Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification (original) (raw)
Abstract
Glycosylation is a key mechanism for orchestrating the bioactivity, metabolism and location of small molecules in living cells. In plants, a large multigene family of glycosyltransferases is involved in these processes, conjugating hormones, secondary metabolites, biotic and abiotic environmental toxins, to impact directly on cellular homeostasis. The red grape enzyme UDP-glucose:flavonoid 3-_O_-glycosyltransferase (_Vv_GT1) is responsible for the formation of anthocyanins, the health-promoting compounds which, in planta, function as colourants determining flower and fruit colour and are precursors for the formation of pigmented polymers in red wine. We show that _Vv_GT1 is active, in vitro, on a range of flavonoids. _Vv_GT1 is somewhat promiscuous with respect to donor sugar specificity as dissected through full kinetics on a panel of nine sugar donors. The three-dimensional structure of _Vv_GT1 has also been determined, both in its ‘Michaelis' complex with a UDP-glucose-derived donor and the acceptor kaempferol and in complex with UDP and quercetin. These structures, in tandem with kinetic dissection of activity, provide the foundation for understanding the mechanism of these enzymes in small molecule homeostasis.
Keywords: catalysis, flavonoid, glycosylation, structure, wine
Introduction
Plants have evolved an extraordinary capacity to perceive changes in their environment and respond rapidly to maximize opportunity and minimize risk. The plasticity of these responses requires the integration of growth, development and metabolism, which in turn has led to the evolution of diverse mechanisms to regulate cellular homeostasis. Glycosylation is one of these mechanisms, with a large multigene family of glycosyltransferases (GTs) able to recognize lipophilic small molecules including hormones, secondary metabolites and both biotic and abiotic toxins in the environment (Lim and Bowles, 2004; Bowles et al, 2005). The plant enzymes belong to the ‘GT1 family' in the CAZy classification of carbohydrate-active enzymes (Couthino et al, 2003), which also includes the mammalian glucuronosyltransferases, similarly involved in cellular homeostasis and the metabolism of pharmaceuticals and dietary compounds of clinical and nutritional relevance (Dutton, 1980; Tukey and Strassburg, 2000).
There is a substantial set of gene sequence data for GT1 enzymes with over 1200 open reading frames currently defined (December 2005). In the case of plants, the phylogenetics of the large multigene family in Arabidopsis thaliana has been analysed and compared to the evolution of substrate recognition (Lim et al, 2003). Studies have revealed that the plant GTs are highly regio- and stereo-selective, but are often capable in vitro of recognizing common features on multiple substrates, whether hormones, secondary metabolites or xenobiotics, a promiscuity which is of direct physiological significance for the plasticity of response (Lim and Bowles, 2004; Bowles et al, 2005). While regio- and stereo-selectivity can be defined using different naturally occurring aglycones and their analogues, it has not been possible to understand the molecular basis of donor and acceptor specificity for these reactions. Structures of several bacterial GTs of the GT1 family involved in vancomycin synthesis have been solved and shown to have the twin Rossmann-domain ‘GT-B' fold (Mulichak et al, 2001, 2003, 2004), but the sequences, of these enzymes of known structure, have less than 5% identity to the plant and mammalian enzymes of the same family.
One of the most significant, and representative, glycosylation reactions in plants is the formation of anthocyanins. Anthocyanins are water-soluble pigments based on a tricyclic flavonoid core. In the plant, the compounds function as colourants determining flower and fruit colour. During red winemaking, anthocyanins are progressively extracted into the aqueous must, which reaches maximum colour intensity at around 6 days. The colour thereafter decreases due to a loss of copigmentation (intramolecular H-bond mediated stacking of anthocyanins with nonpigmented flavonoid or phenolic partners, interrupted by increasing alcoholic concentration as fermentation proceeds) and the formation of pigmented polymeric compounds through chemical reactions with proanthocyanidin (tannin) monomers progressively extracted from skins and seeds as the percentage of alcohol increases. After 6–12 months, almost no free anthocyanins remain in wine and all colour is derived from the presence of pigmented polymeric anthocyanin:tannin compounds (Fulcrand et al, 2004). Increasingly, flavonoids are recognized for their health-related properties, both in the human diet and for the improvement of animal feed. A number of clinical effects have been ascribed to different plant flavonoids, including antitumour, anti-inflammatory and antimicrobial properties (Ross and Kasum, 2002; Halliwell et al, 2005). In the red grape (Vitis vinifera L., cv. Shiraz), cyanidin, an unstable precursor, is glycosylated by a uridine diphospho (UDP)-glucose:flavonoid 3-_O_-glycosyltransferase (_Vv_GT1) to yield the stable anthocyanin, cyanidin 3-_O-_glucoside (Figure 1). Glucosylation leads to transport of the product from the cytosol into the vacuole where levels accumulate as the fruit ripens. _Vv_GT1 is also active on a number of different flavonoids in vitro including quercetin and kaempferol, with rates in vitro ca. 2 and 0.6% of the cyanidin acceptor. The sequence of red grape GT has ca 60% identity with the 78D clade of A. thaliana GTs, and >50% identity to related enzymes in other species including those of petunia and strawberry, and thus serves as a powerful template upon which to illuminate substrate specificity across the whole family.
Figure 1.
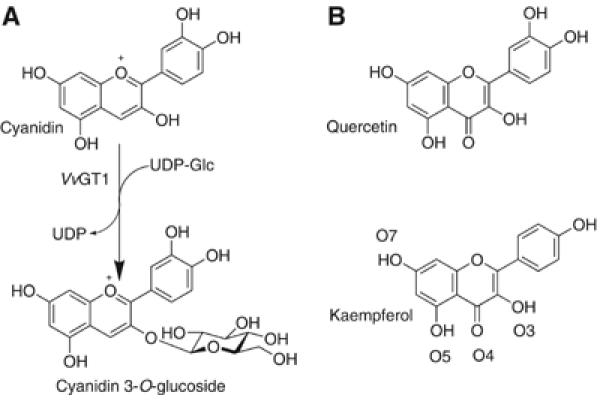
(A) The reaction catalysed by Vitis vinifera UDP-Glucose:flavonoid 3-_O_-glycosyltransferase is the addition of glucose, from a UDP-glucose donor to the 3-O position of cyanidin. The reaction occurs with inversion of anomeric configuration to generate the β-glucoside. (B) Vitis vinifera cyanidin 3-_O_-glycosyltransferase will also accommodate quercetin and kaempferol as acceptors at 2 and 0.6% of the rate with the natural acceptor (Ford et al, 1998). The numbering of the oxygens, as used in the text, is shown for kaempferol.
Here we present the 3-D structure, interpreted in light of ligand complexes, ‘high-throughput' screening of catalytic activity, and kinetics of wild type and mutant enzymes of the plant GT1 involved in the glucosylation of cyanidins in red grapes. The 3-D structure of _Vv_GT1 has been solved at 1.9 Å resolution in a UDP (product) bound form and, subsequently, in its ‘Michaelis' complex with both an intact UDP-Glc donor and the acceptor kaempferol, also at 1.9 Å resolution and in ‘nonproductive' complex with UDP and quercetin, at 2.1 Å. These three-dimensional structures lay the foundation for detailed dissection of catalytic mechanism and specificity, and should, in harness with recent structural work elsewhere (Shao et al, 2005), inform subsequent exploitation of the many plant GT1 enzymes.
Results
Screening for VvGT1 activities in vitro
The gene encoding _Vv_GT1 was expressed as a recombinant protein in Escherichia coli and purified to electrophoretic homogeneity. In vivo, _Vv_GT1 functions as a flavonoid 3-_O_-glycosyltransferase during the formation of red wine pigments. The Green-Amber-Red ‘GAR' screen (Yang et al, 2005a) was used to probe alternative substrates, in vitro, using a large acceptor and nucleotide-sugar donor screen. Liquid Chromatography Mass Spectrometry (LCMS), with an internal guanosine diphosphate (GDP) standard, was thus used to screen a large and representative donor and acceptor library in multi-well format trays. A virtual colour, reflecting DNA micro-array practice, of red (no reaction), amber (total ion-count (TIC) signal/noise >1) and green (TIC signal-to-noise >10) is assigned reflecting the presence of selected ions of mass n+y where y is the mass of the glycosyl moiety transferred (Flint et al, 2005; Yang et al, 2005a). Full, ‘pseudo-single substrate' kinetics, chosen to probe both nucleotide and sugar specificities, were subsequently performed on the panel of viable different sugar donors identified in the high-throughput screen.
VvGT1 native enzyme kinetics
Under the conditions of the mass-spectrometry based kinetic assay, _Vv_GT1 transfers Glc from UDPGlc to Quercetin and kaempferol at around 0.08 s−1 with a _K_m for UDP-Glc of 680 μM and for quercetin and kaempferol of 31 and 42 μM (Table I). In addition to the favoured donor, UDP-Glc, _Vv_GT1 can also harness a wide panel of different UDP-sugars: UDP-5SGlc, UDP-Xyl, UDP-Man, UDP-Gal and UDP-GlcNAc (Table I), as well as GDP-Glc and _d_TDP-Xyl. _Vv_GT1 showed no activity with UDP-6OMeGal, UDP-Ara, UDP-6FGal, UDP-GlcN, UDP-2FGal, UDP-5SAra, GDP-Man and neither GDP-Fuc nor UDP-Fuc. Importantly, _Vv_GT1 does not process the sugar donor UDP-Rha, which is highly important in plant secondary metabolism. The transfer rates for these different sugar donors reflects changes both in _k_cat and _K_m (Table I). Simply in terms of _k_cat, UDP is clearly the favoured nucleotide, with _d_TDP forms of both glucose and xylose transferred approximately five times more slowly than their UDP counterparts. GDP-Glc is accepted at approximately 1/100th the rate of UDP-Glc. The 6-hydroxyl of the donor is of lesser importance for catalysis, with UDP-Xyl transferred sixty times more slowly than UDP-Glc while, in contrast, there is a distinct epimeric preference at O4: UDP-Gal is transferred 210 times more slowly (in terms of _k_cat) than UDP-Glc with _k_cat/_K_m reduced some 16-fold.
Table 1.
Kinetic data of _Vv_GT1 and mutants
| Enzyme | Substrate varied | Substrate fixed | _K_m/μM | _k_cat (s−1) | _k_cat/_K_m (s−1 μM−1) |
|---|---|---|---|---|---|
| WT | UDPGlc | Quercetin | 679±74 | 8.4 × 10−2±9.1 × 10−3 | 124±13 |
| WT | dTDPGlc | Quercetin | 300±35 | 1.6 × 10−2±1.8 × 10−3 | 52±6.0 |
| WT | UDP5SGlc | Quercetin | 166±1.7 | 8.8 × 10−4±9.0 × 10−6 | 5.3±0.05 |
| WT | UDPGlcNAc | Quercetin | 194±23.2 | 1.04 × 10−3±1.2 × 10−4 | 5.3±0.6 |
| WT | UDPGal | Quercetin | 48.2±0.5 | 3.8 × 10−4±3.9 × 10−6 | 7.9±0.08 |
| WT | UDPMan | Quercetin | 50.1±5.0 | 1.0 × 10−5±1.0 × 10−6 | 0.2±0.02 |
| WT | GDPGlc | Quercetin | 167±2.60 | 9.7 × 10−4±1.5 × 10−5 | 5.8±0.09 |
| WT | _d_TDPXyl | Quercetin | 166±8.0 | 2.5 × 10−4±0.12 × 10−5 | 1.5±0.07 |
| WT | UDPXyl | Quercetin | 219±22 | 1.4 × 10−3±1.4 × 10−4 | 6.2±0.6 |
| WT | Quercetin | UDPGlc | 30.8±1.5 | 7.5 × 10−2±3.7 × 10−3 | 2437±118 |
| WT | Kaempferol | UDPGlc | 42.3±2.1 | 1.7 × 10−2±8.9 × 10−3 | 391±19 |
| Q375N | UDPGlc | Quercetin | 529±55 | 7.6 × 10−4±9.0 × 10−5 | 1.6±0.2 |
| Q375H | UDPGlc | Quercetin | 253±30 | 1.0 × 10−4±1.1 × 10−5 | 0.4±0.05 |
| T141A | UDPGlc | Quercetin | 310±28 | 1.5 × 10−2±1.3 × 10−3 | 49±4.5 |
| T141A | UDPGal | Quercetin | 120±15 | 8.1 × 10−4±1.0 × 10−4 | 6.7±0.83 |
| Q375N/T141A | UDPGlc | Quercetin | 120±10 | 7.0 × 10−4±5.8 × 10−5 | 5.8±0.49 |
| Q375N/T141A | UDPGal | Quercetin | 33±2.0 | 6.5 × 10−5±3.9 × 10−6 | 1.9±0.11 |
| H20A | UDPGlc/Gal/Rha | Quercetin | N/D | ||
| D374A | UDPGlc/Gal/Rha | Quercetin | N/D | ||
| N/D, no detectable activity. |
A large panel of acceptors (as described fully in Flint et al, 2005; Yang et al, 2005a) was subsequently used to probe the acceptor tolerance of _Vv_GT1 using the favored UDP-Glc as donor. While, in vivo, _Vv_GT1 is a 3-_O_-glycosyltransferase, it was able to transfer to a spectrum of different compounds (Figure 2), including some whose sole hydroxyl is equivalent to the O7 of the natural acceptors. This strongly suggests that, in vitro, _Vv_GT1 can be persuaded to accept a wide variety of flavonoid and similar acceptors; it is unclear how these preferences would manifest themselves in vivo, where cellular location and the availability and concentration of donors and acceptors would be major factors for catalysis. The relative catalytic promiscuity of _Vv_GT1 adds to the emerging picture (Davies et al, 2005; Flint et al, 2005) that many GTs are less specific, and thus also amenable to exploitation in biocatalysis, than might have, at one time, been assumed (since much of the pioneering work in the glycosyltransferase field was performed on enzymes which show much stricter specificity, exemplified by those enzymes involved in blood group synthesis (Patenaude et al, 2002; Marcus et al, 2003)).
Figure 2.
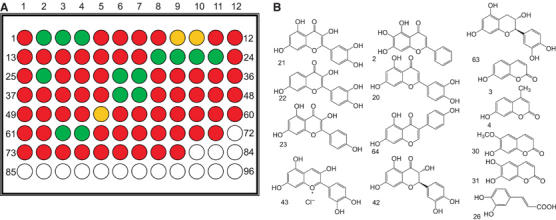
An example of the Green-Amber-Red screen for acceptor specificity using UDP-Glc as the donor. (A) A virtual microarray with red indicating no reaction, amber total ion-count signal/noise >1 and green signal-to-noise >10. (B) The panel of different acceptors identified with TIC signal-to-noise >10. A similar approach led to the identification of a spectrum of potential nucleotide sugar donors (see text).
Structure determination of the UDP-bound form of VvGT1
The structure of _Vv_GT1 was solved initially using 1.9 Å data in complex with UDP in tandem with 2.2 Å selenomethionine data, the latter collected so as to optimize the _f_″ signal from the Se anomalous scatterers (see Materials and methods). Subsequently, crystals were obtained both of the ‘Michaelis' complex of nontransferable UDP-2-deoxy-2-fluoro glucose donor (UDP-2FGlc, synthesized according to Gibson et al, 2004) with the acceptor kaempferol (Figure 1B) and the ‘abortive' complex of UDP with quercetin.
The structure of UDP: _Vv_GT1, a monomer in both crystal and solution, may be traced from Asn7 to Val456 with two regions (His54-Met60; Pro251-Thr259) disordered in electron density. The structure forms two ‘Rossmann-like' (β/α/β) domains, comprising residues 7–250 and 260–437, respectively (Figure 3A) and termed the ‘GT-B' fold. As with other GT-B structures (reviewed in Hu and Walker, 2002), the final C-terminal helix crosses from the C-terminal domain to complete the N-terminal domain fold through residues 437–456. This fold is similar to the GT fold class ‘GT-B' described originally for the T4 DNA β-glucosyltransferase (Vrielink et al, 1994) and subsequently in, for example, the E. coli MurG enzyme involved in peptidoglycan formation (Hu et al, 2003) and the bacterial enzymes involved in vancomycin synthesis (Mulichak et al, 2001, 2003, 2004).
Figure 3.
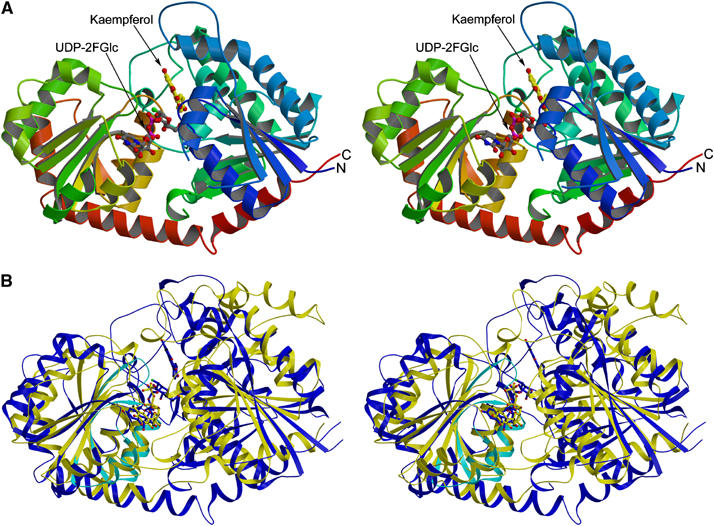
(A) Stereo (divergent) view of the Vitis vinifera UDP-Glucose:flavonoid 3-_O_-glycosyltransferase in Michaelis complex with UDP-2-deoxy-2-fluoro glucose (donor) and kaempferol (acceptor). The molecule is colour-ramped from N-terminus (blue) to C-terminus (red) with the ligands shown in ‘ball-and-stick' representation. (B) Stereo (divergent) view of the overlap of two GT1 enzymes _Vv_GT1 (blue), and the nucleotide complex of the GT1 enzyme GtfA (yellow). The region defining the C-terminal ‘signature' motif, displayed by some GT1 enzymes, is shown in cyan for _Vv_Gt1 only. This figure was generated with MOLSCRIPT (Kraulis, 1991).
To date, glycosyltransferase family GT1 contains over 1200 open reading frames. The sequences of the bacterial GT1 enzymes of known 3-D structure differ substantially from those of the plant enzymes (<5% identity). This has led some authors to consider that family GT1 contains different ‘subfamilies' reflecting, in part, the absence of the C-terminal 44-amino-acid signature motif that is found in the vast majority of plant and mammalian GT1 enzymes (Lim and Bowles, 2004). The 3-D structure of _Vv_GT1 shows that this signature defines the environment of the nucleotide-sugar binding site of the plant (and by implication other higher eukaryotic) enzymes. The ‘consensus' (Lim and Bowles, 2004) sequence, is described by the following residues (with those of _Vv_GT1 highlighted in bold): [FW332]-2X-Q-2X-[LIVMYA]-[LIMV]-46X-[LVGAC]-[LVFYA]-[LIVMF]-[STAGCM]-[HNQ]-[STAGC]-G-2X-[STAG]-3X-[STAGL]-[LIVMFA]-4X-[PQR]-[LIVMT]-3X-[PA]-3X-[DES]-[Q375EHN]. Many of these residues are simply involved in C-terminal fold ‘maintenance', although some play roles in substrate-binding and catalysis (and thus are also present in the bacterial enzymes, below). It is, indeed, the fine details of the nucleotide pocket environment that differ between the plant enzyme (and by implication its close sequence homologs) and the Gtf series of bacterial GT1 enzymes involved in vancomycin synthesis. In particular, the conformation and composition of the loop between the 6th (final) β-strand of the C-terminal domain and its subsequent helix, residues 332–337 in _Vv_GT1, is markedly different in the three families. This loop region contributes Trp332 to form the hydrophobic platform upon which the uracil base stacks in _Vv_GT1. Subsequently, Gln335, while making no direct interactions with the nucleotide-sugar, is involved in maintaining Glu358 in the correct orientation to bind both ribose hydroxyls. In the bacterial enzymes, GtfA, for example, there is no stacking ‘above' the base equivalent to Trp332 (this residue is a Glu in GtfA) nor is there an equivalent to Gln335, the comparable residue being Leu280. Further differences manifest themselves in the ribose binding region of this signature: a glutamate in this position is invariant in those GT-B fold enzymes which use UDP-sugars as donors but replaced by a hydrophobic residue (e.g. Leu301 in GtfA) in _d_TDP-donor enzymes reflecting the more hydrophobic nature of the deoxy-sugar (Mulichak et al, 2001, 2003, 2004). These structural changes are entirely consistent with the moderate reduction in catalytic efficiency of _Vv_GT1 between UDP-Glc and _d_TDP-Glc of some 2.3 (_k_cat/_K_m) to 5 (_k_cat) fold (Table I).
As one approaches the catalytic centre, more of the residues of the ‘signature' motif are conserved within the wider GT1 family, including the bacterial enzymes of vancomycin synthesis. Histidine 350 interacts with O2 of the β-phosphate of UDP and Ser355 with the equivalent atom of the α-phosphate in _Vv_GT1, the equivalents in GtfA are His293 and Thr 298. The Michaelis complex of _Vv_GT1, below, allows us to demonstrate that in _Vv_GT1, Asp374 and Gln375 of this signature motif are key players in sugar recognition whose sequence diversity reflects the use of different sugar donors on different enzymes.
Structure of the VvGT1 Michaelis complex of UDP-2FGlc and kaempferol and its nonproductive complex with UDP and quercetin
The Michaelis complex with the kaempferol acceptor and the nontransferable donor UDP-2FGlc (Figures 4A and 5) reveals the crucial interactions of the flavonoid acceptors and sugar nucleotide donors that are central to catalysis and specificity. Recognition of the O2 and O3 of glucose is conferred through the NH2 of Gln375 interacting with both O2 (F2) and O3 hydroxyls, while Asp374 hydrogen-bonds to both O3 and O4 through its OD1 and OD2 oxygens, respectively. The mutation D374A abolishes detectable catalytic activity (Table I), while the Q375N and Q375H mutations show seriously impaired or abolished catalytic activity, respectively. The O4 hydroxyl of glucose also interacts with the main-chain amide hydrogen of Trp353. The sole direct interaction of the O6 of glucose is with OG of Thr141, mutation of this residue T141A reduces the activity just six-fold, consistent with the lesser importance of the O6 in catalysis (discussed above).
Figure 4.
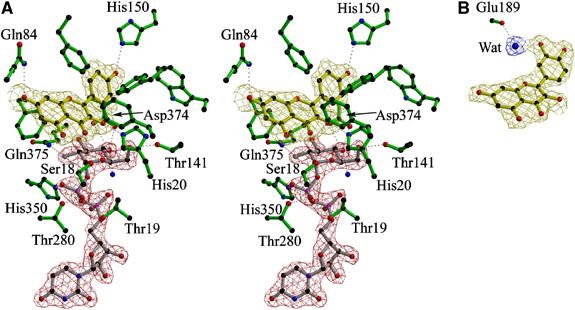
(A) Stereo (divergent) view of the observed electron density (maximum-likelihood weighted 2Fobs−Fcalc contoured at 1σ/0.4 electrons/Å3) of the Michaelis complex of the Vitis vinifera UDP-Glucose:flavonoid 3-_O_-glycosyltransferase with (nontransferrable) UDP-2-deoxy-2-fluoro glucose (donor) and kaempferol (acceptor). (B) Mono view of the observed electron density (as above) for quercetin bound to _Vv_Gt1 showing the single additional interaction it makes with the enzyme, a water-mediated hydrogen bond to the main-chain carbonyl of Glu189. All other interactions of this acceptor are identical to those seen with kaempferol. This figure was drawn with BOBSCRIPT (Esnouf, 1997).
Figure 5.
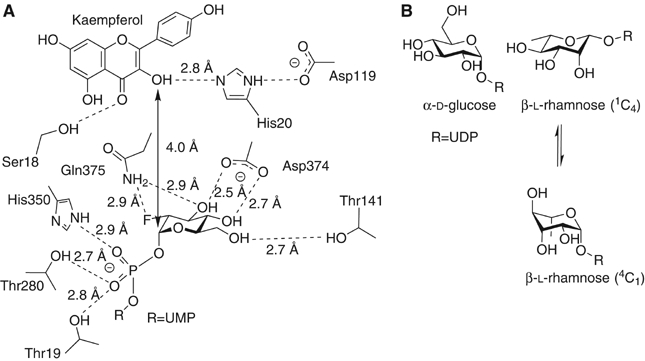
(A) Schematic diagram of active centre of the Vitis vinifera UDP-Glucose:flavonoid 3-_O_-glycosyltransferase showing the role of His20 as a Brønsted base for the activation of the flavonoid O3 hydroxyl for nucleophilic attack at C1 of the donor. (B) Schematic representations of UDP-α-D-glucose in 4_C_1 conformation and UDP-β-L rhamnose in both its favored 1_C_4 conformation and in 4_C_1 conformation. Steric factors dictate that UDP-β-L rhamnose will exist in a conformation skewed between those drawn; one which allows an axial leaving group orientation while fulfilling the stereoelectronic considerations of an incipient oxocarbenium-ion like transition state.
Although many of the donor site interactions are likely to be similar in family GT1 enzymes, the sugar donor site shows distinct differences from that previously described for UDP-GlcNAc binding to the family GT28 enzyme MurG (Hu et al, 2003). While many aspects of the two structures are similar, such as the Glu–ribose interaction (above) and the main-chain amide hydrogens and Ser/Thr hydroxyls binding the phosphates (above), the direct interactions to the sugar itself are significantly different. In both structures, there is a main-chain amide interaction with O4. The interaction equivalent to that of _Vv_GT1 Asp374 with the O3 and O4 hydroxyls is performed in MurG by a similarly located Gln (Gln288) but while the following residue, Gln375 in _Vv_GT1, is also a glutamine in MurG, slight main-chain rearrangements means it interacts only with O3, consistent with the 2-substituent in GlcNAc. O6 interactions of the GlcNAc MurG are quite different (it is flipped through 180 degrees relative to that in _Vv_GT1); they occur through solvent waters and O1 of the α-phosphate, compared to the direct interaction with Thr141 in _Vv_GT1. The _N_-acetyl binding pocket required by MurG, is instead occupied by a phenylalanine, Phe372, in _Vv_GT1. This residue, preceded by an a Pro-Phe motif common to both enzymes, marks a substantial main-chain difference between the two enzymes after strand β-2 of the C-terminal domain; in MurG an extended insertion at the end of this strand prior to the subsequent helix, provides the additional pocket to accommodate the _N_-acetyl moiety of the substrate. No such space is available in _Vv_GT1, which consequently must undergo conformational changes to Phe372 in order to accommodate _N_-acetylglucosamine (which it does with around 4% of the efficiency as with UDP-Glc).
The kaempferol acceptor lies in a hydrophobic, but open-ended, pocket defined by phenylalanine residues at positions 15, 121, 200 and 372 as well as by Ile87 and Val281. The structure clearly demonstrates the difference between the plant GT1 enzymes and the bacterial GTs in terms of acceptor-binding. While the attacking hydroxyl of the kaempferol acceptor lies in exactly the same location as the reacting atom of desvancosaminyl vancomycin in GtfD (Figure 6), no other atom of the flavonoid co-locates with vancomycin and no interactions are conserved. Thus, while the central core of β-strands are topologically equivalent between the _Vv_GT1 and GtfA structures (Figure 3B), all the interactions between acceptor and enzyme are made with the loop regions that are entirely different between the two enzymes. The acceptor binding site of _Vv_GT1 is an open-ended canyon in which the flavonoid O7 group points ‘outwards' into solvent (Figure 7). This topography would permit accommodation of much larger substrates, including flavonol 7-_O_-glycosides, into the active centre. Indeed, we observe a low occupancy of a contaminating 7-O glycoside of kaempferol with the O7 substituent projecting into solvent. Mass-spectrometry (data not shown) confirms that approximately 5% of commercial kaempferol is present as its glycosyl-adduct. This is of special significance since many naturally occuring flavonoids are indeed known to exist with extended glycan decorations at this position (Graham, 1988; Harborne and Willams, 2001) and so it strongly suggests that _Vv_GT1 could glycosylate such compounds in vivo. The open active centre presumably also contributes to the partial acceptor promiscuity defined by the GAR screen (Figure 2), for it would suggest that ‘reversed' binding modes (in which O3 points towards solvent and O7 towards the C1 of the donor sugar) may also be permitted, although given the totally different acceptor binding modes displayed by related GT1 enzymes glycosylating at spatially-close centres (Mulichak et al, 2001, 2003, 2004), further speculation about the exact mode of binding of these different acceptors is dangerous.
Figure 6.
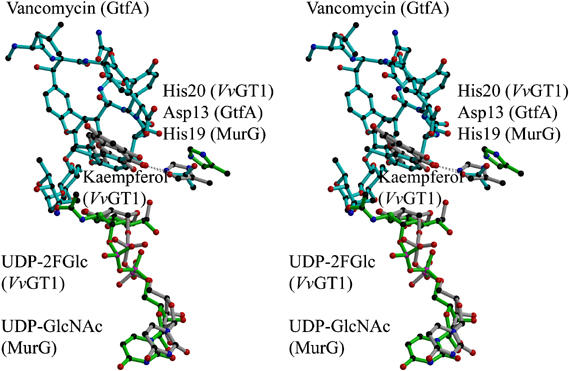
An overlap (divergent stereo) of GtfA (family GT1, in cyan), MurG (family GT28, in green) and _Vv_GT1 (family GT1, in grey). Ligands shown are vancomycin for GtfA, UDPGlcNAc for MurG and both UDP-2FGlc and kaempferol for _Vv_GT1. The proposed catalytic bases are presumed to be His20 of _Vv_GT1, Asp13 of GtfA and His19 of MurG.
Figure 7.
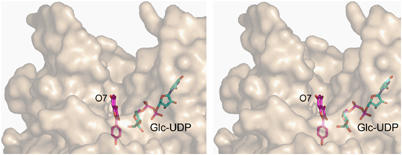
Stereo (divergent) view of the protein surface in the vicinity of the substrate-binding canyon showing that, while UDP-Glc is buried, the kaempferol acceptor is only partially occluded with O7 pointing into solvent. This figure was drawn with PyMOL (DeLano Scientific LLC, http://pymol.sourceforge.net/).
The bicyclic and phenol rings of kaempferol are not co-planar. Instead, the phenolic group lies at about 30° to the bicyclic moiety as would be expected in order to minimize ‘_ortho_-ortho' steric interactions between the two rings. This orientation is favoured by hydrogen-bonding of the flavonol O7 to Gln84 and the phenolic hydroxyl to His150. The attacking O3 hydroxyl of the flavonoid lies 2.7 Å from His20. This residue, and its equivalents on other GT-B fold structures (Figure 6), is believed to play the role of catalytic base for the deprotonation of O3 to allow nucleophilic attack at the anomeric centre of the donor. The geometry is exquisitely poised ‘in-line', with an O3kaempferol-C1UDP-Glc-O1UDP-Glc angle of 160°, as expected for the Michaelis complex in a single displacement mechanism. The _Vv_GT1 complex with both intact donor and acceptor, and the absence of catalytic activity of the H20A mutant (Table I) now confirms the essential role of His20. Interestingly, the role and environment of His20 is reminiscent of serine hydrolases. In the serine hydrolases, the histidine group of the catalytic triad of Ser-His-Asp deprotonates serine for nucleophilic attack at the substrate; protonation of the histidine is subsequently stabilized through interaction with the aspartate. In _Vv_GT1 the equivalent chemistry occurs such that when the O3 of the flavonoid is deprotonated by His20, an aspartate, Asp119 lies in an analogous position (Figure 5). Intriguingly, a histidine base in this position is not invariant across the fold family, with some GT-B enzymes using an aspartate residue as a base (Mulichak et al, 2001, 2003), in a manner more akin to inverting transferases of fold class GT-A (Tarbouriech et al, 2001). There is no simple correlation of the amino acid used as the Brønsted base (His versus Asp) with the ‘chemical' nature of the acceptor nucleophile or its p_K_a.
The structure of _Vv_Gt1 was also determined in its non-productive complex with UDP and the acceptor quercetin. All the interactions of UDP and acceptor are conserved in this complex, with an additional solvent-mediated interaction to the main-chain carbonyl of Glu189 (Figure 4B). With the sole exception of that interaction, quercetin and kaempferol bind in identical positions, as might be expected for closely related acceptors. Together, the two acceptor structures provide the basis for understanding the range of activities shown by _Vv_GT1 towards different natural plant flavonoids. Compared to the natural anthocyanidin substrates, including cyanidin, delphinidin and pelagonidin, the relative rates towards other flavonoids including quercetin and kaempferol are around 100 times lower (Ford et al, 1998). The key structural difference between these two classes of substrates is the presence or absence of a carbonyl group at C4 (Figure 1B). The structure reveals that an O4 carbonyl group on the acceptor would form a partial steric hindrance to nucleophilic attack at the anomeric centre. Furthermore, changes in the electron distribution and any internal H-bond from O3 to O4 might further raise the p_K_a of the O3 proton and hinder reactivity. At low pH (such as in red wine and the plant vacuole where these compounds accumulate), anthocyanidins exist as their coloured flavylium cation (Figure 1A). In these circumstances, the positive charge would lower the p_K_a of the O3 hydroxyl proton thus rendering O3 more nucleophilic, although in the cytoplasm this change in reactivity is unlikely to occur.
Discussion
The structure of _Vv_GT1, in complex with both intact UDP-sugar donor and flavonoid acceptors, provides the basis for identifying the key residues that interact with these ligands. In turn, this informs comparison of the _Vv_GT1 sequence with the sequences of other GT1 enzymes identified both functionally and from plant and mammalian genomic data. For example, of particular importance is the identification of the three residues in _Vv_GT1, Asp374, Gln375 and Thr141, which interact directly with the hydroxyl groups of the glucose moiety of UDP-Glc. Two GT1s of the 78D clade (approximately 60% identical in sequence to the _Vv_GT1 enzyme) of A. thaliana, characterized with respect to sugar-donor specificity, are informative in this context. UGT78D2 is a UDP-Glc transferase (Lim et al, 2004) and indeed, the residues interacting with the glycosyl moiety are invariant between 78D2 and _Vv_GT1. In contrast, we have shown here that _Vv_GT1 does not transfer Rha from UDP-Rha. Arabidopsis UGT78D1 is a UDP-rhamnose:flavonol-3-_O_-rhamnosyltransferase (Jones et al, 2003) (which in plants requires the donor UDP-β-L-rhamnose (Figure 5B), rather than the _d_TDP-Rha donor used by other organisms). In UGT78D1 the threonine residue in _Vv_GT1, that interacts with the O6 of glucose, is replaced with alanine, consistent with the binding of the 6-deoxy-sugar rhamnose. Further speculation of the likely position of the donor sugar, in the case of rhamnose, is clouded by conformational questions. In solution, UDP-β-L-rhamnose exists in a 1_C_4 chair conformation with an equatorial UDP. For nucleophilic attack during catalysis however, an axial leaving-group orientation is expected, which demands distortion of the sugar ring away from its solution form to one of a number of possible alternative ring conformations (Davies et al, 2003). Such distortion necessarily changes the axial versus equatorial nature of the hydroxyl groups and indeed may explain the, counter-intuitive, observation that Asp374 is invariant between the 78D clade glucosyl and rhamnosyltransferases despite the differences in configuration at the O3 and O4 of their respective sugar donors.
Specificity for galactose is also a confusing area. Many of the characterized plant GT1 enzymes such as the Aralia cordata ACGaT and the Scutellaria baicalensis UBGT (Kubo et al, 2004) and _Vv_GT1 (this work), will tolerate both UDP-Glc and UDP-Gal. _Vv_GT1 is able to transfer Gal, from UDP-Gal, with approximately 8% of the efficiency (_k_cat/_K_m) that it harnesses UDP-Glc (although _k_cat is reduced some 200 fold). In the _Vv_GT1 structure, Gln375 interacts with both O2 and O3 hydroxyls of glucose and Asp374 with O4. There is insufficient data to divine which plant enzymes will favor UDP-Gal, although some authors ascribe this to a histidine residue at the O2/O3 interacting position (Kubo et al, 2004). As with work on the A. cordata enzyme, the Q375H mutation of _Vv_GT1 does not confer improved UDP-Gal transfer activity; indeed this mutation in _Vv_GT1 similarly reduces activity over 300-fold. It is thus likely that sugar specificity of GTs will reflect a subtle interplay of stereochemistry and ‘on-enzyme' ring conformation of the donor sugars as they approach the transition-state, as is known to be the case with glycoside hydrolases (Ducros et al, 2002; Davies et al, 2003). In this latter context, knowledge of how enzyme structure dictates the conformational itinerary of the sugars has led to the design of powerful and specific glycosidase inhibitors for specific enzymes. Given the importance of GTs in many disease and infection processes, one would hope that future consideration of the conformational itineraries of GTs may similarly play a part in inhibitor design.
Plant GTs have attracted considerable interest, over many years, for their diverse roles in natural product metabolism (recently reviewed in Lim, 2005). Increasingly, enzymes of this class have become recognized as key components of cellular homeostasis in both the plant and animal kingdoms. Glycosylation is a major mechanism influencing the activity of plant hormones and secondary metabolites and is the mechanism by which environmental toxins are handled by plants. Elucidation of the 3-D structure of a representative of this class now provides the opportunity to inform the design of novel biocatalysts for the synthesis and modification of plant natural products.
Materials and methods
Expression and Purification of the UDP-glucose: flavonoid 3-O-glucosyltransferase, VvGT1
The gene encoding _Vv_GT1 was expressed in E. coli B834 (DE3) cells as described previously (Ford et al, 1998). Prior to crystallization, the histidine affinity tag was removed by thrombin digest, at a ratio of 0.05 units thrombin per mg protein. Selenomethionine-containing _Vv_GT1 was prepared using a similar protocol, following transformation into E.coli strain BL21 (DE3) with SeMet incorporated by metabolic inhibition using standard procedures.
GAR screening and kinetics
GAR screening was performed as described by Yang et al (2005a). Briefly, in a 96-well plate, each well was filled with TRIS buffer (1.0 mM, pH 7.8, 150 μl), donor (10 mM, 2 μl), acceptor (10 mM, 2 μl) and enzyme (5 mg/ml, 5 μl). Solutions and the plate were incubated at 37°C for 16 h and the solutions were analysed by LCMS monitoring the formation of product. Single substrate concentration kinetics were also performed by mass spectrometry, at 25°C with both donor and acceptor fixed at 100 μM by LCMS monitoring the formation of product through the appearance of corresponding ions and relating this to the total ion count with concentration determined through internal calibration. NDP donor substrates were synthesized using a combination of chemical and chemoenzymatic methods using either Gal-1-P uridyltransferase (Errey et al, 2004) or chemical pyrophosphorylation and deprotection (Yang et al, 2005b). UDP-α,β-Rha was synthesized in 19% overall yield from Rha by phosphorylation of Rha-2,3,4-_O_-triacetate with _O_-phenylene phosphorochloridate, deprotection, elaboration to UDP-Rha and purification (full details will be published in due course). Pseudo-single substrate kinetics were also performed by mass spectrometry, at 25°C with acceptor fixed at 100 μM and donor varied at different concentrations, typically from 25 to 2000 μM (i.e. up to 3.5 times _K_m). Product formation was monitored by LCMS through the appearance of corresponding ions and relating this to the total ion count with concentration determined through internal standard, GDP (100 μM, or UDP if GDPGlc as donor).
Crystallization and data collection
_Vv_GT1 was crystallized, initially in the presence of 20 mM UDP-Glc and 0.5 mM kaempferol using the hanging drop method with 4.8 mg ml−1 protein in 5 mM Tris (2-carboxyethyl) phosphine HCl, 10 mM Tris–Cl, pH 8.3 diluted 1:1 with mother liquor which contained 18–22% polyethylene glycol 10 000 (Fluka), 0.1 M Bis-tris propane, pH 7.0 and 0–0.5% v/v Pluronic F-68 (Hampton Research). Crystals appeared within 24 h. Crystals were harvested into nylon-fibre CryoLoops™ (Hampton Research) and transferred to a cryo-protectant solution, consisting of the mother liquor supplemented with ethylene glycol (to 25% v/v) prior to freezing in liquid nitrogen. X-ray diffraction data were collected from single crystals at 100 K. All data were collected at the European Synchrotron Radiation Facility (ESRF); ‘native' derived from the UDP-Glc plus kaempferol complex to 1.9 Å resolution on beamline ID 14.3 and the SeMet data to 2.06 Å resolution, on beamline ID29. All computational analysis used programs from the CCP4 suite (Collaborative Computational Project Number 4, 1994), unless explicitly stated. Data were processed using MOSFLM (Leslie, 1992) and scaled and reduced using SCALA, both from the CCP4 suite. Native and SeMet crystals are in space group P212121 with approximate unit cell dimensions _a_=49.1 Å, _b_=93.1 Å, _c_=106 Å, α=β=γ=90°. The structure was solved using single wavelength anomalous dispersion (SAD) methods, at the _f_″ peak wavelength (0.97930 Å, judged from the X-ray fluorescence scan), from the Se-Met derived version of the protein. SeMet data were 99% complete to 2.2 Å with _R_merge 0.078 (0.25 in 2.3–2.2 Å outer resolution bin) with a multiplicity of 7.2 (7.3) observations per reflection and a mean I/σI of 18 (8 in outer bin). A total of 11 Se sites were found using the programme SOLVE (version 2.08) (Terwilliger and Berendzen, 1999) using the SeMet data only and phases further improved using RESOLVE (Terwilliger, 2000). This yielded an easily interpretable map. ARP-wARP (Perrakis et al, 1999) was used to automatically build the sequence into electron-density using RESOLVE phases with native amplitudes (to 1.9 Å). Manual correction of the model and insertion of solvent was performed using COOT (Emsley and Cowtan, 2004). The structure was refined using REFMAC (Murshudov et al, 1997) (Table II).
Table 2.
Data collection, phasing and refinement statistics for SAD (SeMet) structure determination and complex structure refinement
| | UDP | UDP-2FGlc plus kaempferol | UDP+quercetin | SeMet | | | --------------------------------------------------- | ------------------------- | ----------------- | ----------------- | ----------------- | | Data collection | | | | | | Space group | P212121 | P212121 | P212121 | P212121 | | Cell dimensions | | | | | | a, b, c (Å) | 49.1, 93.1, 106.0 | 49.1, 93.1, 106.0 | 49.2, 93.8, 106.1 | 49.1, 93.1, 106.0 | | α, β, γ (deg) | 90,90,90 | 90,90,90 | 90,90,90 | 90,90,90 | | | | | | | | | | | | | Peak | | | Wavelength | 0.97930 | 0.97900 | 0.93100 | 0.97930 | | Resolution (Å) | 40–1.9 | 40–1.9 | 40–2.1 | 40–2.2 | | R_merge | 0.09 (0.47)* | 0.085 (0.33) | 0.111 (0.41) | 0.078 (0.23) | | I/σ_I | 13 (3.3) | 13 (2.8) | 12.1 (3.0) | 18 (8) | | Completeness (%) | 100 (100) | 99 (98) | 99.9 (100) | 99 (98) | | Redundancy | 5.4 (5.5) | 3.9 (3.8) | 4.4 (4.5) | 7.2 (7.3) | | | | | | | | | Refinement | | | | | | Resolution (Å) | 40–1.9 | 40–1.9 | 40–2.1 | Not refined | | No. of reflections | 37, 371 | 36, 474 | 27, 870 | | | _R_work/_R_free | 0.19/0.23 | 0.19/0.23 | 0.19/0.23 | | | No. of atoms | | | | | | Protein | 3373 | 3429 | 3395 | | | Ligand/ion | 44 | 57 | 46 | | | Water | 371 | 306 | 229 | | | _B_-factors | | | | | | Protein | 22 | 27 | 27 | | | Ligand/ion | 17 (UDP) | 27 (UDP2FGlc) | 24 (UDP) | | | | 40 (Bis-Tris-Propane) | 34 (Kaempferol) | 30 (Quercetin) | | | | Water | 34 | 38 | 33 | | | r.m.s deviations | | | | | | Bond lengths (Å) | 0.017 | 0.017 | 0.016 | | | Bond angles (deg) | 1.5 | 1.6 | 1.5 | | | PDB codes | 2C1X | 2C1Z | 2C9Z | | | *Highest resolution shell is shown in parenthesis. | | | | |
VvGT1 complexes with UDP-2FGlc/kaempferol and UDP/quercetin
Uridine-5′-diphospho-2′-fluoro-glucose (UDP-2FGlc) was synthesized as described in (Gibson et al, 2004). Crystals of _Vv_GT1 complexed with UDP-2FGlc and kaempferol were obtained by co-crystallization with seeding, using the Hanging Drop Method. UDP-2FGlc and kaempferol were added to _Vv_GT1 (4.8 mg/ml in 5 mM TCEP, 10 mM Tris–HCl pH 8.3) to yield final concentrations of 10 mM and 0.5 mM respectively. The protein–ligand sample was mixed with the crystallization-well solution in a 1:1 volume ratio to form the droplet. The well contained 22% v/v PEG (polyethylene glycol) 10 000, 0.1 M Bis-tris propane pH 7.0. Crystallization trays were left at 18°C, and crystals grew within 3 days. The crystals were harvested as above, using a cryo-protectant solution consisting of 22% (v/v) PEG 10 000, 0.1 M Bis-tris propane pH 7.0, 25% (v/v) ethylene glycol, 10 mM UDP-2-fluoro-glucose, 0.5 mM kaempferol. Individual single crystals were mounted as above and data to 1.9 Å resolution collected at the European Synchrotron Radiation Source, beamline ID23 and processed as above. Crystals were sufficiently isomorphous with the UDP-bound structure and so this latter model was used as the starting model for refinement with REFMAC (Murshudov et al, 1997) (with the same reflections maintained for free R calculations). Model-building was performed with COOT (Emsley and Cowtan, 2004). Following refinement of the protein coordinates alone, ligand structures were built into 2Fo-Fc and Fo-Fc electron density maps and refined using stereochemical libraries derived from the CCP4 suite. Water molecules were added automatically using COOT and verified manually. Crystals with 20 mM UDP and quercetin were obtained as for the above but with UDP in place of UDP-2FGlc and quercetin in place of kaempferol. Data were collected on ESRF beamline ID23 and processed and refined exactly as above. All structure and data statistics are given in Table II.
Acknowledgments
We thank The Biotechnology and Biological Sciences Research Council and The Garfield-Weston Foundation for funding. GJD is a Royal Society University Research Fellow. We also thank Professor CJ Schofield and Dr Richard Welford for samples of 2 trans-dihydroquercetin and cyanidin chloride.
Note Added in Proof During the refereeing of this manuscript, Shao and colleagues reported the structure of a plant glycosyltransferase from family GT1: the Medicago truncatula triterpene/flavonoid glycosyltransferase (Shao et al, 2005). This enzyme shares many of the structural features of the _Vitis vinifera Vv_GT1 enzyme described in this publication.
References
- Bowles D, Isayenkova J, Lim E-K, Poppenberger B (2005) Glycosyltransferases: managers of small molecules. Curr Opin Plant Biol 8: 254–263 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The _CCP_4 suite: programs for protein crystallography. Acta Crystallograph D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Couthino P, Deleury E, Davies GJ, Henrissat B (2003) An evolving heirarchical family classification for glycosyltransferases. J Mol Biol 328: 307–317 [DOI] [PubMed] [Google Scholar]
- Davies GJ, Ducros VM-A, Varrot A, Zechel DL (2003) Mapping the conformational itinerary of β-glycosidases by X-ray crystallography. Biochem Soc Trans 31: 523–527 [DOI] [PubMed] [Google Scholar]
- Davies GJ, Gloster TM, Henrissat B (2005) Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr Op Struct Biol 15: 637–645 [DOI] [PubMed] [Google Scholar]
- Ducros V, Zechel DL, Murshudov GN, Gilbert HJ, Szabo L, Stoll D, Withers SG, Davies GJ (2002) Substrate distortion by a β-mannanase: Snapshots of the Michaelis and covalent intermediate complexes suggest a B2,5 conformation for the transition-state. Angewandte Chemie Int Edn 41: 2824–2827 [DOI] [PubMed] [Google Scholar]
- Dutton GJ (1980) Glucuronidation of Drugs and Other Compounds. Boca Raton, FL: CRC [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallograph D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Errey JC, Mukhopadhyay B, Kartha KPR, Field RA (2004) Flexible enzymatic and chemo-enzymatic approaches to a broad range of uridine-diphospho-sugars. Chem Commun 2706–2707 [DOI] [PubMed] [Google Scholar]
- Esnouf RM (1997) An extensively modified version of MolScript that includes greatly enhanced colouring capabilities. J Mol Graph 15: 133–138 [DOI] [PubMed] [Google Scholar]
- Flint J, Taylor E, Yang M, Bolam DN, Tailford LE, Martinez-Flietes C, Dodson EJ, Davis BG, Gilbert HJ, Davies GJ (2005) Structural dissection and high-throughput screening of mannosylglycerate synthase. Nat Struct Mol Biol 12: 608–614 [DOI] [PubMed] [Google Scholar]
- Ford CM, Boss PK, Høj PB (1998) Cloning and characterization of Vitis vinifera UDP-glucose: flavonoid 3-_O_-glucosyltransferase, a homologue of the enzyme encoded by the maize Bronze-1 locus that may primarily serve to glucosylate anthocyanidins in vivo. J Biol Chem 273: 9224–9233 [DOI] [PubMed] [Google Scholar]
- Fulcrand H, Atasanova V, Salas E, Cheynier V (2004) The fate of anthocyanins in wine: are there determining factors? In Red Wine Color, Waterhouse AL, Kennedy JA (eds), pp 68–88. Washington, DC: American Chemical Society [Google Scholar]
- Gibson R, Tarling CA, Roberts S, Withers SG, Davies GJ (2004) The donor subsite of trehalose-6-phosphate synthase: binary complexes with UDP-glucose and UDP-2-deoxy-2-fluoro glucose at 2 Å resolution. J Biol Chem 279: 1950–1955 [DOI] [PubMed] [Google Scholar]
- Graham TL (1988) Flavonoid and flavonol glycoside metabolism in Arabidopsis. Plant Physiol Biochem 36: 135–144 [Google Scholar]
- Halliwell B, Rafter J, Jenner A (2005) Health promotion by flavonoids, tocopherols, tocotrienols, and other phenols: direct or indirect effects? Antioxidant or not? Am J Clin Nutr 81: 268S–276S [DOI] [PubMed] [Google Scholar]
- Harborne JB, Willams CA (2001) Anthocyanins and other flavonoids. Nat Prod Rep 18: 310–333 [DOI] [PubMed] [Google Scholar]
- Hu YN, Chen L, Ha S, Gross B, Falcone B, Walker D, Mokhtarzadeh M, Walker S (2003) Crystal structure of the MurG: UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proc Natl Acad Sci USA 100: 845–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YN, Walker S (2002) Remarkable structural similarities between diverse glycosyltransferases. Chem Biol 9: 1287–1296 [DOI] [PubMed] [Google Scholar]
- Jones P, Messner B, Nakajima JI, Schaffner AR, Saito K (2003) UGT73C6 and UGT78D1, glycosyltransferases involved in flavonol glycoside biosynthesis in Arabidopsis thaliana. J Biol Chem 278: 43910–43918 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 24: 946–950 [Google Scholar]
- Kubo A, Arai Y, Nagashima S, Yoshikawa T (2004) Alteration of sugar donor specificities of plant glycosyltransferases by a single point mutation. Arch Biochem Biophys 429: 198–203 [DOI] [PubMed] [Google Scholar]
- Leslie AGW (1992) Recent changes to the MOSFLM package for processing film and image plate data. In Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography, Vol. 26, Warrington, UK: Daresbury Laboratory [Google Scholar]
- Lim E-K (2005) Plant glycosyltransferases: their potential as novel biocatalysts. Chem Eur J 11: 5486–5494 [DOI] [PubMed] [Google Scholar]
- Lim EK, Ashford DA, Hou BK, Jackson RG, Bowles DJ (2004) Arabidopsis glycosyltransferases as biocatalysts in fermentation for regioselective synthesis of diverse quercetin glucosides. Biotechnol Bioeng 87: 623–631 [DOI] [PubMed] [Google Scholar]
- Lim EK, Baldauf S, Li Y, Elias L, Worrall D, Spencer SP, Jackson RG, Taguchi G, Ross J, Bowles DJ (2003) Evolution of substrate recognition across a multigene family of glycosyltransferases in Arabidopsis. Glycobiology 13: 139–145 [DOI] [PubMed] [Google Scholar]
- Lim EK, Bowles DJ (2004) A class of plant glycosyltransferases involved in cellular homeostasis. EMBO J 23: 2915–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus SL, Polakowski R, Seto NOL, Leinala E, Borisova S, Blancher A, Roubinet F, Evans SV, Palcic MM (2003) A single point mutation reverses the donor specificity of human blood group B-synthesizing galactosyltransferase. J Biol Chem 278: 12403–12405 [DOI] [PubMed] [Google Scholar]
- Mulichak AM, Losey HC, Lu W, Wawrzak Z, Walsh CT, Garavito RM (2003) Structure of the TDP-epi-vancosaminyltransferase GtfA from the chloroeremomycin biosynthetic pathway. Proc Natl Acad Sci USA 100: 9238–9243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulichak AM, Losey HC, Walsh CT, Garavito RM (2001) Structure of the UDP-Glucosyltransferase GtfB that modifies the heptapeptide aglycone in the biosynthesis of vancomycin group antibiotics. Structure 9: 547–557 [DOI] [PubMed] [Google Scholar]
- Mulichak AM, Lu W, Losey HC, Walsh CT, Garavito RM (2004) Crystal structure of vancosaminyltransferase GtfD from the vancomycin biosynthetic pathway: Interactions with acceptor and nucleotide ligands. Biochemistry 43: 5170–5180 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallograph D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Patenaude SI, Seto NOL, Borisova SN, Szpacenko A, Marcus SL, Palcic MM, Evans SV (2002) The structural basis for specificity in human ABO(H) blood group biosynthesis. Nat Struct Biol 9: 685–690 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Ross JA, Kasum CM (2002) Dietary flavonoids: bioavailability, metabolic effects, and safety. Annu Rev Nutr 22: 19–34 [DOI] [PubMed] [Google Scholar]
- Shao H, He X, Achnine L, Blount JW, Dixon RA, Wang X (2005) Crystal structures of a multifunctional triterpene/flavanoid glycosyltransferases from Medicago truncatula. Plant Cell 17: 3141–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbouriech N, Charnock SJ, Davies GJ (2001) Three-dimensional structures of the Mn and Mg dTDP complexes of the family GT-2 glycosyltransferase SpsA: a comparison with related NDP-sugar glycosyltransferases. J Mol Biol 314: 655–661 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr, D 56: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallograph Sect D—Biol Crystallogr 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukey R, Strassburg C (2000) Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol 40: 581–616 [DOI] [PubMed] [Google Scholar]
- Vrielink A, Rüger W, Driessen HPC, Freemont PS (1994) Crystal structure of the DNA modifying enzyme β-glucosyltransferase in the presence and absence of the substrate uridine diphosphoglucose. EMBO J 13: 3413–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Brazier M, Edwards R, Davis BH (2005a) High-throughput mass spectrometry monitoring for multi-substrate enzymes: determining the kinetic parameters and catalytic activities of glycosyltransferases. ChemBioChem 6: 346–357 [DOI] [PubMed] [Google Scholar]
- Yang M, Proctor MR, Bolam DN, Errey JC, Field RA, Gilbert HJ, Davis BG (2005b) Probing the breadth of macrolide glycosyltransferases: in vitro remodeling of a polyketide antibiotic creates active bacterial uptake and enhances potency. J Am Chem Soc 127: 9336–9337 [DOI] [PubMed] [Google Scholar]