The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA (original) (raw)
. Author manuscript; available in PMC: 2006 Jan 25.
Published in final edited form as: Nature. 2003 May 28;424(6944):94–98. doi: 10.1038/nature01707
Abstract
High mutation frequency during reverse transcription has a principal role in the genetic variation of primate lentiviral populations. It is the main driving force for the generation of drug resistance and the escape from immune surveillance. G to A hypermutation is one of the characteristics of primate lentiviruses, as well as other retroviruses, during replication in vivo and in cell culture1–6. The molecular mechanisms of this process, however, remain to be clarified. Here, we demonstrate that CEM15 (also known as apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G; APOBEC3G)7,8, an endogenous inhibitor of human immunodeficiency virus type 1 (HIV-1) replication, is a cytidine deaminase and is able to induce G to A hypermutation in newly synthesized viral DNA. This effect can be counteracted by the HIV-1 virion infectivity factor (Vif). It seems that this viral DNA mutator is a viral defence mechanism in host cells that may induce either lethal hypermutation or instability of the incoming nascent viral reverse transcripts, which could account for the Vif-defective phenotype. Importantly, the accumulation of CEM15-mediated non-lethal hypermutation in the replicating viral genome could potently contribute to the genetic variation of primate lentiviral populations.
HIV-1 Vif protein is required for viral replication in vivo and in some ‘non-permissive’ cells, such as peripheral blood mononuclear cells, macrophages and H9 T cells9–11. The vif_-defective viruses (Δ_vif) from non-permissive cells cannot complete reverse transcription, or the newly synthesized DNA cannot exist in the target cells for a significant time period12–14. Recently, it has been demonstrated that CEM15 is an endogenous inhibitor of HIV-1 that exists only in non-permissive cells. Its inhibitory effect on HIV-1 replication can be counteracted by HIV-1 Vif protein7. As Vif binds to HIV-1 RNA in the cytoplasm of virus-producing cells15–17, we investigated whether CEM15, which shares significant homology with some other cytidine deaminases that edit RNA, could also edit HIV-1 genomic or spliced RNA. We have sequenced the nearly full-length genomic RNA (>98%) of HIV-1 from the Δ_vif_ virions generated from H9 T cells by polymerase chain reaction with reverse transcription (RT–PCR) techniques (primer pairs are listed in Supplementary Table S1). Compared with the sequence of pNL4-3Δvif DNA, the change of genomic RNA in the virions is not significant. We have found an A to G change at positions 2257 and 3608, and a G to T change at position 9418 (data not shown). We have also sequenced several spliced HIV-1(NL4-3Δvif) RNA in H9 cells (Supplementary Fig. S1 and Table S1); however, no mutations were demonstrated.
CEM15, as well as some other homologues such as AID and APOBEC1, can function as a DNA mutator in Escherichia coli18,19. As both CEM15 and Vif can be packaged into HIV-1 virions7,17, we then investigated whether the newly synthesized HIV-1 DNA is a substrate of CEM15. We first compared the sequence of newly synthesized viral DNA in the presence or absence of Vif. The newly synthesized DNA of Δ_vif_ viruses from H9 cells have G to A substitutions in all the sequences (U3-R-U5, V3 and PR regions; primer pairs are listed in Supplementary Table S1) that we have analysed (Fig. 1a, row 2 of top panel, and data not shown). The mutation frequency at various regions is similar (data not shown). Notably, the pattern of G to A hypermutation in Δ_vif_ viruses from H9 cells is consistent with the pattern of G to A hypermutation occurring during wild-type virus passage in cell culture and during in vivo viral growth3,5,6. They both preferentially take place in a GpA or GpG dinucleotide context or in a group of G nucleotides. Besides the G to A substitution, other substitutions have also been found but with much lower frequency. Of note, as C to T substitution is not significantly higher than the rest of substitutions in the analysed regions, it has been classified into the category of ‘other substitutions’. Conversely, the newly synthesized viral DNA from endogenous reverse transcription, which was driven by deoxyribonucleoside triphosphates (dNTPs) at equal amount, also has similar mutations, indicating that all the necessary factors have been packaged within the virions for this process, and that the G to A substitution is not dependent on the imbalance of dNTP pools (Fig. 1a, row 3 of top panel). The mutation frequency in viral DNA isolated from the intravirion reverse transcripts at 4 h after endogenous reverse transcription (ERT) initiation and from intracellular reverse transcripts at 12 h after infection is quite similar (data not shown). Conversely, no G to A substitution was found in Δ_vif_ viruses from permissive SupT1 cells and the wild-type viruses generated from H9 or SupT1 cells (Fig. 1a, top panel, and 1b). Furthermore, we have also examined the effect of Vif on hypermutation in long-term cell culture by passing the wild-type or Δ_vif_ viruses in C8166, a T-cell line that is semi-permissive for Δ_vif_ viruses20 and that expresses small amounts of CEM15 (ref. 7 and data not shown). We found that the G to A hypermutation in the Δ_vif_ viruses is markedly higher than that in the wild-type viruses (Fig 1a, bottom panel, and 1c).
Figure 1.
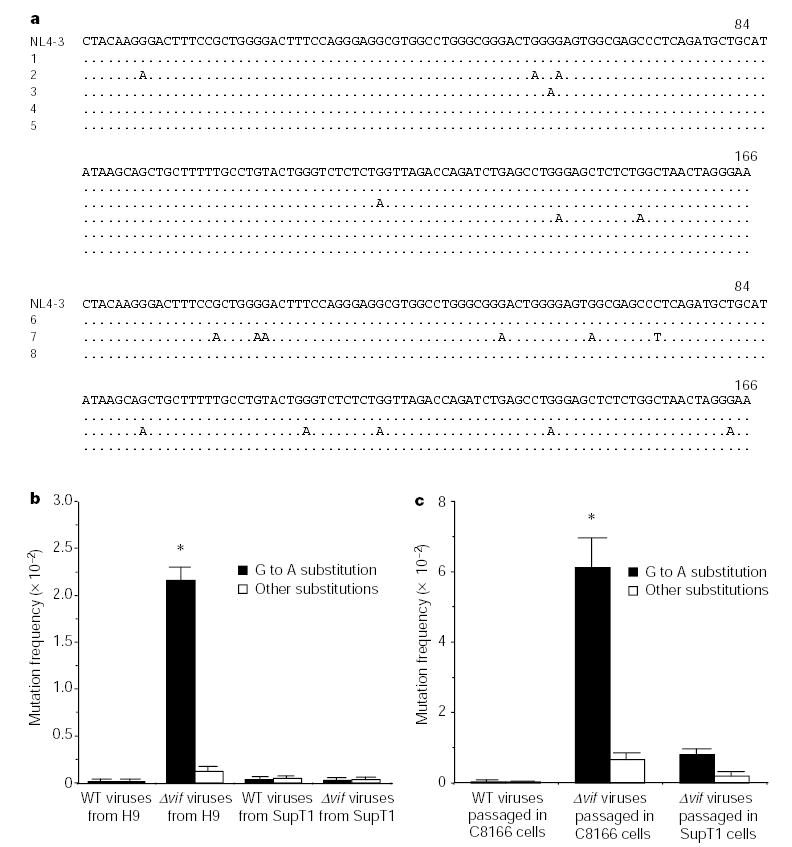
G to A hypermutation in viral DNA of Δ_vif_ viruses from non-permissive cells or semi-permissive cells. a, The DNA sequence alignments in the U3-R region. The clones with the most-identical sequences from the total clones (n = 8) are listed. The newly synthesized viral DNA are listed in the top panel. The rows are: (1) wild-type viruses from H9 cells to infect C8166 cells; (2) Δ_vif_ viruses from H9 cells to infect C8166 cells; (3) Δ_vif_ viruses from H9 cells to process ERT; (4) wild-type viruses from SupT1 cells to infect C8166 cells; (5) Δ_vif_ viruses from SupT1 cells to infect C8166 cells. After two passages, the viral DNA in C8166 cells was sequenced (bottom panel). The rows are: (6) wild-type viruses; (7) Δ_vif_ viruses; (8) Δ_vif_ viruses passaged in SupT1 cells. b, Comparison of mutation frequency of G to A in the newly synthesized DNA of viruses generated from H9 or SupT1 cells (301 nucleotides in U3-R-U5 region, n = 8). The G to A mutation frequency of Δ_vif_ viruses from H9 cells to infect C8166 cells is significantly higher than that of others (asterisk, P < 0.001, t_-test). WT, wild type. c, Comparison of the G to A mutation frequency in the viral DNA after passage in C8166 cells (n = 8). The G to A mutation frequency of Δ_vif viruses passaged in C8166 is significantly higher than that of wild-type viruses (asterisk, P < 0.001, _t_-test).
We then examined the effect of CEM15 on G to A substitution. 293T cells, which are permissive cells and in which no endogenous CEM15 is expressed7, were transfected with wild-type or Δ_vif_ HIV-1 DNA in the presence or absence of CEM15. After 48 h, the viruses in the supernatants were then allowed to infect C8166 cells. We found that the newly synthesized DNA of Δ_vif_ viruses with CEM15 contained markedly more G to A hypermutations than that in the newly synthesized DNA from Δ_vif_ viruses without CEM15, or from wild-type viruses (Fig. 2a, top panel, and 2b). Again, the endogenous reverse transcripts of Δ_vif_ viruses with CEM15 also contained G to A hypermutations (Fig. 2a, row 5 of top panel). Similar phenomena were also found in the newly synthesized viral DNA of Δ_vif_ viruses that were generated from the SupT1 T cells containing retrovirally transduced CEM15 (Fig. 2b).
Figure 2.
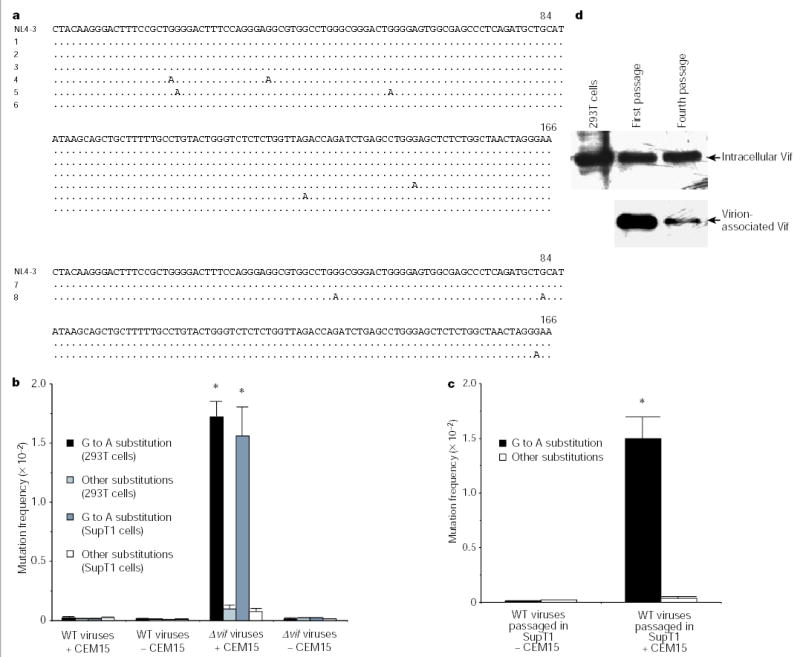
G to A hypermutation in newly synthesized DNA of viruses generated from cells containing CEM15. a, DNA sequence alignment in HIV-1 U3-R region. The clones with the most-identical sequences from the total clones (n = 8) are listed. Viruses were from 293T (top) cells or passaged in SupT1 (bottom) cells. In the top panel, the rows are: (1) wild-type viruses, with CEM15, to infect C8166 cells; (2) wild-type viruses, with CEM15, to process ERT; (3) wild-type viruses, without CEM15, to infect C8166 cells; (4) Δ_vif_ viruses, with CEM15, to infect C8166 cells; (5) Δ_vif_ viruses, with CEM15, to process ERT; (6) Δ_vif_ viruses, without CEM15, to infect C8166 cells. In the bottom panel, the rows are: (7) wild-type viruses, without CEM15; (8) wild-type viruses, with CEM15. b, Comparison of G to A mutation frequency in newly synthesized DNA (301 nucleotides in U3-R-U5 region, n = 8). The G to A mutation frequency of Δ_vif_ viruses from 293T or SupT1 cells containing CEM15 is significantly higher than that of others (asterisk, P < 0.001, _t_-test). c, Comparison of G to A mutation frequency in the viral DNA after passage (n = 8). The G to A mutation frequency of wild-type viruses passaged in SupT1 cells containing CEM15 is significantly higher than that in SupT1 cells without CEM15 (asterisk, P < 0.001, _t_-test). d, The wild-type virions, after various passages in SupT1 cells containing CEM15, were purified29. After normalization with HIV-1 p24, the virion-associated Vif protein was detected by western blot15.
However, it is clear that the G to A hypermutation still occurs in the presence of the vif gene when the viruses replicate in the cell culture for several passages3,21. We then examined the effect of CEM15 on the mutation frequency of wild-type viruses in long-term cultures. The CEM15 gene was transduced into SupT1 cells by a Moloney murine leukaemia virus (MMLV) retroviral vector. The wild-type viruses were then passaged in the SupT1 cells, with or without CEM15 expression. After four passages, the viral DNA was amplified and sequenced. We found that wild-type viruses passaged in SupT1 cells containing CEM15 have significantly more G to A hypermutations in their genomes than those in SupT1 cells without CEM15 (Fig. 2a, bottom panel, and 2c). This result further demonstrated that CEM15 is able to induce G to A hypermutation and could be responsible, at least in part, for the hypermutation in the long-term culture of wild-type viruses. Less Vif incorporation into virions in chronic infection might decrease its counteracting effects on CEM15 in the virions (Fig. 2d)22. It is notable that CEM15 does not significantly affect the dNTP pools in SupT1 cells and C8166 cells (Supplementary Fig. S2), excluding the possibility that CEM15 induces the imbalances in the dNTP pools in cells.
To verify that CEM15 has cytidine deaminase activity, glutathione _S_-transferase (GST)–CEM15 fusion protein was purified from E. coli. Figure 3 illustrates that CEM15 indeed has cytidine deaminase activity that can be inhibited with tetrahydrouridine (THU). By sequence alignment, it is predicted that CEM15 contains two zinc finger domains7. The similar zinc finger domains in other cytidine deaminases have a principal role in cytidine deaminase activity23. To verify that the zinc finger domains in CEM15 are important for cytidine deaminase, several mutations at zinc finger domains were generated. Figure 3b demonstrates that the mutants at either zinc domains have significantly decreased cytidine deaminase activity. To determine whether purified GST–CEM15 contains the enzymatic activity to edit viral RNA, DNA, or DNA–RNA duplexes, PCR-amplified DNA, _in vitro_-transcribed RNA, or in vitro reverse-transcribed RNA–DNA duplexes were incubated with GST–CEM15, respectively. All the nucleic acids were generated from the same HIV-1 fragment (nucleotides 2134–2594). After incubation, the DNA, RNA or RNA–DNA duplexes were used as the templates for PCR or RT–PCR amplification. The PCR products were then inserted into the pGEM-T vector and sequenced. No mutation was found (data not shown). It is probable that, similar to APOBEC1, CEM15 requires additional factor(s) to perform editing23,24.
Figure 3.
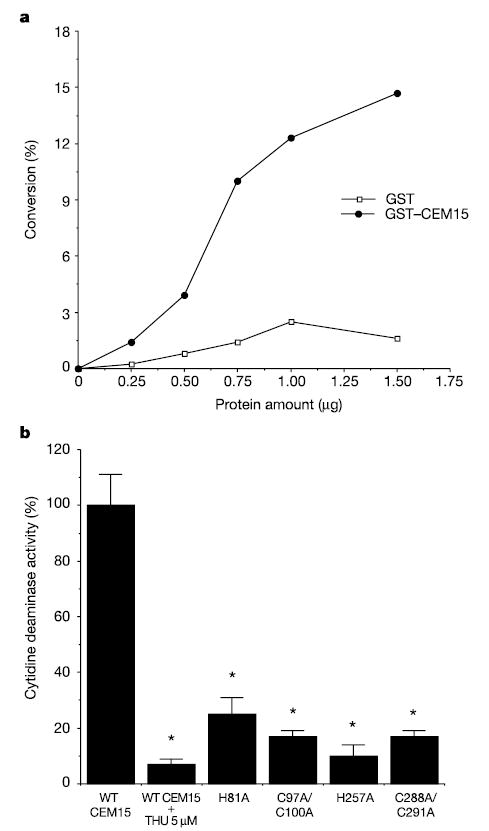
CEM15 has cytidine deaminase activity in vitro. a, GST–CEM15, but not GST alone, converts deoxycytidine to deoxyuridine in vitro in a concentration-dependent manner. This figure represents five independent experiments. b, Mutations at zinc finger domains significantly decrease cytidine deaminase activity (asterisk, P < 0.001, _t_-test). THU significantly inhibits cytidine deaminase activity (asterisk, P < 0.001, _t_-test).
The CEM15 mutants were then co-transfected with pNL4-3Δvif into 293T cells to test their functional activities. After 48 h, the viruses in the supernatants were collected. The virions, normalized for quantity, were then used to infect C8166 cells or HLCD4-CAT cells. Twelve hours after infection, the newly synthesized DNA in C8166 cells was extracted, followed by PCR amplification and sequencing. The lysate of HLCD4-CAT cells was collected for CAT assay after 48 h. Figure 4 shows that, compared with wild-type CEM15, CEM15 mutants that contain less cytidine deaminase activity could not decrease the infectivity of Δ_vif_ viruses in permissive HLCD4-CAT cells (Fig. 4a). Furthermore, the frequency of G to A hypermutation in the newly synthesized DNA induced by the mutants is also much lower than that induced by wild-type CEM15 (Fig. 4b). These data suggest that CEM15 induces G to A hypermutation in the newly synthesized viral DNA through its cytidine deaminase activity.
Figure 4.
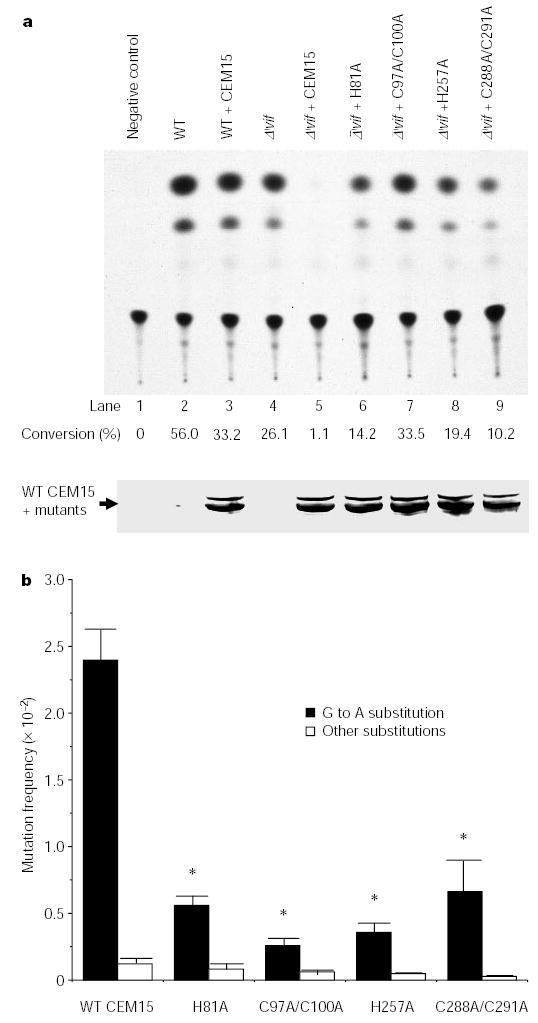
CEM15 without cytidine deaminase activity can neither inhibit the infectivity nor induce hypermutation in the newly synthesized DNA of Δ_vif_ viruses. pNL4-3 or pNL4-3Δvif were transfected into 293T cells with CEM15 or CEM15 mutants. a, The viruses were used to infect HLCD4-CAT cells. CAT assays were performed. This figure represents three independent experiments. b, The newly synthesized viral DNA in C8166 cells was amplified by PCR and sequenced. The G to A mutation frequency of Δ_vif_ viruses from 293T cells containing mutant CEM15 genes is significantly lower than that containing wild-type CEM15 (asterisk, P < 0.01, _t_-test).
Our data have indicated that the genomic RNA of Δ_vif_ viruses from non-permissive cells does not contain the G to A substitution, and CEM15 does not cause a significant imbalance of dNTPs in the cells. We have also found that, in the presence of CEM15, the product of endogenous reverse transcription of Δ_vif_ viruses at high and equal concentration of dNTPs (1 mM) contains the G to A hypermutation. Furthermore, the hypermutation preferentially takes place in a GpA or GpG dinucleotide context or in a group of G nucleotides, which does not occur when the imbalance of the dTTP/dCTP ratio induces dC to dT substitution. On the basis of these observations, we propose that CEM15 may directly deaminate dC in the newly synthesized viral DNA and convert it to dU. The dC to dU conversion in the minus strand DNA of Δ_vif_ viruses would further be base-pair-matched by dA during plus-stranded viral DNA synthesis, generating G to A hypermutation in the newly synthesized viral DNA. If integrated, the Δ_vif_ proviral DNA containing the lethal hypermutation might encode ‘pre-mature stop’ or mutated viral proteins, therefore generating defective viruses. Conversely, the deaminated Δ_vif_ viral DNA may become unstable in the cytoplasm. One of the possibilities is that the uracil-DNA glycosylase (UDG) enzyme, which has been packaged into the HIV-1 virions, could excise the dU from the DNA. Accordingly, a break point in the minus strand of viral DNA might occur, which may no longer be able to serve as the template for full-length viral DNA synthesis and thus would gradually be degraded.
Previous work has found that G to A hypermutation cannot be reproduced in a cell- and virus-free reverse transcription reaction when the concentrations and ratios of dNTPs are similar to those in living cells. When the dTTP/dCTP ratio rises to 10,000 to 1, dC to dT substitution occurs25. Furthermore, there is evidence suggesting that G to A hypermutation occurs in thymidine-treated U937-2 cells26. Here, we have not only indicated that G to A hypermutation occurs in the newly synthesized DNA of Δ_vif_ viruses generated from the non-permissive cells, but we have also shown that CEM15 is responsible for this phenomenon. Furthermore, we have demonstrated that CEM15 induces non-lethal hypermutation in the viral DNA of wild-type viruses passaged in a long-term culture. Therefore, we propose that CEM15 could be responsible for the hypermutation observed in vivo and in cell culture3–6.
It has been reported that interferon-inducible, double-stranded RNA-specific adenosine deaminase can also extensively edit the RNA genome of some RNA viruses in virus-producing cells27,28. We have now demonstrated that G to A hypermutation in newly synthesized reverse transcripts might be induced by a similar host defence factor. Further investigations are needed to determine the precise molecular mechanism(s) of this unique interaction of primate lentiviruses with the host cells.
Methods
Plasmid constructions, mutagenesis and protein purification
The CEM15 gene with a Flag-tag sequence at its 3’ terminus was amplified from the messenger RNA of H9 cells through RT–PCR, and the sequence was confirmed. It was then cloned into various vectors: pcDNA3 for transfection into 293T cells; pGEX for GST fusion protein expression; and pSLX-CMV for retroviral (MMLV) transduction into SupT1 cells. CEM15 mutations were generated by a PCR-based mutagenesis approach15. The GST, GST–CEM15 and other GST fusion CEM15 mutant proteins were produced according to previously described methods15.
Transfections
The 293Tor T-lymphoid cells (H9 or SupT1) were transfected with pNL4-3 or pNL4-3Δvif (2μg), plus various plasmids (0.75μg pcDNA3-CEM15 or its mutants), with FuGene 6 transfection reagents (Roche). The viruses in the supernatants were collected after 48 h. Of note, pNL4-3Δvif was constructed in our laboratory and has been described previously15.
Intracellular reverse transcription
Wild-type or Δ_vif_ viruses (10 ng of p24 equivalents), generated from either permissive cells or non-permissive cells, were treated with RQ1 DNase (5 U) (Promega) at 37 °C for 1 h and then allowed to infect C8166 T cells. Twelve hours after infection, the infected cells (in 150μl TN buffer) were treated again with DNase (5 U) and incubated at 37 °C for 1 h. After washing, the newly synthesized viral DNA was extracted from the cells and amplified by PCR20.
Endogenous reverse transcription
The wild-type or Δ_vif_ viruses (75 ng of p24 antigen equivalents) were concentrated by ultracentrifugation, followed by DNase treatment at 37 °C for 1 h. After washing off DNase, the viruses in the TN buffer were incubated with 1 mM dNTPs, 2.5 mM MgCl2, and 15μg ml−1 melittin at 37 °C for 4 h20,29. The newly synthesized DNA was isolated and amplified with PCR. The PCR products were inserted into the pGEM-T vector and individual clones were sequenced and analysed.
Viral passage experiments
The retroviral vectors (MMLV) carrying the CEM15 gene (pSLX-CMV-CEM15) or the vector alone were first transduced into SupT1 T cells, following methods described previously15. The naive C8166 or G418-resistant SupT1 T cells or C8166 cells were infected by wild-type or Δ_vif_ viruses (0.1 ng of p24 antigen equivalents), which were generated from 293T cells through transfection with pNL4-3 or pNL4-3Δvif. The infected T cells were then cultured in RPMI1640 medium plus 10% fetal bovine serum. After 2 weeks, the progeny viruses in the supernatants were allowed to infect C8166 or SupT1 cells, with or without CEM15, respectively. This procedure was repeated two or four times. The viral DNA in the final round of infected cells was extracted and amplified by PCR, followed by insertion into pGEM-T vector. The individual clones were then analysed by sequencing.
DNA PCR, RT–PCR and DNA/complementary DNA sequencing
The viral DNA was extracted from viruses or infected cells and PCR was performed, as described previously20. The viral RNA was extracted from viruses or infected cells through RNeasy Mini Kits (Qiagen). DNase treatment was performed to eliminate any contaminating DNA. The RT–PCR was performed with ready to go RT–PCR beads (Amersham-Pharmacia). The PCR products were inserted into the pGEM-T vector and individual clones were sequenced. The mutation frequency (f) was calculated as: (number of substitutions)/(number of target nucleotides).
Cytidine deaminase assay
A method described previously30 was performed to determine the cytidine deaminase activity of CEM15, with some modifications. Briefly, GST–CEM15 at various amounts was incubated with 4μCi 3H-deoxycytidine (23 Ci mmol−1; Moravek) and 250μM cytidine in a total volume of 10μl Tris-HCl buffer (45 mM, pH 7.5). After incubation at 37 °C for 4–6 h, the reactions were quenched by adding 2μl of 10μgμl−1 each of deoxycytidine and deoxyuridine. Four microlitres of sample was then added onto a silica-polyester TLC plate (Sigma-Aldrich). The plate was developed in 10:1:0.5 (v/v) ethyl acetate:methanol:trichloroacetic acid (6 M) for 2–3 h. The deoxycytidine and deoxyuridine bands were visualized by exposure to 254 nm ultraviolet light and cut separately into scintillation fluid for 3H-isotope quantifications.
Acknowledgments
This work was supported by research grants from the National Institute of Health (H.Z).
Footnotes
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Pathak VK, Temin HM. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc Natl Acad Sci USA. 1990;87:6019–6023. doi: 10.1073/pnas.87.16.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, et al. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J Virol. 1991;65:3973–3985. doi: 10.1128/jvi.65.8.3973-3985.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vartanian JP, Meyerhans A, Asjo B, Wain-Hobson S. Selection, recombination, and G → A hypermutation of human immunodeficiency virus type 1 genomes. J Virol. 1991;65:1779–1788. doi: 10.1128/jvi.65.4.1779-1788.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vartanian JP, Meyerhans A, Sala M, Wain-Hobson S. G → A hypermutation of the human immunodeficiency virus type 1 genome: evidence for dCTP pool imbalance during reverse transcription. Proc Natl Acad Sci USA. 1994;91:3092–3096. doi: 10.1073/pnas.91.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fitzgibbon JE, Mazar S, Dubin DT. A new type of G → A hypermutation affecting human immunodeficiency virus. AIDS Res Hum Retroviruses. 1993;9:833–838. doi: 10.1089/aid.1993.9.833. [DOI] [PubMed] [Google Scholar]
- 6.Vartanian JP, Henry M, Wain-Hobson S. Sustained G → A hypermutation during reverse transcription of an entire human immunodeficiency virus type 1 strain Vau group O genome. J Gen Virol. 2002;83:801–805. doi: 10.1099/0022-1317-83-4-801. [DOI] [PubMed] [Google Scholar]
- 7.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 8.Jarmuz A, et al. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 9.Strebel K, et al. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature. 1987;328:728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- 10.Fisher AG, et al. The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science. 1987;237:888–893. doi: 10.1126/science.3497453. [DOI] [PubMed] [Google Scholar]
- 11.Gabuzda DH, et al. Role of vif in replication of human immunodeficiency virus type 1 in CD4 + T lymphocytes. J Virol. 1992;66:6489–6495. doi: 10.1128/jvi.66.11.6489-6495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sova P, Volsky DJ. Efficiency of viral DNA synthesis during infection of permissive and nonpermissive cells with vif-negative human immunodeficiency virus type 1. J Virol. 1993;67:6322–6326. doi: 10.1128/jvi.67.10.6322-6326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Schwedler U, Song J, Aiken C, Trono D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J Virol. 1993;67:4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simon JH, Malim MH. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J Virol. 1996;70:5297–5305. doi: 10.1128/jvi.70.8.5297-5305.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Pomerantz RJ, Dornadula G, Sun Y. Human immunodeficiency virus type 1 Vif protein is an integral component of an mRNP complex of viral RNA and could be involved in the viral RNA folding and packaging process. J Virol. 2000;74:8252–8261. doi: 10.1128/jvi.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dettenhofer M, Cen S, Carlson BA, Kleiman L, Yu XF. Association of human immunodeficiency virus type 1 Vif with RNA and its role in reverse transcription. J Virol. 2000;74:8938–8945. doi: 10.1128/jvi.74.19.8938-8945.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan MA, et al. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J Virol. 2001;75:7252–7265. doi: 10.1128/JVI.75.16.7252-7265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10:1247–1253. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 19.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 20.Dornadula G, Yang S, Pomerantz RJ, Zhang H. Partial rescue of the vif-negative phenotype of mutant human immunodeficiency virus type 1 strains from nonpermissive cells by intravirion reverse transcription. J Virol. 2000;74:2594–2602. doi: 10.1128/jvi.74.6.2594-2602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janini M, Rogers M, Birx DR, McCutchan FE. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J Virol. 2001;75:7973–7986. doi: 10.1128/JVI.75.17.7973-7986.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao S, et al. Human immunodeficiency virus type 1 Vif is efficiently packaged into virions during productive but not chronic infection. J Virol. 2003;77:1131–1140. doi: 10.1128/JVI.77.2.1131-1140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chester A, Scott J, Anant S, Navaratnam N. RNA editing: cytidine to uridine conversion in apolipoprotein B mRNA. Biochim Biophys Acta. 2000;1494:1–13. doi: 10.1016/s0167-4781(00)00219-0. [DOI] [PubMed] [Google Scholar]
- 24.Mehta A, Banerjee S, Driscoll DM. Apobec-1 interacts with a 65-kDa complementing protein to edit apolipoprotein-B mRNA in vitro. J Biol Chem. 1996;271:28294–28299. doi: 10.1074/jbc.271.45.28294. [DOI] [PubMed] [Google Scholar]
- 25.Martinez MA, Vartanian JP, Wain-Hobson S. Hypermutagenesis of RNA using human immunodeficiency virus type 1 reverse transcriptase and biased dNTP concentrations. Proc Natl Acad Sci USA. 1994;91:11787–11791. doi: 10.1073/pnas.91.25.11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vartanian JP, et al. HIV genetic variation is directed and restricted by DNA precursor availability. J Mol Biol. 1997;270:139–151. doi: 10.1006/jmbi.1997.1104. [DOI] [PubMed] [Google Scholar]
- 27.Cattaneo R, et al. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;55:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Dornadula G, Pomerantz RJ. Endogenous reverse transcription of human immunodeficiency virus type 1 in physiological microenvironments: an important stage for viral infection of nondividing cells. J Virol. 1996;70:2809–2824. doi: 10.1128/jvi.70.5.2809-2824.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacGinnitie AJ, Anant S, Davidson NO. Mutagenesis of apobec-1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme, reveals distinct domains that mediate cytosine nucleoside deaminase, RNA binding, and RNA editing activity. J Biol Chem. 1995;270:14768–14775. [PubMed] [Google Scholar]