humans to yeasts described here is based on the remarkable conservation of mt tRNALeu(UUR), both in sequence and in structure. In this tRNA, 13 of 14 base substitutions known to be pathogenic are located at conserved positions and could be tested similarly. The situation is more complex for other tRNAs, for which conservation is not as high, but in many cases the discrepancy is due only to different base pairings in stems, which can easily be corrected.
An interesting opening is the possibility of characterizing compensatory effects due to changes in gene dosage. The mitochondrial elongation factor seems to be a universal suppressor of tRNA mutations in yeast: the defective phenotype on glycerol of seven of nine known yeast mutations (either equivalent or not to human pathogenic substitutions) was totally or partly corrected by overexpression of the mt EF-Tu.
This factor is particularly worthy of future explorations even in human cell lines. We must note that the structure of EF-Tu is strongly conserved in evolution (Woriax et al., 1995) and it might therefore be conceivable that a similar suppressing effect might be exerted in cell lines in human patients. However, a first attempt in this direction recently made by Toompu et al. (2002) gave negative results. We think that quantitative effects are probably crucial in these experiments because the human equivalent mutations are only corrected by overexpression of TUF1 on multicopy plasmids, and not on centromeric plasmids as in the yeast random mutations. These latter mutations were isolated as conditional mutations and probably had a less severe phenotype. The conditional expression plasmid for mt EF-Tu would indeed greatly facilitate many kinds of analysis by offering the possibility of studying controlled depletion effects and by allowing intermediate effects that could even mimic in some ways the effects of heteroplasmy.
The approach described here goes far beyond a possible application to human diseases. It opens the way to study in detail the different structural and functional features of mt tRNA genes in a well-known, flexible and controlled system. For example, the fact that EF-Tu can suppress many mutations that are not supposed to be involved in the formation of a ternary complex may indicate that the corrective effect is probably not mediated by its function in the elongation process of protein synthesis, but rather by stabilization of tRNA conformation through the interaction; that is, by a sort of chaperonin-like function. The interaction of tRNAs with other factors, such as modification enzymes or aminoacyl-tRNA synthetases, might also be studied. The yeast system seems well suited to approaching these questions.
It also becomes possible to test whether conserved structural domains of tRNA genes are likely to maintain the same functional role in different organisms, or to study the possible (co)-evolution of tRNAs and their associated factors by comparing functions in the two species and building interspecies functional tests.
In summary, the results described here open the possibility of studying mt tRNAs and their interacting factors, of investigating in the same system the pathogenic potential of some human mutations and of looking for more genes that can either suppress or modify the defective phenotype. This information could have important implications in mitochondrial genetics and physiopathology.
Methods
Media and growth conditions.
Strains were grown on complete medium (1% yeast extract and 1% peptone from Difco) containing 3% glycerol or 2% glucose. Similar media but containing 2% galactose or 0.2% glucose plus 3% glycerol were used to avoid the accumulation of _rho_° cells. Minimal medium was 0.7% yeast nitrogen base (Difco), 5% ammonium sulphate and 2% glucose, supplemented with the necessary auxotrophic requirements. For solid plate tests, 2% agar was added to the above media.
To measure the percentage of _rho_° colonies, cells were taken from galactose medium, inoculated in glucose-containing liquid medium and grown for 4 h before being plated on glucose-containing solid medium. In all cases examined, colonies with a very small size were found to be deprived of mt DNA.
Strains, plasmids, mutagenesis and biolistic transformation.
Plasmids carrying the mitochondrial mutations in the tRNALeu(UUR) gene were constructed from an amplified 1556-base-pair fragment bearing the tRNALeu(UUR) gene of purified mtDNA of yeast strain FF1210-6C (MAT alpha, ura1, ura2), by the procedure described for the Quickchange site-directed mutagenesis kit (Stratagene).
The biolistic procedure was performed as described in Rohou et al. (2001).
Further details can be found in Supplementary Information.
DNA and RNA manipulations.
All methods for nucleic acid preparations, electrophoresis and blotting were as described previously by Francisci et al. (1998) and references therein.
Microscopy.
Wild-type and mutant cells were collected from single colonies growing on galactose-containing plates, selected for their larger size (and hence presumed to contain mitochondrial DNA), and regrown for 4 h in liquid glucose medium. Mutant strains, corrected by TUF1 on a multicopy plasmid, were collected from selective-medium plates and inoculated in glucose medium for 4 h to avoid plasmid loss. Cells were stained with the vital dye DASPMI (2-(4-dimethylaminostyryl)-_N_-methylpyridinium iodide) at a final concentration of 1 μM, as described by Rafael & Nicholls (1984), and were observed immediately by fluorescence microscopy.
Supplementary data are available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor713-s1.pdf).
Supplementary Material
Details of biolistic transformation Plasmids Primers and probes
Acknowledgments
We thank R. Zelikson for her advice in the course of this work. M.F. was in receipt of an AFM (Association Française contre les Myopathies) postdoctoral fellowship. This work was supported by Instituto Pasteur Fondazione Cenci Bolognetti, by AFM, by the Italian Ministry for University and Scientific and Technological Research (COFIN 1999) and by Ateneo La Sapienza.
References
- Bonnefoy N. & Fox T.D. (2001) Genetic transformation of S. cerevisiae mitochondria. Methods Cell Biol., 65, 381–396. [DOI] [PubMed] [Google Scholar]
- Costanzo M.C. & Fox T.D. (1990) Control of mitochondrial gene expression in Saccharomyces cerevisiae. Annu. Rev. Genet., 24, 91–113. [DOI] [PubMed] [Google Scholar]
- Fox T.D., Folley L.S. & Mulero J.J. (1991) Analysis and manipulation of yeast mitochondrial genes. Methods Enzymol., 194, 149–165. [DOI] [PubMed] [Google Scholar]
- Francisci S., Bohn C., Frontali L. & Bolotin-Fukuhara M. (1998) Ts mutations in mitochondrial tRNA genes: characterization and effects of two point mutations in the mitochondrial gene for tRNAphe in Saccharomyces cerevisiae. Curr. Genet., 33, 110–116. [DOI] [PubMed] [Google Scholar]
- Gari E., Piedrafita L., Aldea M. & Herrero E. (1997) A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast, 13, 837–848. [DOI] [PubMed] [Google Scholar]
- Goto Y., Tsugane K., Tanabe Y., Nonaka I. & Horai S. (1994) A new point mutation at nucleotide pair 3291 of the mitochondrial tRNAleuUUR gene in a patient with mitochondrial myophathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Biochem. Biophys. Res. Commun., 202, 1624–1630. [DOI] [PubMed] [Google Scholar]
- Hao H. & Moares C.T. (1996) Functional and molecular mitochondrial abnormalities associated with a C—T transition at position 3256 of the human mitochondrial genome. J. Biol. Chem., 271, 2347–2352. [DOI] [PubMed] [Google Scholar]
- King M.P. & Attardi G. (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science, 246, 500–503. [DOI] [PubMed] [Google Scholar]
- Lestienne P., Bouzidi M.E., Desguerre I. & Ponsot G. (1999) in Mitochondrial Diseases (ed. Lestienne, P.) 33–58. Springer-Verlag, Heidelberg. [Google Scholar]
- Moares C.T. et al. (1993) Two novel pathogenic mitochondrial DNA mutations affecting organelle number and protein synthesis in the tRNAleu(UUR) gene: an etiologic hot spot? J. Clin. Invest., 92, 2906–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A.M., Pape L.K. & Tzagoloff A. (1985) Mitochondrial protein synthesis is required for maintenance of intact mitochondria genomes in S. cerevisiae. EMBO J., 4, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najarian D., Shu H.H. & Martin N.C. (1986) Sequence and expression of four mutant aspartic acid tRNA genes from the mitochondria of Saccharomyces cerevisiae. Nucleic Acids Res., 14, 9561–9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafael J. & Nicholls D.G. (1984) Mitochondrial membrane potential monitored in situ within isolated guinea pig brown adipocytes by a styryl pyridinium fluorescent indicator. FEBS Lett., 170, 181–185. [DOI] [PubMed] [Google Scholar]
- Rinaldi T., Francisci S., Frontali L., Zennaro E. & Bolotin-Fukuhara M. (1994) Suppression of a mitochondrial point mutation in a tRNA gene can cast light on the mechanism of 3′ end processing. Curr. Genet., 25, 451–455. [DOI] [PubMed] [Google Scholar]
- Rinaldi T., Lande R., Bolotin-Fukuhara M. & Frontali L. (1997) Additional copies of the mitochondrial EF-Tu and aspartyl-tRNA synthetase genes can compensate for a mutation affecting the maturation of the mitochondrial tRNAasp. Curr. Genet., 31, 494–496. [DOI] [PubMed] [Google Scholar]
- Rohou H., Francisci S., Rinaldi T., Frontali L. & Bolotin-Fukuhara M. (2001) Reintroduction of a characterized mt tRNA glycine mutation into yeast mitochondria provides a new tool for the study of human neurodegenerative diseases. Yeast, 18, 219–227. [DOI] [PubMed] [Google Scholar]
- Toompu M. et al. (2002) The 7472insC mitochondrial mutation impairs the synthesis and extent of aminoacylation of tRNAser(UCN) but not its structure or rate of turnover. J. Biol. Chem., 277, 22240–22250. [DOI] [PubMed] [Google Scholar]
- Wallace D.C. (1993) Mitochondrial diseases: genotype versus phenotype. Trends Genet., 9, 128–133. [DOI] [PubMed] [Google Scholar]
- Wallace D.C. (1999) Mitochondrial diseases in man and mouse. Science, 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- Woriax V.L., Burkhart W. & Spremulli L.L. (1995) Cloning, sequence analysis and expression of mammalian mitochondrial protein synthesis elongation factor Tu. Biochim. Biophys. Acta, 27, 347–356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of biolistic transformation Plasmids Primers and probes