Characterization of an ERAD Gene as VPS30/ATG6 Reveals Two Alternative and Functionally Distinct Protein Quality Control Pathways: One for Soluble Z Variant of Human α-1 Proteinase Inhibitor (A1PiZ) and Another for Aggregates of A1PiZ (original) (raw)
Abstract
The Z variant of human α-1 proteinase inhibitor (A1PiZ) is a substrate for endoplasmic reticulum-associated protein degradation (ERAD). To identify genes required for the degradation of this protein, _A_1PiZ _d_egradation-_d_eficient (add) yeast mutants were isolated. The defect in one of these mutants, add3, was complemented by VPS30/ATG6, a gene that encodes a component of two phosphatidylinositol 3-kinase (PtdIns 3-kinase) complexes: complex I is required for autophagy, whereas complex II is required for the carboxypeptidase Y (CPY)-to-vacuole pathway. We found that upon overexpression of A1PiZ, both PtdIns 3-kinase complexes were required for delivery of the excess A1PiZ to the vacuole. When the CPY-to-vacuole pathway was compromised, A1PiZ was secreted; however, disruption of autophagy led to an increase in aggregated A1PiZ rather than secretion. These results suggest that excess soluble A1PiZ transits the secretion pathway to the _trans_-Golgi network and is selectively targeted to the vacuole via the CPY-to-vacuole sorting pathway, but excess A1PiZ that forms aggregates in the endoplasmic reticulum is targeted to the vacuole via autophagy. These findings illustrate the complex nature of protein quality control in the secretion pathway and reveal multiple sites that recognize and sort both soluble and aggregated forms of aberrant or misfolded proteins.
INTRODUCTION
Cell function and survival depend on protein quality control to identify and remove aberrant proteins. Although two cellular sites of proteolysis are known, the lysosome/vacuole and cytoplasmic 26S proteasome, the recognition of aberrant proteins and mechanisms for delivery to these sites are still being defined. Endoplasmic reticulum-associated degradation (ERAD) is a protein quality control process in which aberrant or misassembled proteins in the secretory pathway are identified and removed (reviewed in Hampton, 2002; Tsai et al., 2002; Kostova and Wolf, 2003; McCracken and Brodsky, 2003). After entering the endoplasmic reticulum (ER), a nascent protein that fails to fold or assemble properly can be “recognized” by the ER quality control machinery, retained within the ER, and then retrotranslocated to the cytoplasm where it is degraded by the proteasome.
The recognition of ERAD substrates must exhibit flexibility to distinguish slowly folding proteins from aberrant proteins. Molecular chaperones play a critical role in this selection process; for example, the ER heat-shock protein (Hsp70), BiP, is vital for the selection of soluble substrates and is believed to hold the substrates in an unfolded state competent for retrotranslocation (Nishikawa et al., 2001; Kabani et al., 2003).
The complexity of the ERAD pathway has emerged with the identification of new ERAD substrates and the components required for their selection and degradation in yeast. For example, the analysis of topologically distinct ERAD substrates indicates that molecular chaperones in the cytoplasm and in the ER lumen distinguish membrane and soluble substrates, respectively (Huyer et al., 2004), and which chaperones target proteins for degradation is dependent on the site of the misfolded domain (Taxis et al., 2003; Vashist and Ng, 2004; reviewed in Ahner and Brodsky, 2004). Furthermore, the proposal that ERAD substrates are retained in the ER was challenged with the elucidation of the _sec_-dependent ERAD pathway, in which a soluble ERAD substrate, a misfolded form of carboxypeptidase Y (CPY*), was shown to transit between the ER and Golgi before retrotranslocation and degradation (Caldwell et al., 2001; Vashist et al., 2001; Taxis et al., 2002).
Some aberrant soluble proteins in the secretory pathway may escape the ER quality control machinery and are degraded instead in the vacuole (Hong et al., 1996; Holkeri and Makarow, 1998; Jorgensen et al., 1999; Coughlan et al., 2004; reviewed in Arvan et al., 2002). Furthermore, Spear and Ng (2003) reported that the ERAD pathway can be saturated by CPY* overexpression and that excess CPY* is degraded in the vacuole after transiting through the Golgi. In each of these cases, it has been surmised that an ill-defined quality control machinery resides within the Golgi. Overall, because of the potentially catastrophic results of aberrant protein accumulation in the secretory pathway, it is not surprising that the cell possess alternative trafficking schemes to remove unwanted proteins. However, the extent of these alternative schemes and how they are regulated is not clear.
Because the accumulation of misfolded proteins can lead to cellular dysfunction and death, ERAD has biomedical importance and has been linked to numerous disease states, including α1-antitrypsin deficiency (Lin et al., 2001; Perlmutter, 2002; McCracken and Brodsky, 2003). α1-Antitrypsin (or _a_lpha _1_-_p_rotease _i_nhibitor, A1Pi) is a protease inhibitor that suppresses neutrophil-derived proteases in the serum and elastase activity in lung tissue. The classical form of A1Pi deficiency results from the Z variant (A1PiZ) that contains a K342E substitution (Bathurst et al., 1984; Crystal, 1990). A1PiZ homozygous individuals can develop emphysema via a loss-of-function mechanism because the altered conformation of A1PiZ results in its recognition and degradation by ERAD (Wu et al., 1994; Werner et al., 1996; Teckman et al., 2001). A subset of homozygous individuals (12–15%) also experience liver disease (Sveger, 1988) via a gain-of-function mechanism because A1PiZ accumulates and aggregates within the ER of hepatocytes (Lomas et al., 1992, Lomas and Mahadeva, 2002; reviewed in Perlmutter, 2003). Elucidation of the genes required for efficient A1PiZ degradation will help answer whether genetic factors predispose this subset of individuals to liver disease and afford possible therapeutic measures.
Toward this goal, we previously published on the isolation of yeast mutants that exhibited slowed degradation of A1PiZ (McCracken et al., 1996; Palmer et al., 2003) and now report that one of these mutants compromises vacuolar protein targeting via the autophagic pathway. Furthermore, we report that A1PiZ degradation via the autophagic pathway is tied to A1PiZ aggregation and expression levels.
MATERIALS AND METHODS
Strains, Plasmids, Media, and Antisera
The Saccharomyces cerevisiae strains used in this study are listed in Table 1. Electrocompetent Escherichia coli strain HB101 [_F_– mcrB mrr hsdS20(r – B, – mB) recA13 supE44 ara14 galK2 lacY1 proA2 rpsL20(Smr) xyl5 λ– _leu mtl_] were purchased from Invitrogen (Carlsbad, CA). Complementation of A1PiZ degradation-deficient mutant, add3, used the genomic yeast library no. 77162 (American Type Culture Collection, Manassas, VA), constructed with genomic DNA partially digested by Sau3A and subcloned into a LEU2 containing CEN/ARS shuttle vector (Christianson et al., 1992). The cDNA sequence encoding the Z variant of α-1 protease inhibitor (A1PiZ) (McCracken and Kruse, 1993) was cloned either into vectors containing the inducible GAL1 promoter: pYES2.0 (2 μ, Ampr, URA3; Invitrogen) and pBM743 (CEN/ARS, Ampr, URA3; Invitrogen), or into a vector with the repressible MET25 promoter (Mumberg et al., 1994): p426MET25 (2 μ, Ampr, URA3; ATCC, Manassas, VA). Triple-hemagglutinin (HA)-tagged cystic fibrosis transmembrane conductance regulator (CFTR) expression in yeast was driven by the constitutive phosphoglycerate kinase promoter in a 2 μ plasmid (Zhang et al., 2002). The vector p425CyTerm (CEN/ARS, Ampr, LEU2) for constitutive protein expression (see below) was described previously (Palmer et al., 2003). pJC104 (2 μ, Ampr, URA3), encoding the lacZ gene behind four repeats of the unfolded protein response element (Cox et al., 1993) was generously contributed by Dr. Peter Walter (University of California, San Francisco, San Francisco, CA). Yeast cells were cultured in synthetic media containing 0.67% yeast nitrogen base with amino acids, 0.5% casamino acids, and 2% dextrose as a carbon source or with 2% galactose to induce expression of A1PiZ. Antibodies included rabbit anti-human A1Pi (Rockland, Gilbertsville, PA; and DakoCytomation California, Carpinteria, CA), horseradish peroxidase-conjugated goat anti-rabbit (U.S. Biochemical, Cleveland, OH), monoclonal mouse anti-HA (Roche Diagnostics, Indianapolis, IN), monoclonal mouse anti-yeast phosphoglycerate kinase (Invitrogen), rabbit anti-Kar2p (Brodsky and Schekman, 1993), and sheep anti-mouse IgG horseradish peroxidase-conjugated antibody (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom).
Table 1.
Yeast strains used in this study
| Strain | Description/Genotype | Source or reference |
|---|---|---|
| ADD3 | BC212/MATa ura3-52 leu2-3,112 his3Δ1 ade2-1 | McCracken and Kruse (1993) |
| add3 | IK13/MATa ura3-52 leu2-3,112 his3Δ1 ade2-1 add3 | McCracken et al. (1996) |
| WT | BY4742/MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 | Research Genetics; Winzeler et al. (1999) |
| _YPL123c_Δ | MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 YPL123c::KANMX | Research Genetics; Winzeler et al. (1999) |
| _YPL121c_Δ | MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 YPL121c::KANMX | Research Genetics; Winzeler et al. (1999) |
| _vps30_Δ | YPL120wΔ/MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 YPL120w::KANMX | Research Genetics; Winzeler et al. (1999) |
| _YPL119c_Δ | MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 YPL119c::KANMX | Research Genetics; Winzeler et al. (1999) |
| _pep4_Δ | YPL154cΔ/MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 YPL154c::KANMX | Research Genetics; Winzeler et al. (1999) |
| _vps38_Δ | YLR360wΔ/MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 YLR360w::KANMX | Research Genetics; Winzeler et al. (1999) |
| W303 _atg14_Δ | MATα ura3-1 his3-11 leu2-3,112 trp1Δ2 ade2-1 can1-100 YBR128c::KANMX4 | EUROSCARF; Winzeler et al. (1999) |
| _atg14_Δ | YBR128cΔ/MATα his3Δ1 leu2Δ0 lysΔ0 ura3Δ0 YBR128c::KANMX4 | This study |
| VPS10 | SEY6210/MATa leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 | Marcusson et al. (1994) |
| _vps10_Δ | EMY3/MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 YBL017c::HIS3 | Marcusson et al. (1994) |
| ATG7 | SEY6210/MATa leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 | Robinson et al. (1988) |
| _atg7_Δ | VDY101/MATa leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 apg7Δ::Leu2 | Kim et al. (1999) |
Molecular Methods and Transformation
Genomic DNA was isolated from the BY4742 wild-type parent strain as described by Hoffman and Winston (1987). The wild-type VPS30 sequence was amplified from genomic DNA by PCR using oligonucleotides (Invitrogen) specific for the VPS30 locus (YPL120w) and that contained a putative promoter sequence 260 base pairs upstream of the ATG with alterations to introduce unique restriction endonuclease cleavage sites. To design the myc-tagged VPS30 construct, the myc tag sequence (GAACAGAAACTGATTTCCGAAGAGGATCTGTGA) was added in-frame immediately before the VPS30 stop codon. The wild-type VPS30 gene and the myc-tagged VPS30 gene were inserted into the p425CyTerm vector. Correct insertions of each gene were determined by automated DNA sequence analysis following standard protocols using primers specific for the p425CyTerm vector. All primer sequences used are available upon request.
The BY4742 _atg14_Δ strain was created via genomic DNA isolation from the W303 _atg14_Δ strain (accession no. 20412B, EUROSCARF, Frankfurt, Germany) and then amplification of the kanamycin cassette via PCR using oligonucleotides (Invitrogen) specific to the ATG14 locus (YBR128c). The oligonucleotides included sequences 440 base pairs upstream of the ATG (5′-GCATTAAATTGGATCCAATTATCGATTG-3′) and 155 base pairs downstream from the stop codon (5′-CTTATCCTTTTCTGTCGACGGGGTG-3′). Wild-type BY4742 was transformed with the linear PCR product, and candidates deleted for ATG14 were isolated by selection on G418 (Geneticin)-containing medium.
Yeast transformations were performed using a standard lithium acetate procedure (Adams et al., 1997b), and transformants were isolated after growth in selective medium containing 2% dextrose. The Cell-Porator E. coli Pulser (Invitrogen) was used to electroporate competent E. coli. Plasmids were isolated from bacterial transformants using the Quantum Prep Plasmid Miniprep kit (Bio-Rad, Hercules, CA).
Authentication of Yeast Deletion Mutants
Genomic DNA was isolated from the BY4742 _atg14_Δ, _vps30_Δ, and _vps38_Δ strains as described above. A kanamycin cassette for each corresponding locus was amplified from genomic DNA by PCR using oligonucleotides specific for each locus (Invitrogen) as described in the deletion module PCR strategy of the Saccharomyces Genome Deletion Project (available at http://sequence-www.stanford.edu/group/yeast_deletion_project/deletions3.html; Wach et al., 1994). The sequences of the 45 up- and down-stream base pairs and of the kanamycin gene were verified by automated DNA sequencing following standard protocols.
ERAD Assays
The colony-blot immunoassay was a modification of a previously described procedure (McCracken et al., 1996). In brief, 3 μl of cells resuspended to a final optical density (OD) at 600 nm of 0.001 OD/μl was spotted onto a nitrocellulose disk overlaid either on medium containing 2% galactose to induce A1PiZ expression from the GAL1 promoter or onto synthetic medium lacking methionine to induce expression of A1PiZ from the MET25 promoter. After incubation at 30°C for 18 h, cells were lysed with either solution 1 (0.2 M NaOH, 0.1% SDS, and 0.05% 2-mercaptoethanol) or solution 2 (0.2 M NaOH. 1% SDS, 0.5% Triton X-100, 0.5% deoxycholic acid, and 0.05% 2-mercaptoethanol) for 1 h at room temperature, and blots were then assayed as described previously (McCracken et al., 1996). Immunoreactive proteins were detected by developing the blots with SuperSignal West Pico chemiluminescent substrate (Pierce Chemical, Rockford, IL), and Fuji Super RX x-ray film (Fuji, Tokyo, Japan). The density of immunoreactive protein at each colony spot was quantified using Molecular Analyst (Bio-Rad). Modifications to analyze secreted proteins were made by omitting the lysis procedure and placing the filter directly into the 10% nonfat dried milk, Tris-buffered saline blocking solution followed by washing and immunoassay as described above. The presence of phosphoglycerate kinase (PGK), a cytosolic marker protein, was monitored by immunoblotting after blots had been stripped of anti-A1Pi with 0.2 M NaOH. The PGK signal served as a secondary control to measure cell density and to control for cell lysis.
To perform cycloheximide chase analyses, overnight cultures were incubated in medium containing 2% galactose for 4 h at 30°C. Cells were then resuspended to a final OD600 of 2 OD/ml in medium containing 2% dextrose plus 200 μg/ml cycloheximide, incubated at 30°C with shaking, and 500-μl aliquots were collected, and the cells were harvested by centrifugation at the indicated time points. At each time point, the medium was reserved and represented the secreted material. The cell pellets were resuspended in 20 μl of lysis buffer (160 mM Tris, pH 6.8, 4% SDS, 0.2% bromphenol blue, 200 mM dithiothreitol [DTT], and 20% glycerol), incubated at 95°C for 2 min, 0.5-mm glass beads were added, the cells were disrupted via four sequential 60-s bursts on a Vortex mixer at the highest setting followed by cooling on ice for 60 s, and finally an additional 80-μl aliquot of lysis buffer and the reserved media were added.
Lysates or cell fractions were combined with sample buffer (0.125 M Tris, pH 6.8, 4% SDS, 0.004% bromphenol blue, 10% 2-mercaptoethanol, and 20% glycerol), heated to 95°C for 10 min, and resolved by SDS-PAGE, and A1PiZ, CFTR, BiP, or PGK was detected by immunoblot analysis. Results were quantified as described above, with all bands included in the quantification of A1PiZ.
Cell Fractionation
Yeast cultures were grown in medium containing 2% galactose for 24 h at 30°C. Spheroplasts were formed by incubating cells with 10 mg/ml lyticase in 1.2 M sorbitol, 0.1 M K2HPO4, pH 7.2, and were collected after centrifugation through a cushion of 0.8 M sucrose, 1.5% Ficoll, and 20 mM HEPES, pH 7.4, at 4000 × g for 10 min at 4°C. After the spheroplasts were washed with 1.2 M sorbitol, 0.1 M K2HPO4, pH 7.2, they were resuspended in ice-cold spheroplast lysis buffer (0.1 M sorbitol, 50 mM KOAc, 2 mM EDTA, 20 mM HEPES 7.4, 1 mM DTT, and 1 mM phenylmethylsulfonyl fluoride) and lysed by 10 strokes with a Dounce homogenizer on ice. Unbroken cells were removed by centrifugation at 500 × g for 5 min at 4°C. Resulting cell lysates were layered over a cushion of 1 M sucrose, 50 mM potassium acetate, 20 mM HEPES, pH 7.4, and 1 mM DTT, and subjected to 6500 × g centrifugation for 10 min at 4°C. The top fraction, above the interface, was collected and subjected to further centrifugation at 22,000 × g for 10 min at 4°C. The resulting pellet was the microsome fraction, and the supernatant/cytosol was further cleared of small vesicles by centrifugation at 100,000 × g for 1 h at 4°C. Protease protection assays were performed on the microsome fraction by exposing washed microsomes to either 0.4 mg/ml trypsin in buffer 88 (20 mM HEPES, pH 6.8, 150 mM KOAc, 250 mM sorbitol, and 5 mM MgOAc) or to 0.4 mg/ml trypsin and 2% Triton X-100 in buffer 88 for 30 min on ice. Specific proteins were then visualized by immunoblot analysis.
Sucrose Density Gradient Analysis
Microsomes were isolated as described above from yeast producing A1PiZ for 48 h after induction. The microsomes were lysed for2honiceinbuffer88 that had been supplemented with 0.5% Triton X-100 and complete protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The total protein content of the lysates was determined using Coomassie Plus protein assay (Pierce Chemical). An aliquot of 1 mg of total protein for lysates from each yeast strain was loaded on to a 5–60% sucrose gradient (Kabani et al., 2003; Schmidt and Perlmutter, 2005), centrifuged in a Beckman SW50 at 145,000 × g for 20 h at 4°C, and 250-μl fractions were collected from the top of the gradient and proteins were concentrated by precipitation in 10% trichloroacetic acid. Specific proteins were visualized by immunoblot analysis.
Assays for Induction of the Unfolded Protein Response (UPR)
UPR induction was monitored by measuring β-galactosidase activity in cell extracts of pJC104 transformed strains. Cells were grown in selective medium to mid-log phase (OD600 = ∼1) at 30°C and then incubated at 30°C for 1.5 h with or without the addition of tunicamycin to a final concentration of 5 μg/ml. Cell extracts were prepared by agitation of washed cells with glass beads, and β-galactosidase activity was measured using published protocols (Adams et al., 1997a).
RESULTS
Identification of an add Gene as VPS30
Using a classical genetics strategy, wild-type S. cerevisiae expressing human A1PiZ were mutagenized with ethyl methyl sulfonate and screened for colonies that accumulated elevated levels of A1PiZ, with the assumption being that the acquired mutation compromised A1PiZ degradation (McCracken et al., 1996). To identify the locus of the mutated gene from one of these add mutants, add3, the strain was transformed with a yeast genomic library and the resulting colonies were screened using a colony-blot immunoassay that allows quantification of A1PiZ. One complemented colony in which degradation had been restored is shown in Figure 1A. Complementation was verified by isolation of the library plasmid from the complemented clone, retransformation of the plasmid into the add3 mutant, and then rescreening for the add phenotype. The genomic insert of the _add3_-complementing library plasmid was sequenced and the region mapped to chromosome XVI, 317459–328062, which contains five open reading frames, four of which (YPL123c, YPL121c, YPL120w, and YPL119c) were available for analysis using haploid deletion mutants (Winzeler et al., 1999).
Figure 1.

VPS30 complements the A1PiZ degradation-deficient phenotype of the add3 ERAD mutant. (A) Colony-blot immunoassays are shown with vertically placed duplicate samples of ERAD mutant (add3) expressing A1PiZ from pYESA1PiZ high level expression vector, and either lacking (–) or containing (+) a complementing fragment from a yeast genomic library. Controls of the ADD3 parent expressing either A1PiZ (Z) or containing a vector control (O) are also presented. (B) The add3 and _vps30_Δ cells were transformed with the empty vector (–), VPS30 gene (+), or a myc-tagged version of VPS30 (+myc), and colony-blot immunoassays were performed. An immunoblot of the control BY4742 parent expressing pYESA1PiZ (Z), high level expression vector, is also included.
Each deletion mutant and the isogenic wild-type strain (BY4742) were transformed with a 2 μ plasmid containing A1PiZ under the control of a galactose-inducible promoter, and a colony-blot immunoassay was again performed. The strain containing a deletion of YPL123c displayed a growth defect, cells lacking YPL121c and YPL119c displayed a wild-type phenotype, but yeast deleted for YPL120w conferred the add phenotype (our unpublished data). YPL120w encodes Vps30p/Atg6p, a protein known to be involved in both vesicle-mediated protein traffic to the vacuole and autophagy (Tsukada and Ohsumi, 1993; Seaman et al., 1997; Kihara et al., 2001). To confirm that the add phenotype arose from deletion of VPS30/ATG6, plasmids lacking or containing the wild-type VPS30 gene were introduced into the add3 mutant and into a _vps30_Δ strain. VPS30 and a myc-epitope-tagged version of VPS30 complemented the add phenotype of both mutant strains (Figure 1B).
To establish whether the elevated level of A1PiZ seen in the _vps30_Δ strain was because of an A1PiZ degradation defect, a cycloheximide chase assay was performed to examine A1PiZ stability. A1PiZ degradation was significantly reduced in the _vps30_Δ strain compared with the isogenic wild-type strain (Figure 2). These results demonstrate that Vps30p facilitates A1PiZ degradation.
Figure 2.

VPS30 facilitates A1PiZ degradation. Representative blots from a cycloheximide-chase analysis of A1PiZ degradation and a graph of the means ± SE, from at least three experiments, where the BY4742 WT (dotted line and circles), _vps30_Δ (dashed line and crosses), _vps38_Δ (solid line and squares), and _atg14_Δ (dashed line and triangles) strains expressing A1PiZ off the pYESA1PiZ high level expression vector are shown. The multiple bands seen were the expected “ladder” for the ER form of A1PiZ, a glycoprotein that contains three N-linked carbohydrate chain acceptor sites (Moir and Dumais, 1987; McCracken and Kruse, 1993), and all bands were used in quantification.
The UPR Is Constitutively Induced in the vps30Δ Strain
Perturbation of the ERAD machinery can lead to induction of the UPR (Casagrande et al., 2000; Friedlander et al., 2000; Ng et al., 2000; Travers et al., 2000), a cellular response that alleviates the potentially toxic effects of misfolded protein accumulation in the ER (reviewed in Fewell et al., 2001; Rutkowski and Kaufman, 2004). Thus, to further characterize the effect of deleting VPS30 on ER physiology, the UPR was examined in the _vps30_Δ strain.
The _vps30_Δ and the wild-type parent strains were transformed with a reporter plasmid that contains four UPR transcriptional control elements driving β-galactosidase gene expression (Cox et al., 1993). This allows one to measure β-galactosidase levels as a representation of the UPR. Cells were incubated in the absence or presence of tunicamycin to generate unfolded proteins in the ER, and extracts were prepared and assayed for β-galactosidase activity. In the presence of tunicamycin, the _vps30_Δ strain and wild-type strains exhibited a similar UPR induction, but the _vps30_Δ strain exhibited a 1.8-fold induction of UPR compared with wild-type cells in the absence of tunicamycin (Figure 3). This finding suggested that deletion of VPS30 affects ER homeostasis, perhaps by blocking transport of unfolded proteins from the ER to the vacuole.
Figure 3.

The UPR is constitutively induced in cells lacking VPS30. The yeast were transformed with the 2 μ pJC104 vector encoding the lacZ gene behind four repeats of the unfolded protein response element in order to measure the UPR. The relative induction of the UPR was assayed, as described in Materials and Methods, in the presence (+) or absence (–) of 5 μg/ml tunicamycin; BY4742 WT parent (white), _vps30_Δ strain (black), _vps38_Δ (light gray), and _atg14_Δ (dark gray). Data represent the means from at least three independent experiments ± SE.
Vps30p Is Required to Deliver A1PiZ via an Alternative Degradation Pathway to the Vacuole
Based on the role of Vps30p/Atg6p in protein trafficking to the vacuole (see above), we reasoned that A1PiZ overexpression may require targeting of excess A1PiZ for vacuole-mediated proteolysis. This hypothesis is supported by the fact that the ERAD pathway is saturable upon overexpression of another soluble ERAD substrate, CPY* and that in response to ER stress and UPR induction, an overflow pathway for vacuole degradation is initiated (Spear and Ng, 2003). To determine whether excess A1PiZ is trafficked to the vacuole when it is expressed at high levels, we examined the fate of A1PiZ after being produced from high (pYESA1PiZ), medium (pBMA1PiZ), and low (p426Met25A1PiZ) level expression vectors. As displayed in Figure 4A, we found that _VPS30_-dependent accumulation of A1PiZ correlated with increasing levels of expression. No difference in the relative intensity of immunoreactive A1PiZ was seen between wild-type and _vps30_Δ cells expressing low levels of A1PiZ from p426Met25A1PiZ. However, as expression increased the difference between the relative amount of immunoreactive A1PiZ for _vps30_Δ and wild-type cells escalated such that the signal measured for _vps30_Δ cells is 127% of that seen in the wild-type strain with moderate expression. This difference increased further to 175% with high level expression from pYESA1PiZ. These data indicate that lower concentrations of A1PiZ are efficiently targeted to ERAD in a Vps30p-independent manner; however, as the levels of A1PiZ increased so did Vps30p-dependent degradation.
Figure 4.

Vps30p is required for vacuolar degradation of A1PiZ. (A) A colony-blot immunoassay with duplicate samples of _vps30_Δ and the BY4742 WT parent strain expressing high (pYESA1PiZ), medium (pBMA1PiZ), or low (p426Met25A1PiZ) levels of A1PiZ was performed. Each immunoassay was established with equal OD600 units of cells, allowing for the assignment of relative levels of A1PiZ quantified as described in Materials and Methods. The relative amount of A1PiZ in _vps30_Δ compared with the wild type (WT) was determined for each vector type. (B) Representative immunoblot from a cycloheximide-chase analysis and a graph of the means ± SE from at least three experiments monitoring A1PiZ degradation are shown for the _pep4_Δ (solid line and squares) and BY4742 WT (dotted line and circles) strains transformed with the pYESA1PiZ, high level expression vector.
Having demonstrated that Vps30p plays a role in the degradation of A1PiZ when A1PiZ is overexpressed, we examined directly whether vacuole-mediated proteolysis was required for A1PiZ degradation by colony blot immunoassay and cycloheximide chase analysis in a _pep4_Δ mutant that lacks most vacuolar protease activity (Jones et al., 1982). We found that overexpressed A1PiZ was stabilized in the _pep4_Δ strain (Figure 5A) and that degradation was significantly inhibited (Figure 4B). Together, these data demonstrated that wild-type yeast target excess A1PiZ to the vacuole and that the _vps30_Δ strain was defective for vacuole targeting of A1PiZ. This establishes a role for Vps30p in vacuole-dependent degradation of A1PiZ when this ERAD substrate is overexpressed.
Figure 5.

Yeast deleted for VPS30, VPS38, and VPS10, but not PEP4 or ATG14, secrete A1PiZ. (A) Colony-blot immunoassay was performed on the BY4742 parent containing a vector control (WT/O) or expressing A1PiZ (WT/Z) and on the _pep4_Δ, _vps30_Δ, _vps38_Δ, _atg14_Δ, VPS10 parent, and _vps10_Δ strains expressing A1PiZ from the pYESA1PiZ high level expression vector. (B) Secreted A1PiZ was measured by a modified immunoassay analysis of the BY4742 parent containing a vector control (WT/O) or expressing A1PiZ (WT/Z), and the _pep4_Δ, _vps30_Δ, _vps38_Δ, _atg14_Δ, VPS10 parent, and _vps10_Δ strains expressing A1PiZ from the pYESA1PiZ high level expression vector. For both A and B, A1PiZ was visualized in duplicate using antisera against A1Pi (upper two rows), and cell density and lysis were monitored by assaying the cytosolic marker protein PGK (lower two rows). Note that A1PiZ was not detected for the vector control (WT/O) and that PGK was absent in the secretion assay, indicating that cell lysis did not lead to the A1PiZ secretion.
Phosphatidylinositol 3-kinase Complexes I and II Function in A1PiZ Degradation
Vps30p is a component of two distinct phosphatidylinositol 3-kinase (PtdIns 3-kinase) complexes that regulate membrane traffic. Both complexes also contain Vps15p, a membrane associated serine/threonine kinase, and Vps34p, a PtdIns 3-kinase (Kihara et al., 2001; Burda et al., 2002). The two complexes differ in one component: complex I contains Atg14p and this complex is required for macroautophagy and the cytoplasm-to-vacuole targeting pathway (Wurmser and Emr, 2002), whereas complex II contains Vps38p, and this complex is required for retrograde trafficking from the endosome to the Golgi and thus for wild-type CPY-to-vacuole targeting (Burda et al., 2002).
Because complex I and II differ by a single subunit, deletion mutants of ATG14 and VPS38 were used to determine which of these complexes used the A1PiZ overflow pathway. Using both cycloheximide chase analysis and colony blot immunoassay we found that A1PiZ was stabilized in both the _atg14_Δ and the _vps38_Δ strains when expressed at high levels (Figures 2 and 5A, respectively). The requirement for the autophagic pathway was also seen when a second autophagy-specific mutant strain (_atg7_Δ; Kim et al., 1999) was examined by colony-blot immunoassay (our unpublished data).
A1PiZ Is Secreted in Strains Deleted for VPS30, VPS38, and VPS10
The data presented above indicate a requirement for both VPS30 and VPS38 during A1PiZ degradation, and Vps30p and Vps38p are necessary for the recycling of Vps10p, a sorting receptor that plays a role in trafficking CPY (Marcusson et al., 1994; Seaman et al., 1997, 1998; Burda et al., 2002), proteinase A, aminopeptidase Y (Jorgensen et al., 1999), and some misfolded proteins from the Golgi to the vacuole (Hong et al., 1996; Holkeri and Makarow, 1998). We therefore asked whether VPS10 was also required for A1PiZ degradation. The vps10 deletion mutant and the isogenic wild-type strain were transformed with the pYESA1PiZ high-expression vector, and the levels of A1PiZ were examined by colony-blot immunoassay. We observed strong A1PiZ stabilization in the _vps10_Δ mutant (Figure 5A), suggesting Vps10p-dependent trafficking of A1PiZ to the vacuole.
Previous findings demonstrated that when delivery of CPY to the vacuole was blocked by deletion of VPS10, it was secreted (Seaman et al., 1997). Thus, we next asked whether the deletion mutants secreted A1PiZ using a colony-blot immunoassay modified to measure A1PiZ secretion. To ensure our modified immunoassay procedure had not caused cell lysis, the presence of the cytosolic marker PGK was monitored. No PGK was detected; however, we noted significant extracellular A1PiZ from the _vps30_Δ, _vps38_Δ, and _vps10_Δ cells, but not from the wild type, _pep4_Δ, or _atg14_Δ cells (Figure 5B). These results indicated that excess A1PiZ destined for the vacuole can be secreted from the cell when the CPY-to-vacuole pathway is compromised, but A1PiZ that is trafficked to the vacuole via autophagy remains in the cell when this pathway is disrupted.
Atg14p and Vps30p Are Not Required for CFTR Degradation
We next asked whether the add phenotype observed for the _atg14_Δ strain was an indirect effect resulting from perturbed degradation of all ERAD substrates. To this end, we examined the degradation of the cystic fibrosis transmembrane conductance regulator CFTR. CFTR has been extensively studied in yeast and is a well established ERAD substrate. The degradation of CFTR in yeast requires the proteasome (Zhang et al., 2001), E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases (Ubc6, Ubc7, and Der3/Hrd1; Kiser et al., 2001) and the Sar1p/COPII machinery (Fu and Sztul, 2003). Degradation is further catalyzed by the Hsp40 cochaperones Ydj1p and Hlj1p (Youker et al., 2004) and by Hsp70 (Zhang et al., 2001) and slowed proteolysis was observed in several add mutant yeast strains (Palmer et al., 2003). Wild-type, _atg14_Δ, and _vps30_Δ strains were transformed with a CFTR expression vector and degradation was assessed by cycloheximide chase analysis. We found that the mutants displayed the same kinetics of CFTR degradation seen in wild-type yeast (Figure 6), demonstrating that perturbing autophagy does not globally affect ERAD.
Figure 6.

Neither Vps30p nor Atg14p are required for CFTR degradation. Representative blots from a cycloheximide-chase analysis of CFTR degradation and a graph of the means ± SE from at least three experiments are shown. The BY4742 WT (dotted line and circles), _vps30_Δ (solid line and squares), and _atg14_Δ (dashed line and triangles) strains were transformed with the CFTR expression vector, and ERAD was assayed as described in Materials and Methods.
Intracellular A1PiZ Resides in a Protease-protected Membrane Fraction
The A1PiZ degraded by autophagy might have been delivered to the vacuole either directly from ER-derived autophagic vesicles or after its retrotranslocation from the ER and subsequent sequestration into autophagosomes (Garcia-Mata et al., 2002). Consistent with this second scenario, it has been shown in mammalian cell systems that the ERAD substrate CFTR is retrotranslocated to the cytosol and efficiently degraded by the proteasome; however, when overexpressed or when cells are incubated in the presence of proteasome inhibitors a buildup of cytosolic CFTR aggresomes was seen (Johnston et al., 1998; Kopito, 2000). Given the high expression level of A1PiZ from the pYESA1PiZ vector, we asked whether A1PiZ was present in the cytosol or sequestered exclusively within a membrane fraction. High levels of A1PiZ were expressed in wild-type and _atg14_Δ cells for 24 h, after which the cells were collected and microsome and cytosol fractions were isolated. The microsome fractions from both cell lines were then treated with trypsin to determine whether the A1PiZ was protected in the lumen of the microsomes, or exposed on the exterior of the microsomes. Trypsin activity was verified by treating microsomes with trypsin in the presence of Triton X-100, to allow trypsin access to both the interior and exterior pools of A1PiZ. All fractions were examined by immunoblot analysis for cytosolic (PGK) and ER (BiP) marker proteins and for A1PiZ. We found that A1PiZ, like the ER luminal chaperone, BiP, resided exclusively within the microsome fraction prepared from both wild-type cells and from the _atg14_Δ mutant (Figure 7). These data suggested that aggregated A1PiZ forms within the ER and is removed by autophagy in the absence of retrotranslocation.
Figure 7.
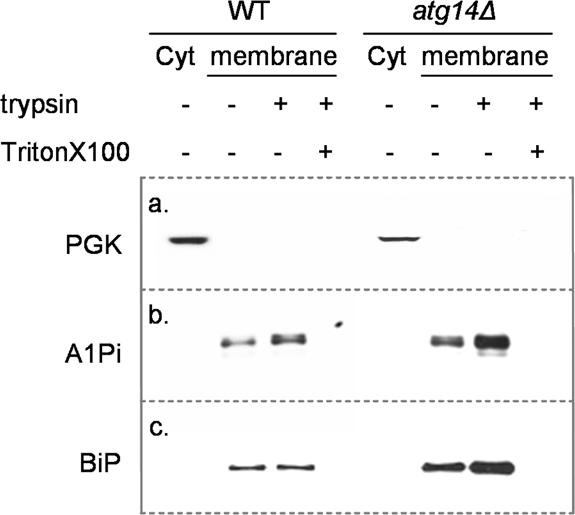
A1PiZ resides only in membrane protected fractions. Cytosol (Cyt) and microsome fractions (membrane) were prepared from BY4742 (WT) and _atg14_Δ strains expressing A1PiZ from the pYESA1PiZ high level expression vector, as described in Materials and Methods. The microsome fractions were left untreated (–) or were treated with trypsin (+) or with trypsin and Triton X-100 (+). Fractions were examined by immunoblot analysis using antisera to either the cytosolic marker protein PGK (a), A1Pi (b), or the ER luminal chaperone, BiP (c).
Deletion of ATG14 Leads to Constitutive UPR
If the A1PiZ that accumulates within the ER is aggregation-prone in yeast, as observed in mammals (Lomas et al., 1992; Dafforn et al., 1999), it is likely that these aggregates could neither be retrotranslocated to the cytosol nor packaged into ER-to-Golgi vesicles. Consistent with the data presented in Figure 7, these aggregates might be delivered directly to the vacuole via autophagy. If this hypothesis is correct, one would predict that disrupting autophagy may lead to an accumulation of proteins in the ER. To begin to test this possibility, we monitored the UPR in the _atg14_Δ strain. We discovered that the UPR was constitutively induced three-fold in the _atg14_Δ yeast compared with the isogenic wild-type strain (Figure 3). Induction of the unfolded protein response in the _atg14_Δ strain was confirmed because approximately threefold more BiP was present in microsomes isolated from the _atg14_Δ strain compared with the wild-type strain (Figure 7). In contrast, the absence of VPS38 did not lead to constitutive UPR induction, suggesting that disruption of the CPY-to-vacuole pathway does not lead to ER stress. This result is in agreement with the finding that A1PiZ is secreted by the _vps38_Δ strain (Figure 5B).
Cells with ATG14 Deleted Accumulate A1PiZ Aggregates
If aggregates are removed from the ER via autophagy, defects in the autophagic pathway might lead to increased levels of aggregated A1PiZ. Thus, we monitored A1PiZ aggregation using sucrose density gradients (see Materials and Methods). We first noted that soluble A1PiZ was spread across several fractions of the gradient (Figure 8), consistent with data obtained by Schmidt and Perlmutter (2005) when A1PiZ was examined in extracts prepared from mammalian cells. The investigators also found that a significant amount of A1PiZ also resided in the bottom (most dense) fraction, and this material was shown to represent A1PiZ aggregates (Schmidt and Perlmutter, 2005). Similarly, we found A1PiZ resided in the bottom fraction of the sucrose density gradient and further, that the amount of aggregated A1PiZ in the bottom fraction isolated from the _atg14_Δ strain was greater than that isolated from the wild-type strain (Figure 8). Together with the findings presented above, these data demonstrate that there is an increase in ER-resident A1PiZ aggregates when autophagy is blocked.
Figure 8.

Aggregates of A1PiZ accumulate in the ER of _atg14_Δ mutant yeast. Microsomes prepared from the BY4742 WT (dotted line and circles) and _atg14_Δ (dashed line and triangles) strains expressing A1PiZ from the pYESA1PiZ high level expression vector were disrupted, and lysates were subjected to 5–60% sucrose density gradients as described in Materials and Methods. A representative blot and a graph of the percentage of A1PiZ found in fractions that were collected from the top (5% sucrose/fraction #1) to the bottom (60% sucrose/fraction #20) are shown.
DISCUSSION
In this report, we identify an ADD gene as VPS30/ATG6, a gene that was uncovered earlier by two independent screens for mutants defective in vacuolar protein sorting (Robinson et al., 1988; Seaman et al., 1997) and in autophagy (Tsukada and Ohsumi, 1993; Kametaka et al., 1998). This result was unexpected given that A1PiZ is an ERAD substrate in both yeast (McCracken and Kruse, 1993; Werner et al., 1996; Brodsky et al., 1999) and human (Qu et al., 1996; Teckman et al., 2000). Because vacuole morphology is largely unperturbed in vps30 yeast (Raymond et al., 1992), these data suggest that the A1PiZ degradation defect does not arise from gross ultrastructural perturbations. Instead, we hypothesized that the ERAD pathway might be overwhelmed when A1PiZ was overexpressed, conditions under which the add3 gene was first isolated (McCracken et al., 1996). We therefore examined whether the Vps30p requirement for A1PiZ degradation was only evident when this ERAD substrate was overexpressed, and the data presented in Figure 4 indicate this to be the case. Thus, we propose that overexpressed A1PiZ, and the inherent ER stress, are sufficient to target A1PiZ to alternate quality control pathway(s).
Interestingly, because Vps30p participates in two distinct PtdIns 3-kinase complexes, we were able to establish that A1PiZ can be targeted to the vacuole via two routes: the CPY-to-vacuole pathway (Horazdovsky et al., 1995; reviewed in Conibear and Stevens, 1998) and the autophagic pathway (reviewed in Klionsky and Emr, 2000; Levine and Klionsky, 2004; Figure 9). These results raise specific questions about the role of each pathway during secretory protein quality control.
Figure 9.
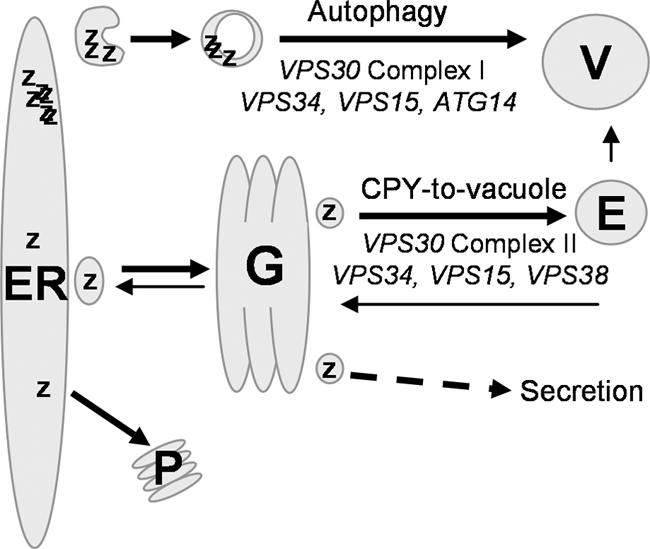
Proposed model for A1PiZ quality control. A1PiZ is targeted to ERAD and thus exits the ER by retrotranslocation with subsequent degradation by the proteasome (P). When overexpressed, excess soluble A1PiZ (Z) exits the ER by vesicle transport, transits the Golgi (G), and is sorted into the CPY-to-vacuole pathway via the endosome (E) to the vacuole (V). If the CPY-to-vacuole pathway is blocked, soluble A1PiZ is secreted. Excess A1PiZ that aggregates (ZZZZ) within the ER is sent to the vacuole via autophagy.
Complex II acts to recruit and stimulate the Vps38p-Vps15p-Vps34p phosphatidylinositol 3-kinase. This creates a specific region of phosphatidylinositol 3-phosphates on the endosome membrane that initiates retromer formation and consequent vesicle trafficking from the endosome to the Golgi. Emr and colleagues found that VPS30 and VPS38 mutant strains missort CPY and the CPY cargo receptor Vps10p when complex II function is abrogated (Seaman et al., 1997; Burda et al., 2002). The resulting absence of _trans_-Golgi network (TGN)-resident Vps10p leads to CPY secretion. Our finding that A1PiZ is secreted when CPY-to-vacuole targeting is blocked by deletion of VPS30, VPS38, or VPS10 (Figure 5B) is consistent with their data and indicates that upon arrival at the TGN, A1PiZ is selected for transport to the vacuole. The existence of a post-ER protein quality control mechanism that is substrate specific is supported further by several reports (Hong et al., 1996; Holkeri and Makarow, 1998; Coughlan et al., 2004). The secretion of A1PiZ in the _vps10_Δ strain suggests that Vps10p may be the cargo receptor that binds and transports A1PiZ from the TGN to the vacuole. Alternatively, because Vps10p is thought to be essential for retromer coat formation (Seaman et al., 1997), A1PiZ secretion by _vps10_Δ cells may simply be due to disruption of CPY-to-vacuole transport.
The site of action for complex I has not been specifically defined, but Nice et al. (2002) proposed that complex I plays a role similar to that seen for complex II: stimulation of phosphatidylinositol 3-kinase activity within a specific region of the presumptive autophagosome membrane, which is required to initiate vesicle formation. The source of autophagosomal membrane has yet to be determined; however, a dissection of the role of complex I and its site of action may help to resolve this question. Should autophagosomes form directly from a specified region of the ER as a means of clearing unwanted aggregates from the ER, then the lack of ATG14 would compromise ER homeostasis and result in constitutive UPR induction, which is in agreement with our data (Figure 3). Moreover, differing levels of UPR induction when comparing the _vps30_Δ and the _atg14_Δ strains (Figure 3) suggest that autophagy may proceed in the _vps30_Δ strain, albeit inefficiently, but it is completely blocked in the _atg14_Δ strain. This premise is supported by the fact that ATG14 overexpression suppresses the autophagic defect associated with a VPS30 mutant, atg6-1 (Kametaka et al., 1998). If autophagy continues in the absence of Vps30p, then it is possible that Vps30p facilitates recruitment of complex I to the site of autophagosome formation but is not essential for complex I phosphatidylinositol 3-kinase activity.
One possible model for the stabilization of A1PiZ in the _atg14_Δ strain is that A1PiZ is not targeted to the vacuole via autophagy but rather that blocking autophagy severely perturbs the homeostasis of the ER, impairing ERAD and the cell's ability to degrade A1PiZ. In turn, this might lead to aggregate accumulation. However, we disfavor this model for several reasons. First, the analysis of the degradation of a known ERAD substrate, CFTR, showed that CFTR is degraded normally in the _atg14_Δ strain (Figure 6); this provides evidence that ERAD is functioning despite perturbed ER homeostasis and UPR induction. Second, BiP is upregulated in the _atg14_Δ strain approximately threefold (Figure 7). Thus, the resulting increase in molecular chaperones is not likely to promote A1PiZ aggregation but rather should slow the aggregation of A1PiZ, as observed for other substrates (Kabani et al., 2003). In addition, the analyses of aggregate formation in microsomes showed that A1PiZ aggregates accumulate in the ATG14 delete strain (Figure 8). Together, our data support the model, summarized in Figure 9, that A1PiZ aggregates are removed from the ER via a mechanism dependent upon autophagy.
Finally, the results of our study are applicable to the mechanism of A1PiZ-associated liver disease. As noted in the Introduction, within the population of A1PiZ homozygotes is a subpopulation (15–20%) that develops liver disease due to A1PiZ aggregate accumulation in hepatocytes (reviewed in Teckman et al., 1996; Perlmutter, 2003). The autophagic pathway has been implicated in the removal of ER-aggregated A1PiZ. Specifically, there is an increase in the number of autophagosomes in human liver cells and fibroblasts engineered to express A1PiZ, and the A1PiZ degradation rate is slowed in the presence of chemical inhibitors of autophagy (Teckman and Perlmutter, 2000). Our results are not only in accordance with these findings but provide more direct evidence that A1PiZ aggregates are cleared by autophagy (Figures 7 and 8). Therefore, we conclude that the yeast system is an appropriate model to study the mechanisms of liver damage caused by A1PiZ accumulation. Nevertheless, an unresolved question is whether A1PiZ homozygotes with liver disease carry a second mutation or polymorphism that prevents efficient removal of A1PiZ from the ER. Based on our and other's work, such genetic modifiers of A1PiZ-associated liver disease might include either genes involved in ERAD or in autophagy. The answer to this important question remains to be delineated.
Acknowledgments
We thank National Science Foundation Research Experience for Undergraduates summer students Jillian Collins, Sarah Henderson-Hall, Brandan Crum, and Erin Kaltenbrun for technical assistance. We thank Drs. Bill Courchesne, Beth Jones, Dan Klionsky, and Peter Walter for supplying reagents. We also thank Drs. Dan Klionsky and Scott Emr for critical reading of the manuscript and useful suggestions. This work was supported by National Science Foundation Grants MCB-011079 to A.A.M. and MCB-0110331 to J.L.B.
References
- Adams, A., Gottschling, D. E., Kaiser, C. A., and Stearns, T. (1997a). Techniques and protocols: assay of β-galactosidase in yeast. In: Methods in Yeast Genetics, ed. M. M. Dickerson, Cold Spring Harbor, NY: Cold Spring Harbor Press, 123–127.
- Adams, A., Gottschling, D. E., Kaiser, C. A., and Stearns, T. (1997b). Techniques and protocols: high-efficiency transformation of yeast. In: Methods in Yeast Genetics, ed. M. M. Dickerson, Cold Spring Harbor, NY: Cold Spring Harbor Press, 99–102.
- Ahner, A., and Brodsky, J. L. (2004). Checkpoints in ER-associated degradation: excuse me, which way to the proteasome? Trends Cell Biol. 14, 474–478. [DOI] [PubMed] [Google Scholar]
- Arvan, P., Zhao, X., Ramos-Castaneda, J., and Chang, A. (2002). Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 3, 771–780. [DOI] [PubMed] [Google Scholar]
- Bathurst, I. C., Travis, J., George, P. M., and Carrell, R. W. (1984). Structural and functional characterization of the abnormal Z alpha 1-antitrypsin isolated from human liver. FEBS Lett. 177, 179–183. [DOI] [PubMed] [Google Scholar]
- Brodsky, J. L., and Schekman, R. (1993). A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 123, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, J. L., Werner, E. D., Dubas, M. E., Goeckeler, J. L., Kruse, K. B., and McCracken, A. A. (1999). The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 274, 3453–3460. [DOI] [PubMed] [Google Scholar]
- Burda, P., Padilla, S. M., Sarkar, S., and Emr, S. D. (2002). Retromer function in endosome-to-Golgi retrograde transport is regulated by the yeast Vps34 PtdIns 3-kinase. J. Cell Sci. 115, 3889–3900. [DOI] [PubMed] [Google Scholar]
- Caldwell, S. R., Hill, K. J., and Cooper, A. A. (2001). Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J. Biol. Chem. 276, 23296–23303. [DOI] [PubMed] [Google Scholar]
- Casagrande, R., Stern, P., Diehn, M., Shamu, C., Osario, M., Zuniga, M., Brown, P. O., and Ploegh, H. (2000). Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell 5, 729–735. [DOI] [PubMed] [Google Scholar]
- Christianson, T. W., Sikorski, R. S., Dante, M., Shero, J. H., and Hieter, P. (1992). Multifunctional yeast high-copy-number shuttle vectors. Gene 110, 119–122. [DOI] [PubMed] [Google Scholar]
- Conibear, E., and Stevens, T. H. (1998). Multiple sorting pathways between the late Golgi and the vacuole in yeast. Biochim. Biophys. Acta 1404, 211–230. [DOI] [PubMed] [Google Scholar]
- Coughlan, C. M., Walker, J. L., Cochran, J. C., Wittrup, K. D., and Brodsky, J. L. (2004). Degradation of mutated bovine pancreatic trypsin inhibitor (BPTI) in the yeast vacuole suggests post-endoplasmic reticulum protein quality control. J. Biol. Chem. 279, 15289–15297. [DOI] [PubMed] [Google Scholar]
- Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Crystal, R. G. (1990). alpha 1-Antitrypsin deficiency, emphysema, and liver disease: genetic basis and strategies for therapy. J. Clin. Investig. 85, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafforn, T. R., Mahadeva, R., Elliott, P. R., Sivasothy, P., and Lomas, D. A. (1999). A kinetic mechanism for the polymerization of alpha1-antitrypsin. J. Biol. Chem. 274, 9548–9555. [DOI] [PubMed] [Google Scholar]
- Fewell, S. W., Travers, K. J., Weissman, J. S., and Brodsky, J. L. (2001). The action of molecular chaperones in the early secretory pathway. Annu. Rev. Genet. 35, 149–191. [DOI] [PubMed] [Google Scholar]
- Friedlander, R., Jarosch, E., Urban, J., Volkwein, C., and Sommer, T. (2000). A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2, 379–384. [DOI] [PubMed] [Google Scholar]
- Fu, L., and Sztul, E. (2003). Traffic-independent function of the Sar1p/COPII machinery in proteasomal sorting of the cystic fibrosis transmembrane conductance regulator. J. Cell Biol. 160, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata, R., Gao, Y. S., and Sztul, E. (2002). Hassles with taking out the garbage: aggravating aggresomes. Traffic 3, 388–396. [DOI] [PubMed] [Google Scholar]
- Hampton, R. Y. (2002). ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 14, 476–482. [DOI] [PubMed] [Google Scholar]
- Hoffman, C. S., and Winston, F. (1987). A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57, 267–272. [DOI] [PubMed] [Google Scholar]
- Holkeri, H., and Makarow, M. (1998). Different degradation pathways for heterologous glycoproteins in yeast. FEBS Lett. 429, 162–166. [DOI] [PubMed] [Google Scholar]
- Hong, E., Davidson, A. R., and Kaiser, C. A. (1996). A pathway for targeting soluble misfolded proteins to the yeast vacuole. J. Cell Biol. 135, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horazdovsky, B. F., DeWald, D. B., and Emr, S. D. (1995). Protein transport to the yeast vacuole. Curr. Opin. Cell Biol. 7, 544–551. [DOI] [PubMed] [Google Scholar]
- Huyer, G., Piluek, W. F., Fansler, Z., Kreft, S. G., Hochstrasser, M., Brodsky, J. L., and Michaelis, S. (2004). Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble lumenal protein. J. Biol. Chem. 279, 38369–38378. [DOI] [PubMed] [Google Scholar]
- Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, E. W., Zubenko, G. S., and Parker, R. R. (1982). PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics 102, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, M. U., Emr, S. D., and Winther, J. R. (1999). Ligand recognition and domain structure of Vps10p, a vacuolar protein sorting receptor in Saccharomyces cerevisiae. Eur. J. Biochem. 260, 461–469. [DOI] [PubMed] [Google Scholar]
- Kabani, M., Kelley, S. S., Morrow, M. W., Montgomery, D. L., Sivendran, R., Rose, M. D., Gierasch, L. M., and Brodsky, J. L. (2003). Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Mol. Biol. Cell 14, 3437–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kametaka, S., Okano, T., Ohsumi, M., and Ohsumi, Y. (1998). Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 273, 22284–22291. [DOI] [PubMed] [Google Scholar]
- Kihara, A., Noda, T., Ishihara, N., and Ohsumi, Y. (2001). Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 152, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J., Dalton, V. M., Eggerton, K. P., Scott, S. V., and Klionsky, D. J. (1999). Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell. 10, 1337–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser, G. L., Gentzsch, M., Kloser, A. K., Balzi, E., Wolf, D. H., Goffeau, A., and Riordan, J. R. (2001). Expression and degradation of the cystic fibrosis transmembrane conductance regulator in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 390, 195–205. [DOI] [PubMed] [Google Scholar]
- Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. [DOI] [PubMed] [Google Scholar]
- Kostova, Z., and Wolf, D. H. (2003). For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 22, 2309–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, B., and Klionsky, D. J. (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477. [DOI] [PubMed] [Google Scholar]
- Lin, L., Schmidt, B., Teckman, J., and Perlmutter, D. H. (2001). A naturally occurring nonpolymerogenic mutant of alpha 1-antitrypsin characterized by prolonged retention in the endoplasmic reticulum. J. Biol. Chem. 276, 33893–33898. [DOI] [PubMed] [Google Scholar]
- Lomas, D. A., Evans, D. L., Finch, J. T., and Carrell, R. W. (1992). The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 357, 605–607. [DOI] [PubMed] [Google Scholar]
- Lomas, D. A., and Mahadeva, R. (2002). α1-Antitrypsin polymerization and the serpinopathies: pathobiology and the prospects for therapy. J. Clin. Investig. 110, 1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcusson, E. G., Horazdovsky, B. F., Cereghino, J. L., Gharakhanian, E., and Emr, S. D. (1994). The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell 77, 579–586. [DOI] [PubMed] [Google Scholar]
- McCracken, A. A., and Brodsky, J. L. (2003). Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD). Bioessays 25, 868–877. [DOI] [PubMed] [Google Scholar]
- McCracken, A. A., Karpichev, I. V., Ernaga, J. E., Werner, E. D., Dillin, A. G., and Courchesne, W. E. (1996). Yeast mutants deficient in ER-associated degradation of the Z variant of alpha-1-protease inhibitor. Genetics 144, 1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken, A. A., and Kruse, K. B. (1993). Selective protein degradation in the yeast exocytic pathway. Mol. Biol. Cell 4, 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir, D. T., and Dumais, D. R. (1987). Glycosylation and secretion of human alpha-1-antitrypsin by yeast. Gene 56, 209–217. [DOI] [PubMed] [Google Scholar]
- Mumberg, D., Muller, R., and Funk, M. (1994). Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22, 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, D. T., Spear, E. D., and Walter, P. (2000). The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice, D. C., Sato, T. K., Stromhaug, P. E., Emr, S. D., and Klionsky, D. J. (2002). Cooperative binding of the cytoplasm to vacuole targeting pathway proteins, Cvt13 and Cvt20, to phosphatidylinositol 3-phosphate at the pre-autophagosomal structure is required for selective autophagy. J. Biol. Chem. 277, 30198–30207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa, S. I., Fewell, S. W., Kato, Y., Brodsky, J. L., and Endo, T. (2001). Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 153, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, E. A., Kruse, K. B., Fewell, S. W., Buchanan, S. M., Brodsky, J. L., and McCracken, A. A. (2003). Differential requirements of novel A1PiZ degradation deficient (ADD) genes in ER-associated protein degradation. J. Cell Sci. 116, 2361–2373. [DOI] [PubMed] [Google Scholar]
- Perlmutter, D. H. (2002). Liver injury in alpha1-antitrypsin deficiency: an aggregated protein induces mitochondrial injury. J. Clin. Investig. 110, 1579–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter, D. H. (2003). alpha1-Antitrypsin deficiency: liver disease associated with retention of a mutant secretory glycoprotein in the endoplasmic reticulum. In: Protein Misfolding and Disease, Principles and Protocols, ed. P. Bross and N. Gregersen, Totowa, NJ: Humana Press, 39–56. [DOI] [PubMed]
- Qu, D., Teckman, J. H., Omura, S., and Perlmutter, D. H. (1996). Degradation of mutant secretory protein, α1-antitrypsin Z, in the endoplasmic reticulum requires proteosome activity. J. Biol. Chem. 271, 22791–22795. [DOI] [PubMed] [Google Scholar]
- Raymond, C. K., Howald-Stevenson, I., Vater, C. A., and Stevens, T. H. (1992). Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vps mutants. Mol. Biol. Cell 3, 1389–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. S., Klionsky, D. J., Banta, L. M., and Emr, S. D. (1988). Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 8, 4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski, D. T., and Kaufman, R. J. (2004). A trip to the ER: coping with stress. Trends Cell Biol. 14, 20–28. [DOI] [PubMed] [Google Scholar]
- Schmidt, B. Z. and Perlmutter, D. H. (2005). Grp78, Grp94 and Grp170 interact with alpha-1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am. J. Physiol. 289, G444–G455. [DOI] [PubMed] [Google Scholar]
- Seaman, M. N., Marcusson, E. G., Cereghino, J. L., and Emr, S. D. (1997). Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J. Cell Biol. 137, 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman, M. N., McCaffery, J. M., and Emr, S. D. (1998). A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J. Cell Biol. 142, 665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear, E. D., and Ng, D. T. (2003). Stress tolerance of misfolded carboxypeptidase Y requires maintenance of protein trafficking and degradative pathways. Mol. Biol. Cell 14, 2756–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveger, T. (1988). The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr. Scand. 77, 847–851. [DOI] [PubMed] [Google Scholar]
- Taxis, C., Hitt, R., Park, S. H., Deak, P. M., Kostova, Z., and Wolf, D. H. (2003). Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J. Biol. Chem. 278, 35903–35913. [DOI] [PubMed] [Google Scholar]
- Taxis, C., Vogel, F., and Wolf, D. H. (2002). ER-Golgi traffic is a prerequisite for efficient ER degradation. Mol. Biol. Cell 13, 1806–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teckman, J. H., Burrows, J., Hidvegi, T., Schmidt, B., Hale, P. D., and Perlmutter, D. H. (2001). The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J. Biol. Chem. 276, 44865–44872. [DOI] [PubMed] [Google Scholar]
- Teckman, J. H., Gilmore, R., and Perlmutter, D. H. (2000). Role of ubiquitin in proteasomal degradation of mutant alpha(1)-antitrypsin Z in the endoplasmic reticulum. Am. J. Physiol. 278, G39–G48. [DOI] [PubMed] [Google Scholar]
- Teckman, J. H., and Perlmutter, D. H. (2000). Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am. J. Physiol. 279, G961–G974. [DOI] [PubMed] [Google Scholar]
- Teckman, J. H., Qu, D., and Perlmutter, D. H. (1996). Molecular pathogenesis of liver disease in alpha1-antitrypsin deficiency. Hepatology 24, 1504–1516. [DOI] [PubMed] [Google Scholar]
- Travers, K. J., Patil, C. K., Wodicka, L., Lockhart, D. J., Weissman, J. S., and Walter, P. (2000). Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258. [DOI] [PubMed] [Google Scholar]
- Tsai, B., Ye, Y., and Rapoport, T. A. (2002). Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell. Biol. 3, 246–255. [DOI] [PubMed] [Google Scholar]
- Tsukada, M., and Ohsumi, Y. (1993). Isolation and Characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174. [DOI] [PubMed] [Google Scholar]
- Vashist, S., Kim, W., Belden, W. J., Spear, E. D., Barlowe, C., and Ng, D. T. (2001). Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 155, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist, S., and Ng, D. T. (2004). Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 165, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach, A., Brachat, A., Pohlmann, R., and Philippsen, P. (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Werner, E. D., Brodsky, J. L., and McCracken, A. A. (1996). Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA 93, 13797–13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler, E. A., et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. [DOI] [PubMed] [Google Scholar]
- Wu, Y., Whitman, I., Molmenti, E., Moore, K., Hippenmeyer, P., and Perlmutter, D. H. (1994). A lag in intracellular degradation of mutant alpha-1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ individuals. Proc. Natl. Acad. Sci. USA 91, 9014–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser, A. E., and Emr, S. D. (2002). Novel PtdIns(3)P-binding protein Etf1 functions as an effector of the Vps34 PtdIns 3-kinase in autophagy. J. Cell Biol. 158, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youker, R. T., Walsh, P., Beilharz, T., Lithgow, T., and Brodsky, J. L. (2004). Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol. Biol. Cell 15, 4787–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., Michaelis, S., and Brodsky, J. L. (2002). CFTR expression and ER associated degradation in yeast. In: Cystic Fibrosis Methods and Protocols, Methods in Molecular Medicine, ed. W. R. Skach, Totowa, NJ: Humana Press, 257–265. [DOI] [PubMed]
- Zhang, Y., Nijbroek, G., Sullivan, M. L., McCracken, A. A., Watkins, S. C., Michaelis, S., and Brodsky, J. L. (2001). Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12, 1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]