Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage (original) (raw)
Abstract
Cells maintain genomic stability by the coordination of DNA-damage repair and cell-cycle checkpoint control. In replicating cells, DNA damage usually activates intra-S-phase checkpoint controls, which are characterized by delayed S-phase progression and increased Rad53 phosphorylation. We show that in budding yeast, the intra-S-phase checkpoint controls, although functional, are not activated by the topoisomerase I inhibitor camptothecin (CPT). In a CPT-hypersensitive mutant strain that lacks the histone 2A (H2A) phosphatidylinositol-3-OH kinase (PI(3)K) motif at Ser 129 (h2a-s129a), the hypersensitivity was found to result from a failure to process full-length chromosomal DNA molecules during ongoing replication. H2A Ser 129 is not epistatic to the RAD24 and RAD9 checkpoint genes, suggesting a non-checkpoint role for the H2A PI(3)K site. These results suggest that H2A Ser 129 is an essential component for the efficient repair of DNA double-stranded breaks (DSBs) during replication in yeast, particularly of those DSBs that do not induce the intra-S-phase checkpoint.
Introduction
A stable genome is characterized by the passing from generation to generation of essentially identical DNA. Genomic instability and chromosomal abnormalities often result from the defective repair of DNA double-stranded breaks (DSBs). One of the earliest responses to the formation of DNA DSBs in mammals is the rapid phosphorylation of histone 2AX (H2AX) molecules adjacent to the break site (Rogakou et al., 1999). The phosphorylated derivative, γ-H2AX, forms foci and is necessary for subsequent recruitment of DNA repair factors (Paull et al., 2000; Celeste et al., 2002). The phosphorylated serine is in a Ser-Gln-Glu consensus target motif, which is recognized by the PI(3)Ks DNA-PK, ATM, and ATR (Shiloh, 2001). Mice lacking H2AX have unstable genomes and primary mouse embryonic fibroblasts and T cells from these mice contain numerous chromosome abnormalities (Celeste et al., 2002).
The yeast histone species that are orthologous to H2AX, H2A1 and H2A2 (hereafter referred to as H2A(X)), comprise approximately 95% of the yeast H2A complement. Yeast H2A(X)s are phosphorylated on the homologous serine that is located four residues from the carboxyl terminus (Ala-Ser(129)-Gln-Glu-Leu*; Ala-Ser(139)-Gln-Glu-Tyr* in mammalian H2AX; where asterisks indicate C termini) in response to DNA DSBs (Rogakou et al., 1999; Downs et al., 2000). We report that DNA DSBs induced by the topoisomerase 1 (Top1) inhibitor CPT do not lead to intras-phase delay in yeast, although CPT functions by increasing the incidence of collisions between Top1–DNA complexes and replication forks. The collisions generate DSBs, which trap the Top1 molecules on the DNA in irreversible suicide complexes. Yeast strains that lack the PI(3)K serine in H2A(X) show CPT hypersensitivity, which is manifested by defective maturation of the large, newly replicated chromosomes. These results suggest that phosphorylation of yeast H2A(X) Ser 129 is not involved in activating the intra-S-phase checkpoint, but rather in the efficiency of DNA repair. Thus, H2A Ser 129 is essential for the efficient repair of DNA DSB damage, particularly that which is unrecognized by cell-cycle checkpoints.
Results and Discussion
As in mammalian cells, γ-H2A(X) is formed in yeast in the 30 min immediately after exposure to bleomycin or ionizing radiation. Using two-dimension gel electrophoresis and a specific antibody (Rogakou et al., 1998, 1999; supplementary information online), γ-H2A(X) formation can be quantified (Table I). The H2A(X) species with the PI(3)K site comprise 95% of chromatin H2A in yeast and 10% in mammals, and are randomly distributed throughout the chromatin; hence, the fraction of H2A(X) that is phosphorylated is a measure of the fraction of chromatin involved in DSB recognition. This fraction is similar in yeast and mammals, although mammalian cells contain 400 times the amount of chromatin found in haploid yeast cells (see column 5 in Table 1). γ-H2A(X) is also formed in yeast in response to several agents that have not been examined in mammalian cells, including methylmethanesulphonate (MMS) and phleomycin (Fig. 1A; Downs et al., 2000). CPT, the cellular target of which is Top1 (Pourquier & Pommier, 2001), also induces γ-H2A(X) formation in a concentration- and time-dependent manner (Fig. 1A–C).
Table 1.
Fraction of the yeast and mammalian genomes involved in each DNA double-stranded break
| | | Species | | | | --- | ------------------------------------------------- | ----- | ----- | | Row | | Sc | Hs | | (1) | DNA content (Mbp) (known) | 17 | 6,600 | | (2) | γ-H2A(X)/total H2A(X) (%) (experimental) | 4 | 25 | | (3) | Gy (experimental) | 2,000 | 25 | | (4) | DSBs/Gy | 0.1 | 33 | | (5) | γ-H2A(X)/DSB (%) ((row 2)/[(row 3) × (row 4)]) | 0.02 | 0.03 | | (6) | Focus size (Mbp) ((row 1) × (row 5)) | 0.004 | 2 | | (7) | H2A(X) × 106 molecules per cell (experimental) | 0.17 | 6 | | (8) | Molecules of γ-H2A(X) per DSB ((row 1) × (row 5)) | 34 | 1,800 |
Figure 1.
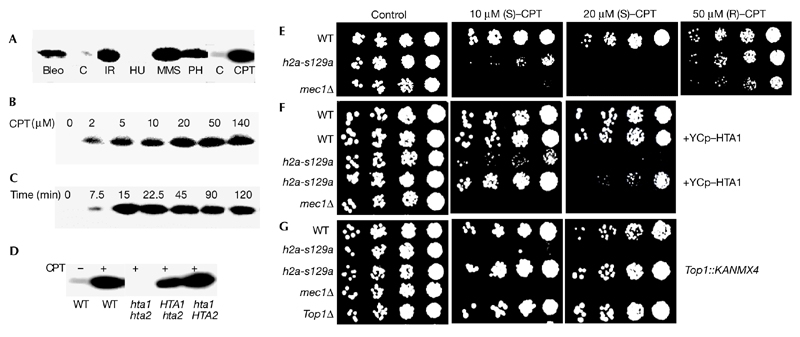
H2A(X) Ser 129 is essential for survival in the presence of topoisomerase-1-mediated DNA damage. (A) γ-H2A(X) formation by various DNA-damaging agents in wild-type (WT) yeast. Cultures were exposed to the agents for 1 h (14 min for IR) at 30 °C. (B) γ-H2A(X) formation in wild-type yeast incubated with various concentrations of camptothecin (CPT) or (C) incubated with 20 μM CPT for various durations. (D) γ-H2A(X) formation in strains in which Ser 129 had been altered to alanine in either one or both H2A(X) genes and which had been incubated with 20 μM CPT for 15 min. (E–G) h2a-s129a cells are sensitive to CPT. (E) Cells were treated with active (S) or inactive (R) CPT. Insertion of a functional HTA1 gene (F) and deletion of topoisomerase 1 (TOP1) (G) rescue the h2a-s129a mutation from CPT-induced death. Bleo, 20 μg bleomycin ml−1; C, untreated controls; H2A(X), histone 2A(X); IR, irradiation with 200 Gy; HU, 200 mM hydroxyurea; MMS, 0.2% methylmethanesulphonate; Ph, 2.5 μg phleomycin ml−1.
To determine a biological role for γ-H2A(X) formation, we altered HTA1 and HTA2 to encode proteins with Ser 129 of the motif Ala-Ser-Gln-Glu-Leu* changed to alanine (hta1-s129a and hta2-s129a; referred to hereafter as h2a-s129a). The presence of either of the wild-type H2A(X) protein species is sufficient for yeast γ-H2A(X) formation (Fig. 1D). The h2as129a mutant was found to be hypersensitive to CPT (Fig. 1E). The relationship between H2A(X) γserine and Top1 functions was confirmed by the requirements for the active S-enantiomer of CPT (Fig. 1E; Jaxel et al., 1989), for the absence of HTA1 and HTA2 (Fig. 1F) and for the presence of TOP1 (Fig. 1G). The sensitivity to CPT was not due to altered Top1 expression in the histone mutants, as a functional fusion of Top1 with a green fluorescent protein showed similar fluorescent protein levels in wild-type and h2as129a cells (data not shown).
Sensitivity to CPT is reported to occur primarily during S-phase, when CPT stabilization of Top1–DNA intermediate covalent complexes increases their probability of collision with replication forks (Strumberg et al., 2000). These collisions may result in DSB- containing Top1 suicide complexes that are difficult to repair. However, a preliminary analysis of G1synchronized yeast cells suggested arrest or blockage in G2/M rather than in S-phase. CPT-treated h2a-s129a cultures at 9 h post-release underwent mitotic catastrophe with abnormal cell and nuclear morphology, whereas wild-type cultures seemed normal (Fig. 2A). Fluorescence-activated cell sorting (FACS) analysis of these cultures also did not show any difference in G1-phase and S-phase progression, but did confirm that h2as129a cultures exposed to CPT were blocked at the first mitosis for at least 9 h, whereas wild-type cultures exposed to CPT resumed cycling (Fig. 2B). More detailed FACS analysis showed that all four G1synchronized cultures (wild-type and h2a-s129a; with or without CPT) progressed through S phase and entered G2 at indistinguishable rates (Fig. 2C). Again, differences were not observed until the cultures attempted to progess past mitosis. While CPT-treated wild-type cultures progressed through cell division after a 10–15 min delay compared with cultures that were not treated with CPT, the CPT-treated h2as129a cultures seemed to be blocked at mitosis (Fig. 2D).
Figure 2.
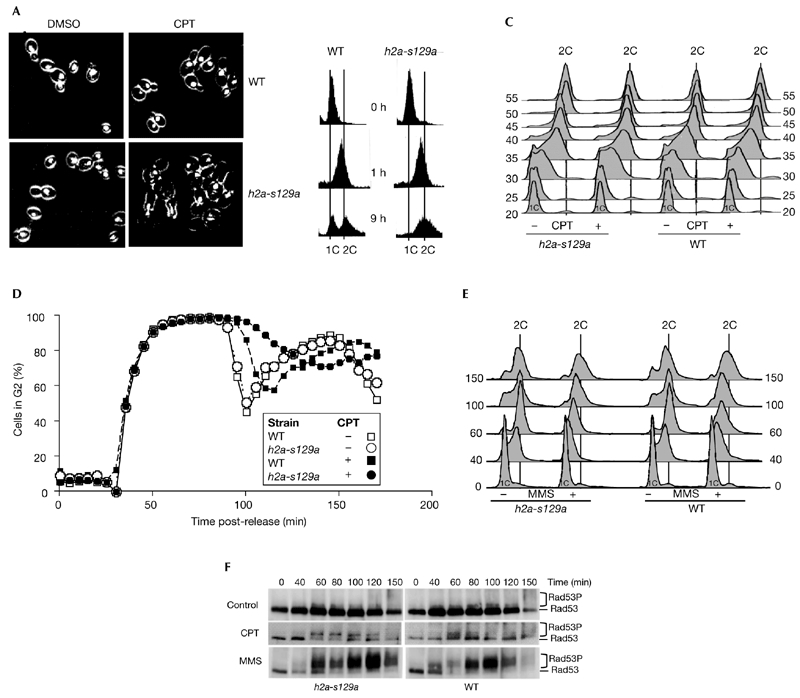
Cell-cycle analysis. Wild-type (WT) and h2a-s129a cells were synchronized in G1 phase with α-factor and then released in the presence of 20 μM camptothecin (CPT) or 0.03% methylmethanesulphonate (MMS), as indicated. Aliquots were taken at the indicated times for analysis by fluorescence microscopy (A), fluorescence-activated cell-sorting analysis (B–E) or immunoblotting with an anti-Rad53 polyclonal antibody (F). DMSO, dimethylsulphoxide; Rad53P, phosphorylated Rad53. 1C and 2C indicate haploid and diploid DNA content, respectively.
As CPT treatment did not induce any decrease in the rate of DNA replication in these cultures, the status of the intra-S-phase checkpoint was examined. The intra-S-phase checkpoint, which functions to slow the rate of DNA replication when DNA is damaged during S phase, is dependent on a number of genes, including RAD53 (Weinert, 1998). Chronic MMS treatment of yeast cultures is reported to inhibit the firing of late replication origins in a _RAD53_-dependent manner (Shirahige et al., 1998). When the effect of 0.03% MMS on the progression through S-phase of wild-type and h2as129a G1-synchronized cultures was examined, both cultures showed delays in S-phase, requiring about twice as long as control cultures to reach G2 (Fig. 2E). These results suggest that control and h2as129a cells have functional intra-S-phase checkpoints, but that these are not activated by CPT.
Rad53 hyperphosphorylation is necessary for the activation of cell-cycle checkpoints in response to DNA damage (Sanchez et al., 1996). When wild-type and h2as129a cultures were examined for the presence of hyperphosphorylated Rad53 species, those exposed to MMS strongly induced extensively phosphorylated Rad53 species, but those exposed to CPT contained smaller amounts of partially phosphorylated species (Fig. 2F). In addition, tenfold higher concentrations of CPT failed to induce Rad53 phosphorylation further (data not shown). The increased lethality in those cultures indicates that the lack of Rad53 activation by CPT is due to the accumulation of checkpoint-invisible CPT-induced DNA DSBs rather than rapid repair.
As CPT-induced DNA damage fails to activate the intra-S-phase checkpoint or delay DNA synthesis, the presence of DNA damage during S phase was examined by pulsed-field gel electrophoresis (PFGE; Desany et al., 1998; Fig. 3A). The gels show that when the yeast cultures begin replication 20 min after G1 release there is a decrease, as expected, in the amount of mature DNA molecules and an increase in the amount of immature DNA replication intermediates retained in the plug. However, as newly matured DNA molecules begin to reappear at 50 min, densitometry shows consistent differences among the cultures. h2as129a cells treated with CPT show substantially decreased rates and extents of reappearance of mature DNA, and these decreases are more pronounced with the longer DNA molecules (Fig. 3C). By contrast, the wild-type cultures and those without CPT show only slightly decreased reappearance of the large DNA molecules. Interestingly, the smaller DNA molecules reappear at the same time, irrespective of whether CPT or the γserine (Ser 129) was present (Fig. 3B). Furthermore, when the extents of replication of all the chromosomal DNA bands are compared at 80 min post-G1-release (Fig. 3D), it is apparent that there is an incremental decrease in the extent of faithful replication with increasing DNA length. Whereas this decrease is small in wild-type cells and those grown without CPT, this increment becomes critically large in h2as129a cells grown with CPT, resulting in a substantial deficit of large chromosomes in these cells. However, this deficit does not alter the progress of the CPT-containing h2a-s129a cultures through the cell cycle until mitosis, indicating that CPT-induced DNA damage and the presence of γ-H2A(X) do not activate the intra-S-phase checkpoint.
Figure 3.

Pulsed-field gel electrophoresis analysis of yeast chromosomes. (A) G1-synchronized cultures were released in 20 μM camptothecin (CPT). Aliquots of equal volume were taken at the indicated times, mixed immediately with equal volumes of melted agarose, and formed into plugs. The plugs were subjected to pulsed-field gel electrophoresis. The ethidium-bromide-stained gel was imaged, and the image subjected to densitometry. (B,C) Time courses of density changes of the smallest and largest chromosome bands (chromosome (Chr) I and Chr IV/XII) from the pulsed-field gel. The insets show the gel image. The horizontal lines indicate the initial density (1X) and complete population doubling (2X). (D) Relative extent of chromosome doubling. The density at 80 min post-release relative to the initial density was calculated for each chromosomal DNA band in the four cultures and was plotted against chromosome size. The horizontal lines denote the initial density (1X) and complete population doubling (2X). The symbols used on the graph are the same as in (B) and (C). The slopes of the wild-type cultures are −0.023 without CPT and −0.035 with CPT. The slopes of the mutant cultures are −0.037 without CPT and −0.67 with CPT. h2A, h2a-s129a; Kbp, kilobase pairs; WT, wild type.
To clarify the relationship of H2A(X) Ser 129 to other factors that function in checkpoint activation and DNA repair, h2a-s129a strains were crossed with strains defective in these pathways. Strains lacking both Mec1 and Tel1 were unable to form γ-H2A(X) (Fig. 4A), but the presence of either led to substantial γ-H2A(X) formation, indicating that the two kinases are partially redundant for this function. Thus, the mec1 tel1 double deletion is epistatic to H2A Ser 129 (Fig. 4B). Other in vitro evidence suggests that the PI(3)Ks interact directly with the H2A(X) γser (Rogakou et al. 1998; Downs et al., 2000). When crossed into rad52Δ or mre11Δ repair-deficient strains, h2as129a did not show greater sensitivity to CPT (Fig. 4C), a result consistent with H2A(X) Ser 129 being in the same pathways as Rad52 and Mre11. When crossed into rad24Δ or rad9Δ checkpoint-deficient strains, h2as129a substantially decreased survival, even in the rad24Δ rad9Δ double-mutant strain. This lack of epistasis indicates that the function of H2A(X) Ser 129 is independent of the RAD9 and RAD24 checkpoint pathways (Fig. 4D).
Figure 4.
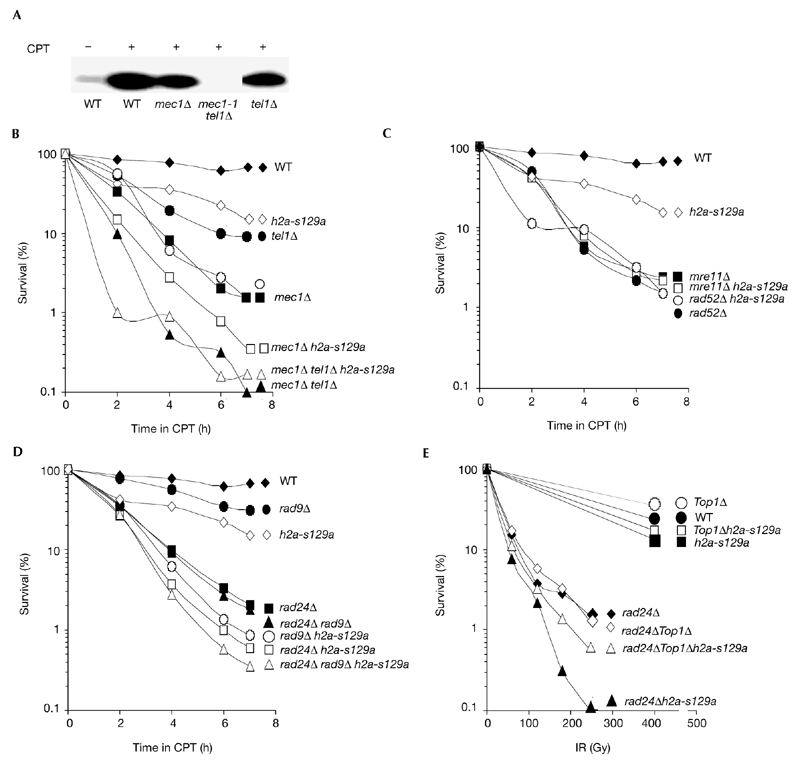
Interactions of h2a-s129a with other mutations. (A) γ-H2A(X) does not form in response to camptothecin (CPT) treatment in the mec1-1 tel1Δ strain. (B–D) Cultures of the indicated strains were synchronized in G1 and released in the presence of 20 μM CPT. At the times indicated, aliquots were removed, diluted and plated on YPD without CPT to determine viability. Strains are listed in the supplementary information online. (E) Cultures of the indicated strains were irradiated (IR) and examined for survival. H2A(X), histone 2A(X); WT, wild type.
The increased CPT-induced killing of h2a-s129a combined with rad24Δ and/or rad9Δ suggested that the same combination of mutations may also increase yeast sensitivity to γ-irradiation. h2a-s129a cells have only a low sensitivity to γ-irradiation; however, rad24Δ h2a-s129a cells are 15 times more sensitive to γ-irradiation than are rad24Δ cells (Fig. 4E), indicating that radiation-induced DNA damage that is unrepaired during S phase in h2as129a cells can be repaired while the Rad24 checkpoint is activated, whereas CPT-induced DNA damage cannot be completely repaired. Thus, in the absence of the Rad24 checkpoint function, H2A(X) Ser 129 has a more important function in yeast survival after exposure to γ-irradiation. It is possible that some of the radiation-induced DNA damage may also be Top1-mediated. When this was analysed by disrupting TOP1 in rad24Δ h2a-s129a cells, survival increased sixfold (Fig. 4E), suggesting that Top1-mediated DNA damage comprises a substantial fraction of the damage induced by agents other than CPT.
These findings show that CPT-induced Top1-mediated DNA damage fails to activate the intra-S-phase checkpoint, allowing the damage to accumulate until a mitotic catastrophe occurs. As CPT functions to stabilize Top1–DNA complexes, which then interfere with replication and other chromatin-related processes, these complexes may form to various extents during normal metabolism, and may be a prevalent and constant threat to cell survival. The removal of suicide complexes and DNA repair appears to be a complex process that occurs by several different pathways (Pourquier & Pommier, 2001).
Interestingly, treatment with either MMS or CPT induces H2A(X) γ-serine phosphorylation, and h2a-s129a cells are sensitive to both reagents, suggesting that γ-H2A(X) is necessary for the efficient repair of both types of lesions, even though MMS-induced lesions activate the intra-S-phase checkpoint, whereas CPT-induced lesions do not. As MMS is a more promiscuous reagent, γ-H2A(X) may participate in the repair of a subset of MMS-induced lesions, possibly those involving Top1. Other subsets of MMS-induced lesions may activate the intra-S-phase checkpoint. Thus, γ-H2A(X) may have a similar checkpoint- independent function in the repair of DNA lesions induced by many reagents. This model is supported by the finding that cell survival in CPT-treated cultures is much lower for the rad9Δ h2a-s129a double-mutant than for either mutation alone, and cell survival of irradiated cultures is much lower for the rad24Δ h2a-s129a double-mutant than for either mutation alone. Thus, H2A(X) γ-serine-related repair seems to be partially redundant with the Rad24 and Rad9 checkpoint pathways. It is possible that DNA damage that is usually repaired by a mechanism involving H2A(X) γ-serine can be partially repaired during _RAD9-RAD24_-related G2 arrest when the γ-serine is absent. Conversely, the fact that DNA damage is efficiently repaired by a mechanism involving H2A(X) γ-serine in rad9Δ or rad24Δ cells makes the lack of these G2 checkpoints less crucial. H2A(X) Ser 129 is essential for efficient checkpoint-invisible 'on-the-fly' repair of DNA damage during ongoing replication. As some of these lesions do not activate the intra-S-phase checkpoint, they must be repaired by a pathway that functions during ongoing DNA synthesis. These considerations indicate that H2A(X) Ser 129 has a central and important function in the S-phase repair of DNA lesions that do not activate the intra-S-phase checkpoint.
Methods
Yeast methods and strains.
All methods for the manipulation of yeast were performed according to standard methods (Burke et al., 2000). The strains used in this study are derived from a W303 background and are listed in the supplementary information online.
Sensitivity assays on plates.
Stock solutions of CPT (Sigma) were prepared at 6 mg ml−1 in dimethylsulphoxide (DMSO). Aliquots of the stock were spread on YPD plates, giving a final concentration of 20 μM CPT; control plates received the same volume of DMSO. Overnight cultures were diluted 1:10, grown for 3 h at 30 °C and diluted to 1 × 107 cells ml−1. Fivefold dilution series were spotted on the plates and grown at 30 °C for 2 days.
Cell viability assays in liquid cultures with camptothecin.
Overnight cultures were diluted 1:10, grown for 3h at 30 °C, diluted to 2 × 106 cells ml−1 in YPD containing DMSO or 20 μM CPT, and grown at 30 °C. At the times indicated, aliquots were diluted, sonicated briefly, spread onto plates and allowed to grow for 2 days at 30 °C, after which colonies were counted. For G1-release studies, cultures were arrested in G1 with 5 μg ml−1 α-factor (US Biological) for 2.25 h. Efficiency of arrest was monitored microscopically (>95% unbudded cells).
Cell-cycle and microscopy analyses.
Yeast samples were prepared for flow-cytometric analysis (using a Becton Dickinson FACScan machine) using propidium iodide (Sigma) as described in Heichman & Roberts (1996) or with SYBR Green I (Margarida et al., 2000). The propidium-iodidestained samples were also visualized using a PCM2000 laser-scanning confocal microscope (Nikon, Inc.).
Ionizing radiation.
Cells were collected by centrifugation, placed in ice and exposed to the indicated amount of ionizing radiation from a 137Cs source (Mark I irradiator; J.L. Shepherd and Associates) at a rate of 15.7 Gy min−1.
Protein extraction and immunoblotting.
Cell extracts were prepared with glass beads (0.45 g of 0.5-mm-diameter beads, and 0.4 ml of 0.2 M H2SO4 as extraction solution added to the cell pellets) in 1.5-ml tubes, which were placed in a mini-beadbeater-8™ cell disrupter (Biospec Products) for 5 min at its highest setting. After centrifugation, supernatants (340 μl each) were collected and trichloroacetic acid was added to a final concentration of 20% for 20 min on ice. The precipitate was pelleted, washed with cold ethanol and dried. Pellets were then dissolved in the appropriate loading buffer for either histone gels or SDS gels. Proteins were also analysed by high-resolution two-dimension AUT-AUC gel electrophoresis, as previously described (Rogakou et al. 1998). Procedures for Rad53 analysis have been described previously (de la Torre-Ruiz et al., 1998). For H2A(X) western blot analysis, proteins were separated using 13% SDS–polyacrylamide gels and blotted onto a polyvinylidene difluoride membrane. Membranes were incubated with the γ-H2A(X) antibody (Rogakou et al., 1999).
32P labelling.
Cells from exponential cultures were pelleted, washed once with distilled water, irradiated, and resuspended in 1 ml of solution containing 2% glucose and H3[32P]O4 (ICN Biomedicals, Inc.) with a total activity of 1 mCi. After incubation for 15 min at 30 °C, proteins were analysed as described above.
Pulsed-field gel electrophoresis analysis of yeast chromosomes.
Early log-phase cultures were synchronized in G1 with α-factor as described above. After synchronization, 20 μM CPT was added, and cultures were incubated for 30 min at 30 °C. Cultures were washed once with YPD, diluted to 7.5 × 106 cells ml−1 and released in 20 μM CPT. 5-ml aliquots were made at a concentration of 70% in ethanol and kept at 23 °C. Sample preparation for PFGE analysis of yeast chromosomal DNA was performed as described in Louis (1998).
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor871-s1.pdf).
Supplementary Material
Supplementary information
Acknowledgments
We thank K.W. Kohn and M. Lichten for reading the manuscript, and P. Pourquier and Y. Pommier for helpful discussions.
References
- Burke D., Dawson D. & Sterans T. (2000) Methods in Yeast Genetics. A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA. [Google Scholar]
- Celeste A. et al. (2002) Genomic instability in mice lacking histone H2AX. Science, 296, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desany B.A., Alcasabas A.A., Bachant J.B. & Elledge S.J. (1998) Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev., 12, 2956–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs J.A., Lowndes N.F. & Jackson S.P. (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature, 408, 1001–1004. [DOI] [PubMed] [Google Scholar]
- Heichman K.A. & Roberts J.M. (1996) The yeast CDC16 and CDC27 genes restrict DNA replication to once per cell cycle. Cell, 85, 39–48. [DOI] [PubMed] [Google Scholar]
- Jaxel C., Kohn K.W., Wani M.C., Wall M.E. & Pommier Y. (1989) Structure–activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: evidence for a specific receptor site and a relation to antitumor activity. Cancer Res., 49, 1465–1469. [PubMed] [Google Scholar]
- Louis E.J. (1998) in Methods in Microbiology (eds Brown, A.J.P. & Tuite, M.F.), 15–32. Academic, London, UK. [Google Scholar]
- Margarida M., Sousa M.J., Corte-Real M., Leao C., Salvador A. & Sansonetty F. (2000) in Current Protocols in Cytometry (eds Robinson, J.P., Darzyniewicz, Z., Dean, P.H., Dressler, L.G., Robinovitch, P.S., Stewart, C.C., Tanke, H.J. & Wheeless, L.), 11.13.1–11.13.9. John Wiley & Sons, Inc., Edison, New Jersey, USA. [Google Scholar]
- Paull T.T., Rogakou E.P., Yamazaki V., Kirchgessner C.U., Gellert M. & Bonner W.M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol., 10, 886–895. [DOI] [PubMed] [Google Scholar]
- Pourquier P. & Pommier Y. (2001) Topoisomerase I-mediated DNA damage. Adv. Cancer Res., 80, 189–216. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S. & Bonner W.M. (1998) DNA doublestranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem., 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Boon C., Redon C. & Bonner W.M. (1999) Megabase chromatin domains involved in DNA doublestrand breaks in vivo. J. Cell Biol., 146, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y., Desany B.A., Jones W.J., Liu Q., Wang B. & Elledge S.J. (1996) Regulation of RAD53 by the _ATM_-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science, 271, 357–360. [DOI] [PubMed] [Google Scholar]
- Shirahige K., Hori Y., Shiraishi K., Yamashita M., Takahashi K., Obuse C., Tsurimoto T. & Yoshikawa H. (1998) Regulation of DNA-replication origins during cell-cycle progression. Nature, 395, 618–621. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. (2001) ATM and ATR: networking cellular responses to DNA damage. Curr. Opin. Genet. Dev., 11, 71–77. [DOI] [PubMed] [Google Scholar]
- Strumberg D., Pilon A.A., Smith M., Hickey R., Malkas L. & Pommier Y. (2000) Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5'-phosphorylated DNA doublestrand breaks by replication runoff. Mol. Cell. Biol., 20, 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ruiz M.A., Green C.M. & Lowndes N.F. (1998) RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J., 17, 2687–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T. (1998) DNA damage checkpoints update: getting molecular. Curr. Opin. Genet. Dev., 8, 185–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information