Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron (original) (raw)
Abstract
The biogenesis of spliceosomal small nuclear ribonucleoproteins (snRNPs) requires the cytoplasmic assembly of the Sm-core complex, followed by the hypermethylation of the small nuclear RNA (snRNA) 5′ cap. Both the Sm-core complex and the snRNA trimethylguanosine cap are required for the efficient nuclear import of snRNPs. Here, we show that trimethylguanosine synthase 1 (TGS1), the human homologue of the yeast snRNA cap hypermethylase, interacts directly with the survival of motor neuron (SMN) protein. Both proteins are similarly distributed, localizing in the cytoplasm and in nuclear Cajal bodies. The interaction between TGS1 and SMN is disrupted by a mutation in SMN that mimics the predominant isoform of the protein that is expressed in patients with the neurodegenerative disease, spinal muscular atrophy. These data indicate that, in addition to its function in cytoplasmic Sm-core assembly, the SMN protein also functions in the recruitment of the snRNA cap hypermethylase.
Introduction
Small nuclear ribonucleoproteins (snRNPs) are essential eukaryotic molecules that are required for the splicing of nuclear pre-messenger RNA (Yu et al., 1999). In metazoans, the assembly of spliceosomal snRNPs is an ordered, multistep process that involves both nuclear and cytoplasmic phases (for a review, see Will & Lührmann, 2001). After transcription, the Sm-class small nuclear RNAs (snRNAs; U1, U2, U4 and U5) are exported to the cytoplasm. This step requires the binding of the snRNA 7-methylguanosine (m7G) cap structure to the PHAX/CBC complex (Izaurralde et al., 1995; Ohno et al., 2000). In the cytoplasm, the snRNAs then associate with the survival of motor neuron (SMN) complex, which directs the assembly of the seven related Sm proteins, SmB/B', SmD1–3, SmE, SmF and SmG (Paushkin et al., 2002; Meister et al., 2002; Kambach et al., 1999). The m7G cap is then hypermethylated to form a 2,2,7-trimethylguanosine (m3G) cap structure. Both the assembled Sm core and the m3G cap structure of the snRNAs provide signals for the efficient nuclear import of the newly assembled snRNPs (Fischer & Lührmann, 1990; Hamm et al., 1990; Plessel et al., 1994). Last, the m3G cap structure is recognized by the snurportin1 (SPN1) protein, which enhances the nuclear import of U snRNPs (Huber et al., 1998).
In mammals, the SMN complex is required for the assembly of the Sm core. Mutations in the human SMN1 gene are responsible for the neuromuscular disease, spinal muscular atrophy (SMA; for a review, see Frugier et al., 2002). SMN interacts directly with three of the Sm proteins by binding to the Arg- and Gly-rich carboxy-terminal tails of the SmB, SmD1 and SmD3 proteins (Friesen & Dreyfuss, 2000). Interestingly, these domains are dimethyl-arginine-modified, and such modifications are essential for strong interactions with SMN (Friesen et al., 2001; Brahms et al., 2000, 2001).
The methyltransferase that is responsible for m3G cap formation has recently been identified (Mouaikel et al., 2002; Verheggen et al., 2002), but the mechanism linking Sm-core assembly to cap hypermethylation is unknown. The observation that snRNA cap hypermethylation takes place in the cytoplasm after Sm-core assembly, and the crucial function of the SMN complex in bringing snRNAs and Sm proteins together (Fischer et al., 1997; Meister et al., 2001; Pellizzoni et al., 2002), led us to examine whether the SMN protein might also be involved in snRNA m3G cap formation. Here, we show that the SMN protein and the human snRNA cap hypermethylase associate in vivo and in vitro, indicating that, in addition to its role in cytoplasmic Sm-core protein assembly, the SMN protein also functions in the cap hypermethylation process.
Results
SMN and TGS1 proteins form a complex in vivo and in vitro
To analyse the relationship between the SMN protein and snRNA cap hypermethylation, we first cloned the human orthologue of the yeast trimethylguanosine synthase, Tgs1 (data not shown). The full-length TGS1 open reading frame encodes an 852 amino-acid protein, which comprises a carboxy-terminal region that contains conserved methyltransferase motifs, and a large amino-terminal domain that has no obvious similarities to other known proteins (Mouaikel et al., 2002). In yeast, the cap hypermethylase associates preferentially with the C-terminal extension of the SmB protein (Mouaikel et al., 2002). We therefore tested the interactions of TGS1 using in vitro glutathione-_S_-transferase (GST) pull-down assays. [35S]methionine-labelled TGS1 strongly bound a GST fusion that contained the C-terminal tail of human SmB, bound less efficiently to a fusion containing the SmD1 C terminus, and did not bind significantly to a fusion containing the C terminus of SmD3 (Fig. 1A). These data indicate that TGS1, like its yeast counterpart, binds preferentially to the C-terminal extension of SmB.
Figure 1.

Interaction of human trimethylguanosine synthase 1 with the carboxyl tail of SmB and the survival of motor neuron protein. (A) The glutathione-_S_-transferase (GST) fusion proteins indicated were incubated with [35S]-labelled human trimethylguanosine synthase 1 (TGS1), which was prepared by in vitro transcription and translation using rabbit reticulocyte lysate. Bound proteins were analysed by SDS–polyacrylamide gel electrophoresis (SDS–PAGE). 'Input' indicates aliquots of radioactive proteins that corresponded to 10% of that used in each binding reaction. (B) Anti-TGS1 recognizes endogenous TGS1 as well as the green fluorescent protein (GFP)–TGS1 fusion protein and a fusion protein consisting of the carboxyl terminal of TGS1 tagged with GFP (GFP–Cter). The GFP fusion proteins indicated were transfected into HeLa cells and the extracts were separated by SDS–PAGE. Western blotting analysis was performed using anti-TGS1 antibodies. (C) The survival of motor neuron (SMN) protein was immunoprecipitated from a cytoplasmic extract using anti-TGS1 antibodies. Immuno-precipitation was performed using HeLa S100 extract and anti-TGS1 or preimmune serum (PI), and the proteins were immunoblotted with anti-TGS1 (upper panel) and anti-SMN (lower panel) antibodies. 'Tot' indicates 10% of the input. (D) TGS1 was immunoprecipitated from a cytoplasmic extract using anti-SMN antibodies. Immunoprecipitation was performed using HeLa cytoplasmic S100 extract and anti-SMN or PI, and the proteins were immunoblotted with anti-TGS1 (upper panel) and anti-SMN (lower panel) antibodies.
Next, we used antibodies against the C-terminal domain of the human hypermethylase to characterize a possible interaction between SMN and TGS1 in mammalian cells. As expected, western blot analysis (Fig. 1B) showed that these antibodies specifically recognize the endogenous TGS1 protein as well as green fluorescent protein (GFP)–TGS1 and the GFP–Cter fusion, which carrys the C-terminal domain of TGS1 (amino acids 478–852). By contrast, the GFP–Nter fusion protein, which carries the amino-terminal domain (amino acids 1–477), was not detected. Immunoprecipitation experiments that used these anti-TGS1 antibodies and a cytoplasmic extract showed that the SMN protein coprecipitated with TGS1 (Fig. 1C). The TGS1 protein was also co-immunoprecipitated by an antisMN antibody (Fig. 1D). Thus, these proteins form part of an interrelated complex in vivo.
To show that TGS1 and SMN interact directly, we performed in vitro GST pull-down assays. As shown in Fig. 2A, [35S]-labelled SMN bound quantitatively to GST–TGS1 and GST–Cter, whereas no significant interaction was detected with the GST–Nter fusion. By contrast, the N-terminal domain of TGS1 interacted strongly with the human SmB protein, whereas the C-terminal domain did not (Fig. 2B). The separability of the two binding activities suggested that the interaction between TGS1 and SMN was not mediated by Sm proteins. To definitively rule out the possibility that the interaction between TGS1 and SMN was due to other proteins in the rabbit reticulocyte lysate, we performed pull-down assays using [35S]-labelled SMN that was translated from a bacterial extract. As shown in Fig. 2C, [35S]-labelled SMN bound efficiently to GST–TGS1. These results show that TGS1 can interact directly and simultaneously with the SMN and SmB proteins, as the C-terminal domain of TGS1 binds to SMN and to the N-terminal domain of SmB.
Figure 2.
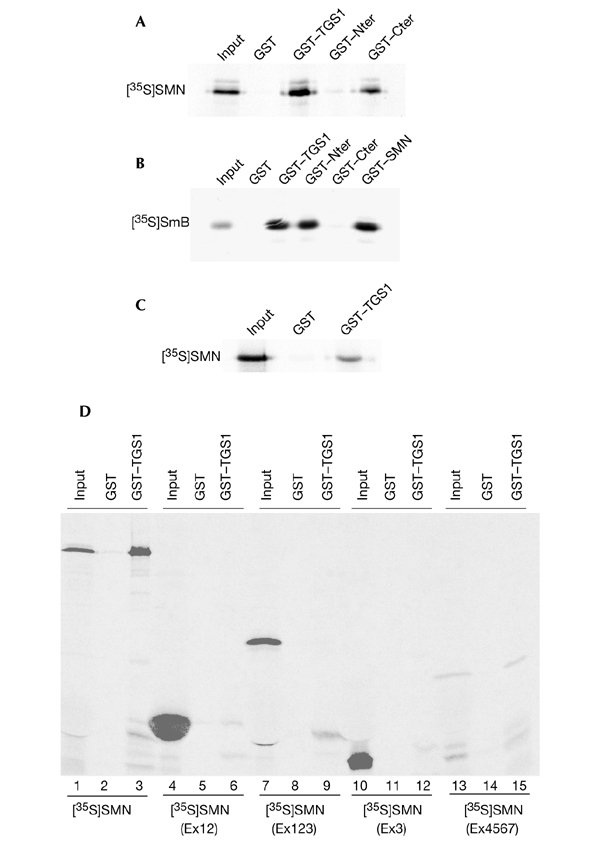
Direct binding of human trimethylguanosine synthase 1 to the survival of motor neuron protein. (A) Binding assays using recombinant glutathione-_S_-transferase (GST)–trimethylguanosine synthase 1 (TGS1) and fusions of GST to the amino and carboxyl termini of TGS1 (GST–Nter and GST–Cter, respectively) with [35S]-labelled survival of motor neuron (SMN) that was in vitro translated using rabbit reticulocyte lysate. Bound proteins were analysed by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and autoradiography. (B) The GST fusion proteins indicated were tested for binding with [35S]-labelled SmB protein and treated as described for (A). (C) Binding of the GST–TGS1 protein to in vitro translated [35S]-labelled SMN using an S30 bacterial extract. Proteins bound to GST beads were analysed by SDS–PAGE and autoradiography. (D) The C-terminal region of SMN mediates the interaction with TGS1. The indicated [35S]-labelled SMN proteins were mixed with the GST–TGS1 or GST proteins, and bound proteins were analysed by SDS–PAGE and autoradiography. Note that the amount of [35S]-labelled SMN(Ex4567) input is lower compared with the other [35S]-labelled SMN domains. 'Input' indicates aliquots of radioactive proteins that corresponded to 10% of that used in each binding reaction.
To characterize the domains of the SMN protein that are involved in the interaction with TGS1, we tested the ability of various SMN deletion mutants to bind recombinant GST–TGS1. Fig. 2D shows that both wild-type SMN (lane 3) and the SMN(Ex4567) mutant (lane 15) bind the GST–TGS1 fusion. By contrast, the SMN(Ex12), SMN(Ex123) and SMN(Ex3) constructs (Fig. 2D, lanes 6, 9 and 12, respectively) failed to bind efficiently to GST–TGS1. These experiments show that the region containing exons 4–7 of the SMN protein is sufficient for interaction with TGS1.
Similar subcellular distributions of SMN and TGS1
As described above, SMN is distributed diffusely throughout the cytoplasm, and also accumulates in nuclear Cajal bodies (Liu & Dreyfuss, 1996; Matera & Frey, 1998; Carvalho et al., 1999). Fluorescence microscopy of HeLa cells after transient transfection of a GFP–TGS1 construct shows that the fusion protein localizes similarly to SMN: it is present in Cajal bodies and is distributed diffusely in the cytoplasm (Fig. 3A–D). As expected, a GFP–SMN fusion also localizes diffusely throughout the cytoplasm, and also accumulates in nuclear Cajal bodies (Fig. 3E). Immunocytochemistry using TGS1 antibodies also shows that the hypermethylase distributes similarly to SMN, localizing in the cytoplasm and in nuclear Cajal bodies (Fig. 3F; Verheggen et al., 2002). The colocalization of the SMN and TGS1 proteins was confirmed using a dominant-negative SMN mutant (SMNΔN27), which causes a rearrangement of snRNPs and gives rise to cytoplasmic aggregates that contain endogenous SMN complexes, Sm proteins and m7G-capped snRNAs (Pellizzoni et al., 1998). We investigated whether the expression of SMNΔN27 had an effect on the subcellular localization of TGS1 in HeLa cells. As shown in Fig. 3I–L, TGS1 accumulated with the GFP–SMNΔN27 protein in cytoplasmic aggregates, showing that this mutant construct prevents the formation of subnuclear bodies containing TGS1 and SMN. It is interesting to note that expression of the GFP–SMNΔN27 fusion did not generate the nuclear inclusions seen by Pellizzoni et al. (1998) using MycsMNΔN27. The differences might be because of variations in protein expression levels due to the vectors used in both studies.
Figure 3.
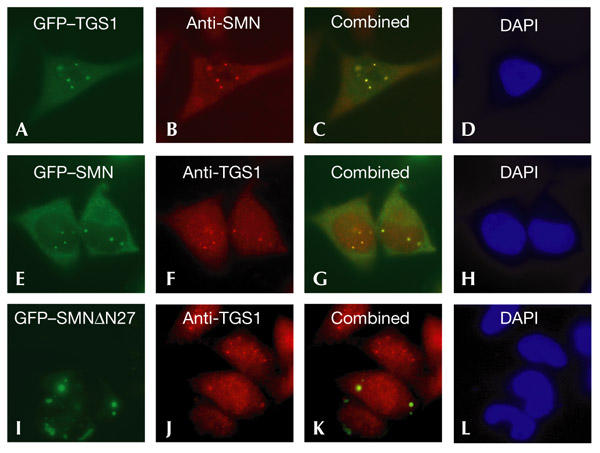
Colocalization of the human trimethylguanosine synthase 1 and survival of motor neuron proteins. HeLa cells carrying green fluorescent protein (GFP)–trimethylguanosine-synthase-1 (TGS1; upper row), GFP–survival-of-motor-neuron (SMN; middle row) or GFP–SMNΔN27 (lower row) constructs were examined by immunofluorescence microscopy using anti-SMN or anti-TGS1 antibodies. Colocalization of the GFP fusion proteins (green) and SMN or TGS1 (red) is indicated by the yellow colour in the combined panels. DNA was stained with 4,6-diamidino-2-phenylindole (DAPI; blue).
TGS1 does not form a complex with snurportin 1
It has been reported recently that the SMN complex is associated with m3G-capped snRNPs bound to the SPN1 protein (Narayanan et al. 2002; Massenet et al., 2002). We therefore tested whether TGS1 is also part of this complex. To test this, we transfected HeLa cells with GFP–TGS1, immunoprecipitated the total cell lysates with monoclonal antibodies against GFP, and probed a western blot with anti-GFP and antisPN1 antibodies. As shown in Fig. 4A, no SPN1 was co-immunoprecipitated, indicating that TGS1 and SPN1 do not form a complex in vivo. Similarly, in cells transfected with a GFP–SPN1 construct, immunoprecipitation with anti-GFP antibodies did not recover TGS1 (Fig. 4B). By contrast, in both experiments, the SMN protein was immunoprecipitated with anti-GFP antibodies (Fig. 4A,B), confirming that SMN associates with both GFP–TGS1 and GFP–SPN1 in vivo. The idea that TGS1 and SPN1 do not form a complex in vivo is further supported by the inability of anti-TGS1 antibodies to pull down endogenous SPN1. As shown in Fig. 4C (lane 3), anti-TGS1 antibodies failed to immunoprecipitate SPN, whereas in control experiments antisMN (Narayanan et al., 2002; Massenet et al., 2002) recovered both TGS1 and SPN1 (Fig. 4C, lane 4).
Figure 4.

Snurportin 1 does not form a complex with human trimethylguanosine synthase 1 in vivo. (A) HeLa cells were transiently transfected with green fluorescent protein (GFP)–trimethylguanosine-synthase-1 (TGS1) and total cell lysates were immunoprecipitated with anti-GFP antibodies. The immunoprecipitates were then analysed by western blotting using the antibodies indicated. Normal mouse serum (NMS) was used as a non-immune control. (B) HeLa cells were transfected with GFP–snurportin 1 (SPN1) and immunoprecipitation experiments were performed either with NMS or with anti-GFP antibodies. Blots were probed with the indicated antibodies. (C) Cytoplasmic S100 extract was immunoprecipitated with anti-TGS1, anti-survival-of-motor-neuron (SMN) and preimmune serum (PI). Immunoprecipitates were analysed by SDS–polyacrylamide gel electrophoresis and immunoblotted with anti-TGS1 (upper panel) and anti-SPN1 (lower panel) antibodies. 'Tot' indicates 2% of the input. 'Input' indicates aliquots of radioactive proteins that corresponded to 2% of that used in each binding reaction.
Disrupted SMNDEx7 and TGS1 interaction
In view of the pathology of SMA and the interaction between SMN and TGS1, we tested whether the hypermethylase is able to form a complex with SMN mutant proteins in vitro and in vivo. As shown in Fig. 5A, GST pull-down assays showed that binding of the [35S]-labelled SMNΔEx7 construct to a GST–TGS1 protein was greatly impaired. Moreover, the [35S]-labelled SMNΔN27 mutant had a binding activity similar to that of wild-type SMN. To determine whether the association between the TGS1 and SMNΔEx7 proteins was disrupted in vivo, we transiently transfected plasmids carrying SMN, SMNΔN27 and SMNΔEx7 constructs tagged with ZZ (a fragment of protein A). Whole-cell extracts were prepared from these cells and, as shown in Fig. 5B (lower panel), the ZZ–SMN, ZZ–SMNΔN27 and ZZ–SMNΔEx7 fusion proteins were immunoprecipitated with rabbit IgG. The TGS1 protein was co-immunoprecipitated from cells expressing the ZZ–SMN and ZZ–SMNΔN27 constructs (Fig. 5B, upper panel), but only a very small amount was detected in the pellet of extracts prepared from cells expressing ZZ–SMNΔEx7 (Fig. 5B). These results show that the interaction of the hypermethylase with SMN is disrupted by the SMN mutation that is most commonly present in SMA patients.
Figure 5.
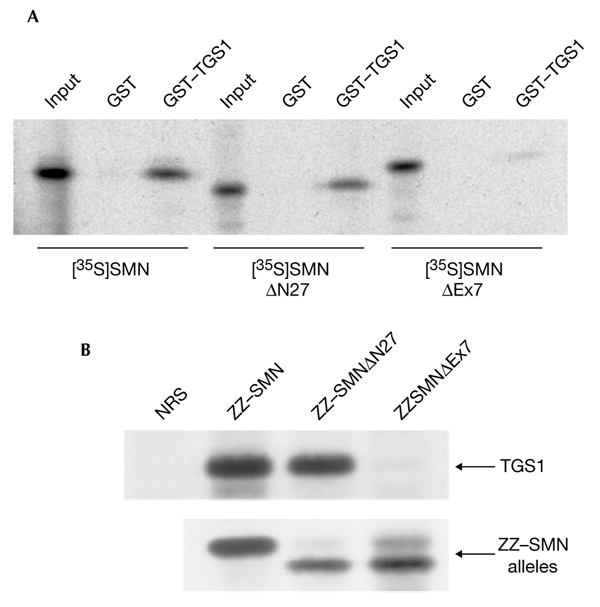
Disruption of the interaction between the SMNΔEx7 mutant and human trimethylguanosine synthase 1. (A) In vitro translated [35S]-labelled SMN, [35S]-labelled SMNΔN27 and [35S]-labelled SMNΔEx7 were incubated with recombinant glutathione-_S_-transferase (GST)–trimethylguanosine-synthase-1 (TGS1) or GST. Bound proteins were analysed by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and autoradiography. (B) Hela cells were transfected with the ZZ-tagged SMN alleles indicated and immunoprecipitation experiments were performed with either normal rabbit serum (NRS) or rabbit IgG and protein-A–sepharose beads. Immunocomplexes were then separated by SDS–PAGE and immunoblotted with anti-TGS1 (upper panel) or anti-SMN (lower panel) antibodies. SMN, survival of motor neuron protein. 'Input' indicates aliquots of radioactive proteins that corresponded to 10% of that used in each binding reaction.
Discussion
We show here, for the first time, that the SMN protein binds directly to the human snRNA cap hypermethylase both in vitro and in vivo. Our data suggest a role for the SMN protein complex in the recruitment of TGS1, suggesting a function for the SMN complex in the formation of the snRNA m3G cap structure. As SMN is essential for the assembly of the Sm core complex in higher eukaryotes, our results also suggest that the SMN complex can be seen as a machine that participates in the generation of the bipartite nuclear localization signal of snRNPs.
The human snRNA cap hypermethylase, like its yeast counterpart, binds preferentially to the C-terminal extension of SmB (Fig. 1A). These findings are consistent with a previous report that suggested that the human snRNA-(guanosine-N2)-methyltransferase recognizes U1 snRNP by binding to the SmB/B' proteins (Plessel et al., 1994). As the C-terminal extension of SmB also interacts with SMN (Friesen & Dreyfuss, 2000; Friesen et al., 2001; Brahms et al., 2001), our results show that both TGS1 and SMN recognize, at least in part, a similar interaction determinant in the heptameric Sm-core-complex. Whether SMN and TGS1 interact consecutively or simultaneously with the C-terminal extension of SmB cannot be concluded from our results and will require further competition experiments.
The interaction between the SMN and TGS1 proteins that we describe in this report are consistent with two recent studies that showed that the SMN complex is associated with snRNPs during the various cytoplasmic steps of snRNP maturation (Narayanan et al. 2002; Massenet et al., 2002). Indeed, SMN was shown to associate with both m7G- and m3G-capped snRNAs, suggesting that the SMN complex is present before and after the snRNA cap hypermethylation process. Consistent with these results, we show that SMN interacts directly with the hypermethylase. A role for SMN in m3G cap formation is further supported by the fact that the dominant-negative mutant, SMNΔN27, blocks the snRNP maturation pathway in the cytoplasm in a step that precedes cap hypermethylation (Pellizzoni et al., 1998). A model that shows the relationships between SMN and TGS1 in snRNP assembly is shown in Fig. 6. We suggest that after promoting the formation of the Sm ring, the SMN complex allows the engagement of TGS1 with the m7G-capped snRNP particle. As shown in the model, a conformational change might then allow dissociation of the interaction between the C terminus of SmB and the SMN complex, followed by the association of the SmB C terminus with the hypermethylase. Such a step could also allow the concomitant release of the PHAX/CBC complex (see below) and proper folding of the TGS1 protein in order to bring the m7G cap structure to the catalytic centre and allow m3G cap formation. The association of the SPN1 protein with the m3G cap structure could then induce the loss of the hypermethylase from this complex and generate import-competent snRNPs. This idea is consistent with our finding that no SPN1–TGS1 complex was detected in HeLa cells in co-immunoprecipitation experiments (Fig. 4).
Figure 6.

Model of TGS1 and SMN interactions in cytoplasmic snRNP biogenesis. Filled red circles represent the carboxy-terminal extensions of SmB, SmD1 and SmD3 proteins. The defect of the SMNΔN27 mutant is also indicated. See text for more details. m3G, 2,2,7-trimethylguanosine cap; m7G, 7-methylguanosine cap; PHAX/CBC; SMNc, survival of motor neuron complex; snRNP, small nuclear ribonucleoprotein; SPN1, snurportin 1; TGS1, trimethylguanosine synthase 1.
Our subcellular localization studies (Fig. 3) show that TGS1 is found in the cytoplasmic accumulations that are generated by the expression of the SMNΔN27 mutant in HeLa cells (Pellizzoni et al., 1998). The fact that PHAX is also found in similar aggregates (Massenet et al., 2002) indicates that both PHAX and TGS1 could also be part of the same functional complex. It is not yet known how and when the PHAX/CBC complex is released from m7G-capped snRNPs. According to our model (Fig. 6), following the initial binding of TGS1 to SMN, a conformational change is required both for the release of the PHAX/CBC complex and for the correct positioning of the hypermethylase with respect to the m7G cap structure. The hypermethylation defect of the SMNΔN27 allele could therefore be a consequence of an inability of this mutant to undergo such a conformational change.
It is not yet known whether the observed weak in vitro and in vivo interactions of the hypermethylase with the SMNΔEx7 isoform result in a defect in snRNA cap hypermethylation. The correlation between the binding of TGS1 with SMN and snRNA m3G cap formation remains to be demonstrated directly and will require the development of an in vitro hypermethylation assay. However, our finding that SMN and TGS1 interact directly strengthens the view of the SMN complex as a machine that is involved in the formation of import-competent snRNPs (Massenet et al., 2002; Narayanan et al., 2002). Accordingly, it has recently been shown that reduced SMN/Gemin 2 protein levels lead to reduced nuclear accumulation of U snRNPs and to enhanced motoneuron degeneration (Jablonka et al., 2002). These observations suggest that impaired generation of the UsnRNP bipartite nuclear localization signal represents an important biochemical defect that is responsible for the SMA disease.
Methods
Cloning of complete trimethylguanosine synthase 1 complementary DNA and plasmid constructs.
The full-length TGS1 cDNA was obtained by PCR and RT–PCR (PCR after reverse transcription) using oligonucleotides based on the sequence of the human orthologue (GenBank accession number AY028423) of the yeast trimethylguanosine synthase, Tgs1 (Mouaikel et al., 2002). A human cDNA library and Image clone 4513665 (GenBank accession number BG287526) were used to amplify the corresponding DNA fragments.
GFP-tagged and ZZ-tagged wild-type and deletion SMN constructs were generated by PCR using appropriate oligonucleotides and were cloned into the pEGFP vector (Clontech) and a version of the pEGFP vector that lacks the GFP coding region (for the ZZ-tagged constructs).
Cell culture and immunocytochemistry.
HeLa cells were grown at 37 °C in 5% CO2, in DMEM containing 10% FCS. Cells were transfected using the calcium phosphate coprecipitation procedure as described previously (Verheggen et al., 2002). HeLa cell extracts were prepared as described in Narayanan et al. (2002).
Immunofluorescence was performed on cells grown on coverslips, washed in PBS and fixed in 4% (w/v) paraformaldehyde in PBS for 15 min at 25 °C. Permeabilization was performed using 0.5% Triton-X100 in PBS for 5 min at 25 °C or in acetone (3 min at −20 °C). Anti-SMN (Transduction Laboratories) and anti-TGS1 rabbit polyclonal antibodies (Verheggen et al., 2002) were diluted 1 in 500. Incubations were performed for 1 h at 25 °C in PBS containing 2% BSA, and slides were then washed three times for 10 min in PBS. Secondary antibodies, which were coupled to Cy3, were diluted to 1 in 2000 and were used in the same conditions. Coverslips were mounted on glass slides using mounting medium (Vectorshield) and were observed as described previously (Verheggen et al., 2002).
Glutathione-_S_-transferase pull-down assays and immunoprecipitations.
Plasmids carrying GST fusions were transformed into Escherichia coli BL21 cells, which were grown in LB medium containing ampicillin and 20 mM glucose; expression was induced by the addition of 0.5 mM isopropyl-_D_-thiogalactoside. Recombinant proteins were purified using GST beads (Amersham Pharmacia). In vitro transcriptions using T7 RNA polymerase and translations using rabbit reticulocyte lysate (TnT) or bacterial extract (S30) were performed in the presence of [35S]methionine, in accordance with the manufacturer's instructions (Promega). In vitro protein–protein interaction studies were carried out as already described (Mouaikel et al., 2002). Immunoprecipitation experiments were performed as described previously (Mouaikel et al., 2002; Narayanan et al., 2002). Immunocomplexes were detected with horseradish-peroxidase-conjugated sheep-anti-mouse or peroxidase-conjugated immunopure protein A/G (Pierce), followed by chemiluminescence.
Acknowledgments
We thank J. Soret for discussion and critical comments on the manuscript and the UK Human Genome Project Resource Centre for the Image clone. J.M. was supported by a graduate fellowship from MENRT and the Association pour la Recherche contre le Cancer (ARC). This work was supported by grants from the ARC, the Association Française contre les Myopathies (AFM), the Centre National de la Recherche Scientifique, the US National Institutes of Health and the Muscular Dystrophy Association.
References
- Brahms H., Raymackers J., Union A., de Keyser F., Meheus L. & Lührmann R. (2000) The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for antism autoantibodies. J. Biol. Chem., 275, 17122–17129. [DOI] [PubMed] [Google Scholar]
- Brahms H., Meheus L., de Brabandere V., Fischer U. & Lührmann R. (2001) Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B' and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA, 7, 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho T., Almeida F., Calapez A., Lafarga M., Berciano M.T. & Carmo-Fonseca M. (1999) The spinal muscular atrophy disease gene product, SMN: a link between snRNP biogenesis and the Cajal (coiled) body. J. Cell. Biol., 147, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U. & Lührmann R. (1990) An essential signaling role for the m3G cap in the transport of U1 snRNP to the nucleus. Science, 249, 786–790. [DOI] [PubMed] [Google Scholar]
- Fischer U., Liu Q. & Dreyfuss G. (1997). The SMNsIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell, 90, 1023–1029. [DOI] [PubMed] [Google Scholar]
- Friesen W.J. & Dreyfuss G. (2000) Specific sequences of the Sm and Sm-like (Lsm) proteins mediate their interaction with the spinal muscular atrophy disease gene product (SMN). J. Biol. Chem., 275, 26370–26375. [DOI] [PubMed] [Google Scholar]
- Friesen W.J., Massenet S., Paushkin S., Wyce A. & Dreyfuss G. (2001) SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell, 7, 1111–1117. [DOI] [PubMed] [Google Scholar]
- Frugier T., Nicole S., Cifuentes-Diaz C. & Melki J. (2002) The molecular bases of spinal muscular atrophy. Curr. Opin. Genet. Dev., 12, 294–298. [DOI] [PubMed] [Google Scholar]
- Hamm J., Darzynkiewicz E., Tahara S.M. & Mattaj I.W. (1990) The trimethylguanosine cap structure of U1 snRNA is a component of a bipartite nuclear targeting signal. Cell, 62, 569–577. [DOI] [PubMed] [Google Scholar]
- Huber J., Cronshagen U., Kadokura M., Marshallsay C., Wada T., Sekine M. & Lührmann R. (1998) Snurportin1, an m3G-capspecific nuclear import receptor with a novel domain structure. EMBO J., 17, 4114–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E., Lewis J., Gamberi C., Jarmolowski A., McGuigan C. & Mattaj I.W. (1995) A cap-binding protein complex mediating U snRNA export. Nature, 376, 709–712. [DOI] [PubMed] [Google Scholar]
- Jablonka S., Holtmann B., Meister G., Bandilla M., Rossoll W., Fischer U. & Sendtner M. (2002) Gene targeting of Gemin2 in mice reveals a correlation between defects in the biogenesis of U snRNPs and motoneuron cell death. Proc. Natl Acad. Sci. USA., 99, 10126–10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambach C., Walke S., Young R., Avis J.M., de la Fortelle E., Raker V.A., Lührmann R., Li J. & Nagai K. (1999) Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell, 96, 375–387. [DOI] [PubMed] [Google Scholar]
- Liu Q. & Dreyfuss G. (1996) A novel nuclear structure containing the survival of motor neurons protein. EMBO J., 15, 3555–3565. [PMC free article] [PubMed] [Google Scholar]
- Massenet S., Pellizzoni L., Paushkin S., Mattaj I.W. & Dreyfuss G. (2002) The SMN complex Is associated with snRNPs throughout their cytoplasmic assembly pathway. Mol. Cell. Biol., 22, 6533–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera A.G. & Frey M.R. (1998) Coiled bodies and gems: Janus or gemini? Am. J. Hum. Genet., 63, 317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G., Buhler D., Pillai R., Lottspeich F. & Fischer U. (2001) A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nature Cell Biol., 11, 945–949. [DOI] [PubMed] [Google Scholar]
- Meister G., Eggert C. & Fischer U. (2002) SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol., 12, 472–478. [DOI] [PubMed] [Google Scholar]
- Mouaikel J., Verheggen C., Bertrand E., Tazi J. & Bordonné R. (2002) Hypermethylation of the cap structure of both yeast snRNAs and snoRNAs requires a conserved methyltransferase that is localized to the nucleolus. Mol. Cell, 9, 891–901. [DOI] [PubMed] [Google Scholar]
- Narayanan U., Ospina J.K., Frey M.R., Hebert M.D. & Matera A.G. (2002) SMN, the spinal muscular atrophy protein, forms a pre-import snRNP complex with snurportin1 and importin β. Hum. Mol. Genet., 11, 1785–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M., Segref A., Bachi A., Wilm M. & Mattaj I.W. (2000) PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell, 101, 187–198. [DOI] [PubMed] [Google Scholar]
- Paushkin S., Gubitz A.K., Massenet S. & Dreyfuss G. (2002) The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell. Biol., 14, 305–312. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L., Kataoka N., Charroux B. & Dreyfuss G. (1998) A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell, 95, 615–624. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L., Yong J. & Dreyfuss G. (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science, 298, 1775–1779. [DOI] [PubMed] [Google Scholar]
- Plessel G., Fischer U. & Lührmann R. (1994) m3G cap hypermethylation of U1 small nuclear ribonucleoprotein (snRNP) in vitro: evidence that the U1 small nuclear RNA-(guanosine-N2)-methyltransferase is a nonsnRNP cytoplasmic protein that requires a binding site on the Sm core domain. Mol. Cell. Biol., 14, 4160–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheggen C., Lafontaine D.L., Samarsky D., Mouaikel J., Blanchard J.M., Bordonné R. & Bertrand E. (2002) Mammalian and yeast U3 snoRNPs are matured in specific and related nuclear compartments. EMBO J., 21, 2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will C.L. & Lührmann R. (2001) Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell. Biol., 13, 290–301. [DOI] [PubMed] [Google Scholar]
- Yu Y.T., Scharl E.C., Smith C.M. & Steitz J.A. (1999) in The RNA World 2nd edn. (eds Gesteland, R.F., Cech, T.R. & Atkins, J.F.), 487–524. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA. [Google Scholar]