Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence (original) (raw)
Abstract
The human T-lymphotropic virus type 1 (HTLV-1) Tax binds the anaphase promoting complex (APC) and activates it ahead of schedule. Here, we show that APC activation by Tax induces rapid senescence (tax_-IRS) independently of p53 and pRB. In response to tax, cyclin A, cyclin B1, securin, and Skp2 becomes polyubiquitinated and degraded starting in S phase. This is followed by a surge in p21_CIP1/WAF1 and p27_KIP1_ in mid to late S and G2/M leading to a permanent G1 arrest. Tax-positive HTLV-1-transformed T-cell lines express elevated levels of p21_CIP1/WAF1_, but low levels of p27_KIP1_. Finally, Tax can be stably expressed in p27_KIP1_-null NIH3T3 cells. These results indicate that APC activation by Tax causes inactivation of SCFSkp2 and stabilization of p21_CIP1/WAF1_ and p27_KIP1_. The build-up of p21_CIP1/WAF1_ and especially p27_KIP1_ commits cells to senescence. Evading tax_-IRS through a loss of p27_KIP1 function is likely to be critical for cell transformation by Tax and development of adult T-cell leukemia after HTLV-1 infection. Finally, activation of APC ahead of schedule may be exploited to arrest cancer cell growth.
Keywords: anaphase promoting complex, cyclin-dependent kinase inhibitor, HTLV-1 Tax, senescence, Skp1-Cullin-F-box
Introduction
Human T-lymphotropic virus type I (HTLV-1) is the etiological agent of adult T-cell leukemia/lymphoma (ATL) and a neurological disorder called HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). How HTLV-1 infection progresses to T-cell malignancy and HAM/TSP is not well understood but is thought to involve the viral transactivator/oncoprotein, Tax. The effects that Tax exerts on cells are pleiotropic, and include potent NF-κB activation, cell cycle perturbation, and cell transformation. How Tax influences ATL development is incompletely understood. A significant fraction of ATL cells contain deletions in HTLV-1 proviral DNAs. In these cells, tax coding sequence is preferentially retained, implicating its role in ATL development (Korber et al, 1991). ATL cells in general do not express HTLV-1 sequence, suggesting that tax likely affects the early stage of the disease and persistent tax expression is not needed for maintenance of the neoplasm (Franchini et al, 1984). This distinguishes tax from viral oncogenes such as the human papilloma virus E6 and E7 whose constitutive expression is needed for cell transformation.
Tax activates viral transcription by interacting with CREB/ATF-1 and by recruiting transcriptional co-activators, CBP/p300, to three CRE-containing 21-bp repeat enhancer elements in the viral long terminal repeat. The activation of NF-κB by Tax is a result of the constitutive activation of I-κB kinase (IKK) by Tax, due in part to a direct interaction between Tax and the γ-subunit of IKK, IKKγ (Chu et al, 1998; Yamaoka et al, 1998; Jin et al, 1999; Sun and Ballard, 1999; Xiao and Sun, 2000). Recent data indicate that via a tripartite interaction, Tax, protein phosphatase 2A (PP2A), and IKKγ form a stable ternary complex, wherein PP2A activity is inhibited or diminished by Tax (Fu et al, 2003), thereby maintaining IKK in a phosphorylated and active state.
Tax perturbs critical steps in cell cycle progression (see Jeang et al (2004) for a review). Some of these effects of Tax are thought to lead to cell transformation. We have recently observed that Tax causes a reduction in the levels of cyclin B1 and securin prior to mitosis (Liu et al, 2003). This activity of Tax is seen in both Saccharomyces cerevisiae and HeLa cells. Analyses of S. cerevisiae mutants have indicated that the Cdc20-anaphase promoting complex (APCCdc20) mediates this function of Tax (Liu et al, 2003). APCCdc20 is an E3 ubiquitin ligase that becomes active during mitosis, and controls metaphase to anaphase transition by targeting the destruction of cyclin A, securin, and cyclin B1. Recent data indicate that Tax directly binds and activates APC during S phase (Liu et al, 2005), causing polyubiqutination and degradation of cyclin A, cyclin B1, and securin before the onset of M phase (Liu et al, 2005). The Tax-induced loss of mitotic regulators is associated with delay in cell cycle progression, DNA aneuploidy, and formation of micro-, bi-, and multinucleated cells (Liang et al, 2002; Liu et al, 2003).
We now show that the cell cycle dysregulation induced by tax does not end with mitotic abnormalities. HeLa cells transduced with tax, after passage through a faulty cell division cycle, immediately became arrested with G1 DNA content (termed G1 arrest or tax_-induced rapid senescence, tax_-IRS herein). They expressed high levels of Cdk2 inhibitors: p21_CIP1/WAF1 and p27_KIP1 and displayed phenotypes indistinguishable from cells in senescence (Dimri et al, 1995). Expression of tax during G1 or early G1/S does not lead immediately to a cessation of the cell cycle; rather, the senescence caused by Tax depends upon transit through S/G2/M wherein a dramatic and persistent rise in p21_CIP1/WAF1_ and p27_KIP1_ occurs in mid to late S, G2/M, leading up to the arrest.
The destruction of p21_CIP1/WAF1_ and p27_KIP1_ occurs during S phase and is regulated by the multisubunit E3 ubiquitin ligase, SCF (Skp-Cullin-F box), in association with its substrate-targeting subunit, Skp2 (Carrano et al, 1999; Nakayama et al, 2000, 2004; Bornstein et al, 2003; Bashir et al, 2004; Wei et al, 2004) and the cell cycle regulatory protein, Cks1 (Ganoth et al, 2001; Bashir et al, 2004; Wei et al, 2004). We found that in tax_-expressing cells, Skp2 also became polyubiquitinated and degraded during S phase. The decline in Skp2 appears to cause inhibition of SCFSKP2 and stabilization of p21_CIP1/WAF1 and p27_KIP1_, thereby committing HeLa cells to rapid senescence. Transcriptional activation of p21_CIP1/WAF1_ by Tax has been previously reported (Akagi et al, 1996; Chowdhury et al, 2003; de la Fuente et al, 2003; Kawata et al, 2003). Despite abundant Tax and p21_CIP1/WAF1_ expression, HTLV-1-transformed T cells grow and proliferate normally. They invariably express low levels of p27_KIP1_ mRNA and protein, in contrast to HeLa cells newly transduced with tax. The difference in the p27_KIP1_ status in HTLV-1-transformed cells and tax_-transduced HeLa cells strongly implicates loss or inactivation of p27_KIP1 as a critical step in T-cell transformation by Tax and HTLV-1. Indeed, Tax could be stably expressed in p27_KIP1_-null NIH3T3 cells but not in p21_CIP1/WAF1_-null cells. We propose that a loss of p27_KIP1_ functions is necessary for _tax_-expressing cells to evade _tax_-IRS. This allows the transforming properties of Tax, including potent activation of IKK-NF-κB, activation of Cdk4, and induction of chromosome instability to take effect, leading to cell transformation and ATL.
Results
Tax-transduced HeLa cells become permanently G1-arrested
We have shown recently that Tax directly interacts with APC and activates it in an unscheduled manner, causing the polyubiquitination and degradation of cyclin A, cyclin B1, and securin in S. cerevisiae, HeLa, and human T cells before the onset of mitosis (Liu et al, 2003, 2005). The premature APC activation by Tax is accompanied by a delay in S/G2/M progression, and severe mitotic abnormalities including DNA aneuploidy, cytokinesis failure, and formation of micro-, bi-, and multinucleated cells (Liang et al, 2002; Liu et al, 2003).
To explore the biological effects of tax further, we produced a tax lentivirus vector, LV-Tax, and a control green fluorescence protein vector, LV-GFP, using the HR′-CMV plasmid (Naldini et al, 1996). A packaging plasmid, pCMVΔR8.2Δ_vpr_, (kindly provided by Dr Irvin Chen) deleted for env and vpr, together with a VSV-G (vesicular stomatitis virus glycoprotein) expression plasmid were used for vector production. The deletion of vpr in pCMVΔR8.2Δ_vpr_ eliminated Vpr from vector particles and prevented Vpr-induced G2/M arrest and apoptosis. LV-Tax and LV-GFP particles were produced as in Materials and methods. Asynchronously growing HeLa cells were then transduced with LV-Tax or LV-GFP at a multiplicity of infection (m.o.i.) of 5. Interestingly, LV-Tax-transduced cell population ceased growing within 3–4 days, and accumulated many binucleated cells (marked with arrows, Figure 1 LV-Tax panel, see also Figure 3B). The LV-Tax cells were enlarged, granulated, and assumed a flattened morphology (Figure 1). By contrast, LV-GFP cells continued to proliferate (note the round and refractive mitotic cells) with typical HeLa morphology (Figure 1 LV-GFP panel).
Figure 1.
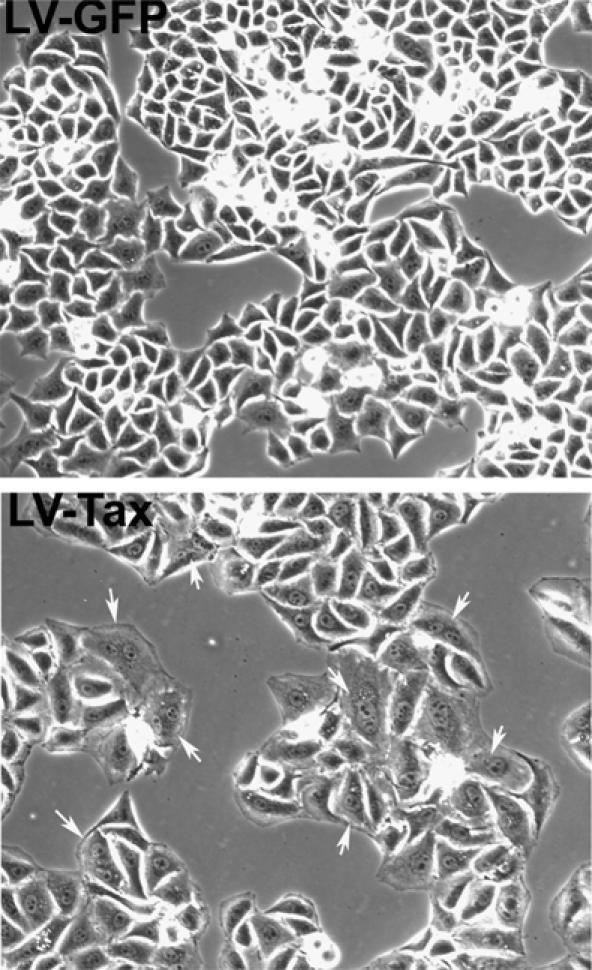
Altered morphology of HeLa cells transduced by LV-Tax. Asynchronously growing HeLa cells were plated at a density of 2.5 × 104 cells/well in six-well plates and transduced with LV-Tax or LV-GFP at an m.o.i. of 5, grown for 3 days, and photographed. Binucleated or micronucleated cells are marked with arrows.
Figure 3.
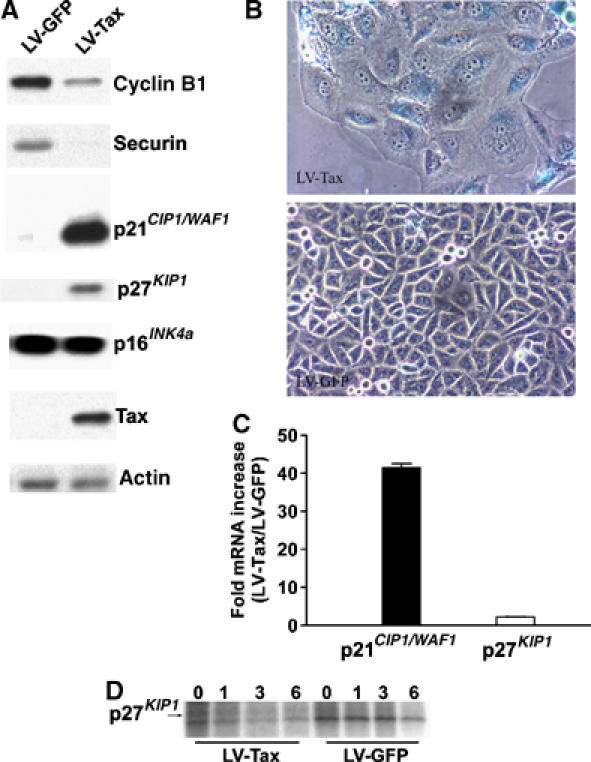
G1-arrested tax_-expressing cells are in senescence. (A) Immunoblots of HeLa cells transduced with LV-Tax or LV-GFP. Cell lysates were prepared and immunoblotted using cyclin B1, human securin (Securin), p21_CIP1/WAF1, p27_KIP1_, p16INK4a, Tax, and actin antibodies as described. (B) Expression of the senescence-associated β-galactosidase (SA-β-Gal) in HeLa cells transduced with LV-Tax. Asynchronously growing HeLa cells (2.5 × 104 cells/well in six-well plates) were transduced with LV-Tax or LV-GFP at an m.o.i. of 5, grown for 3 days, and stained with X-Gal overnight at 37°C (see Materials and methods). (C) Ratios of p21_CIP1/WAF1_ and p27_KIP1_ mRNA levels in LV-Tax- versus LV-GFP-transduced cells. Quantitative real-time RT–PCR was as in Materials and methods. Four RT–PCRs were carried out for each RNA species (p21_CIP1/WAF1_, p27_KIP1_, and β-actin) in LV-Tax and LV-GFP cells. The relative mRNA levels of p21_CIP1/WAF1_ and p27_KIP1_ were quantified by normalizing against that of β-actin. The fold increases of p21_CIP1/WAF1_ and p27_KIP1_ mRNAs (LV-Tax/LV-GFP) were then calculated and plotted. (D) Pulse chase of p27_KIP1_ in LV-Tax- versus LV-GFP-transduced HeLa cells. Pulse chase and immunoprecipitation were carried out as in Materials and methods. The times after chase (0, 1, 3, 6 h) are indicated.
To characterize further the LV-Tax cells, we examined their progression through successive cell cycles. HeLa cells were first synchronized at G1/S border with a double-thymidine block (DTB), released from the arrest for 1 h, and then infected with LV-Tax or LV-GFP. Because reverse transcription, like cellular DNA synthesis, is inhibited by the thymidine block, entry into S is necessary for lentivirus vector-mediated gene transduction. The LV-transduced cells were then collected at the indicated times after release for flow cytometry. The LV-GFP cells maintained synchrony within 62 h after release, and cycled normally (note the undulation of G1 populations in Figure 2A LV-GFP panel). In striking contrast, and consistent with microscopic observations (Figure 1), most LV-Tax cells reached G1 at or before 38 h after release and ceased cycling beyond that point (note the lack of undulation of G1 populations in Figure 2A LV-Tax panel). A portion of the LV-Tax cells (20%) also accumulated with 4N DNA content (Figure 2B, 40 & 56 h). The appearance of this cell population coincided with the emergence of binucleated LV-Tax cells (Figure 1).
Figure 2.
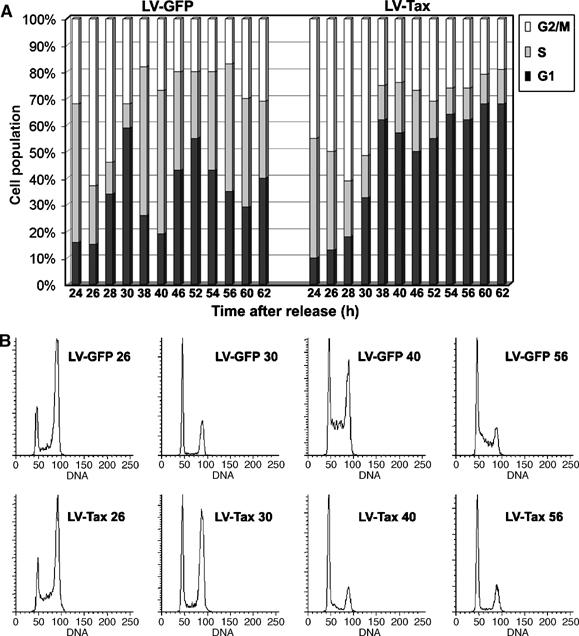
LV-Tax-transduced cells became G1-arrested after passage through an abnormal cell cycle. (A) Comparison of cell cycle progression of LV-GFP- and LV-Tax-transduced HeLa cells by flow cytometry. HeLa cells were first synchronized by DTB, released from the arrest for 1 h, and then infected with LV-Tax or LV-GFP at an m.o.i. of 5. The cells were then collected at the indicated times (24, 26, 28, 30, 38, 40, 46, 52, 54, 56, 60, and 62 h) after release for flow cytometry. The percentages of cells in G1 (solid), S (gray), and G2/M (open) phases of the cell cycle at a given time were computed using the ModFit LT software package. (B) Flow cytometry histogram of propidium iodide-stained HeLa cells transduced with LV-GFP or LV-Tax vector (samples collected at 26, 30, 40, and 56 h post-release are shown, ordinate: cell numbers; abscissa: DNA content).
As reported previously (Liu et al, 2003), LV-Tax-transduced cells showed a delay in G2/M progression prior to the G1 arrest, as evidenced by a considerable G2/M population at 30 h post-release, a time when most LV-GFP cells had exited mitosis (Figure 2B, compare the G2 populations at 26 and 30 h after release: LV-GFP 26 versus LV-Tax 26, and LV-GFP 30 versus LV-Tax 30). After the mitotic delay, the bulk of LV-Tax cells were arrested with G1 DNA content (Figure 2B compare LV-GFP 40 versus LV-Tax 40 and LV-GFP 56 versus LV-Tax 56). Based on the 16-h doubling time of control HeLa cells, we calculated the point of the cell cycle at which LV-Tax cells became arrested (at or prior to 38 h post-release) to be the second G1 phase after release from DTB. That LV-Tax cells became arrested at the second, but not the first G1 phase immediately after tax transduction is most likely because 12–24 h is needed for reverse transcription, integration, and tax expression. Indeed, HeLa cells transduced with Ad-Tax, an adenovirus vector that allows immediate tax expression after infection, arrested in the first G1 phase (Figure 5).
Figure 5.
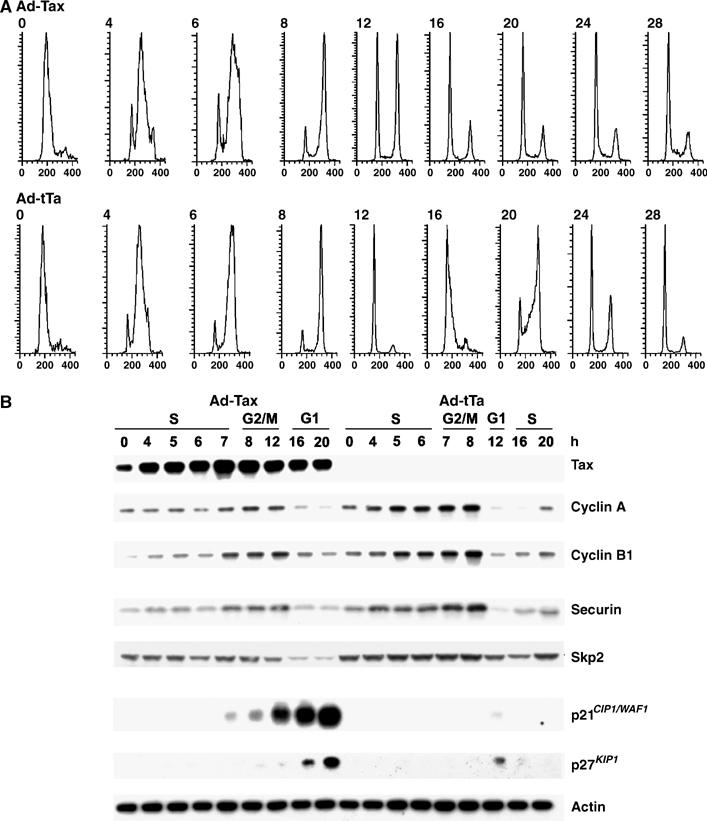
Unscheduled activation of the APC by Tax leads to a loss of Skp2 and stabilization of p21_CIP1/WAF1_ and p27_KIP1_. HeLa cells were synchronized and infected with Ad-Tax or Ad-tTa as in Figure 4. Cells were then collected at the indicated times after release for flow cytometry (0, 4, 6, 8, 12, 16, 20, 24, and 28 h) and immunoblotting (0, 4, 5, 6, 7, 8, 12, 16, and 20 h). (A) Flow cytometry of propidium iodide-stained HeLa cells transduced with Ad-Tax and Ad-tTa, respectively. Inclusion of later time points (24 and 28 h post-release) is made to highlight the G1 arrest of tax_-expressing cells. For each panel, the ordinate represents cell numbers; the abscissa, DNA content. (B) Immunoblots of cyclin A, cyclin B1, securin, Skp2, p21_CIP1/WAF1, p27_KIP1_, and actin. Cell lysates from Ad-Tax (left panels)- or Ad-tTa (right panels)-transduced cells were collected at the indicated times and immunoblotted. The stage of the cell cycle for each chosen post-release time point was determined based on the flow cytometry results in (A) and indicated at the top of the immunoblots.
Tax-induced G1 arrest is indistinguishable from cellular senescence
The phenotypes of tax_-transduced cells—cessation of cell proliferation, permanent G1 arrest with enlarged, granulated and flattened cell shape—resemble cellular senescence seen in aging cells, cells treated with chemotherapeutic chemicals (Chang et al, 1999), primary cells expressing activated form of ras (Serrano et al, 1997), and HeLa cells whose p53 and pRB pathways have been activated by a repression of HPV-18 (human papilloma virus type 18) E6 and E7 expression (Wells et al, 2000, 2003). To characterize the LV-Tax cells further, we determined their levels of p16_INK4a, p21_CIP1/WAF1_, and p27_KIP1_, CDK inhibitors, and stained them for the senescence-associated β-galactosidase (SA-β-Gal) (Dimri et al, 1995). The level of p16_INK4a_ in LV-Tax cells was similar to that of LV-GFP cells 3 days after transduction (Figure 3A). By contrast, the LV-Tax cells expressed prodigious amounts of p21_CIP1/WAF1_ and p27_KIP1_ (Figure 3A), and stained positive for the SA-β-Gal (Figure 3B). The levels of cyclin B1 and securin in LV-Tax cells were low or undetectable, consistent with their degradation induced by Tax and/or the cessation of cell proliferation. As anticipated, LV-GFP cells showed no sign of senescence (Figure 3B). The growth-arrested LV-Tax cells persisted in culture and remained metabolically active for at least 2 weeks (not shown). These results strongly suggest that immediately after an aberrant cell cycle, _tax_-transduced cells entered into senescence.
Tax has been reported to activate p21_CIP1/WAF1_ expression previously (Akagi et al, 1996; Chowdhury et al, 2003; de la Fuente et al, 2003; Kawata et al, 2003); however, transcriptional activation of p27_KIP1_ by Tax has not been observed (Cereseto et al, 1999). To confirm these data, we performed quantitative real-time RT–PCR on total RNA samples prepared from LV-GFP and LV-Tax cells 3 days after infection. Indeed, the mRNA level of p21_CIP1/WAF1_ in LV-Tax-transduced HeLa cells was 40-fold of that in LV-GFP-transduced control, while the level of p27_KIP1_ mRNA in LV-Tax cells was only two-fold than that of the control (Figure 3C). We infer that the great surge in p21_CIP1/WAF1_ level is due, in part, to activation of mRNA transcription by Tax, but the increase in p27_KIP1_ in LV-Tax cells cannot be accounted for by transcriptional activation alone. A pulse-chase experiment showed that the half-life of p27_KIP1_ in HeLa cells became greatly increased in the presence of Tax (Figure 3D). As indicated, p27_KIP1_ was barely detectable 1 h after chase in LV-GFP cells, but persisted at 6 h after chase in LV-Tax cells. Therefore, stabilization of p27_KIP1_, and possibly p21_CIP1/WAF1_, plays a critical role in their build-up in _tax_-transduced cells.
Tax-induced rapid, senescence (tax-IRS) occurs independently of p53 and pRB
Cellular senescence induced by the oncogenic ras is p53- and p16INK4a/pRB-mediated (Wells et al, 2000, 2003). The tax_-mediated G1 arrest/senescence observed here occurred in HPV-18-transformed HeLa cells whose p53 is targeted for degradation by the HPV-18 E6 (Scheffner et al, 1990), and whose pRB is inactivated by HPV-18 E7 (reviewed in Helt and Galloway, 2003). This suggests that tax_-induced rapid senescence (tax_-IRS) occurs in a p53- and pRB-independent manner. To rule out the possibility that Tax indirectly stabilized p53 or pRb in HeLa cells, we examined their protein levels throughout cell cycle progression. HeLa cells were again arrested at G1/S with DTB. At the start of the second thymidine treatment, cells were infected with Ad-Tax or control Ad-tTa (an adenovirus vector for tax or a control vector for tet-transactivator) for 16 h, released from the arrest, and blotted for p53, pRB, p21_CIP1/WAF1, and p27_KIP1. Despite a sharp rise in p21_CIP1/WAF1 and p27_KIP1_ in Ad-Tax cells (Figure 4), the levels of p53 in Ad-tTa- and Ad-Tax HeLa cells were low or undetectable throughout the cell cycle progression (not shown). The level of pRB in control Ad-tTa cells rose slightly during G2/M/G1 and fell during S. By contrast, pRB cycling was not observed in Ad-Tax cells, possibly due to the cessation of cell cycle activities caused by Tax. Importantly, the level of pRB in Ad-Tax-infected cells remained comparable to that in the Ad-tTa control (Figure 4). Finally, consistent with the data in Figure 3C, Tax greatly induced p21_CIP1/WAF1_ mRNA transcription but had only a minor effect on p27_KIP1_ mRNA levels (Figures 3C and 4B). The Tax-mediated increase in p27_KIP1_ is therefore exerted at the level of protein stabilization as indicated in Figure 3D. These results indicate that the rapid cellular senescence induced by Tax occurs in a p53- and pRB-independent manner, apparently via a mechanism distinct from that executed by oncogenic ras, and involves transactivation of p21_CIP1/WAF1_ and stabilization of both p21_CIP1/WAF1_ and p27_KIP1_. As expected, tax also induced rapid senescence in p53-null human SKOV ovarian cancer cells and proliferative arrest in S. cerevisiae (not shown).
Figure 4.
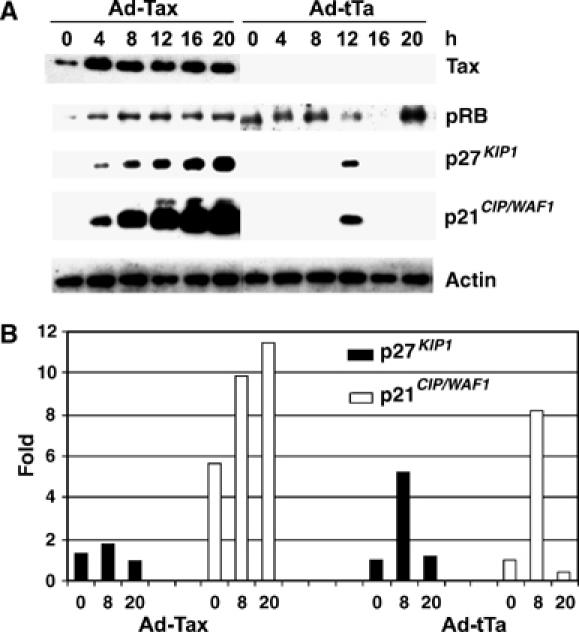
Tax-induced surge in p21_CIP1/WAF1_ and p27_KIP1_ occurs independently of p53 and pRB. (A) Immunoblots of HeLa cells infected with Ad-Tax or Ad-tTa. HeLa cells were synchronized as described for Figure 1. At the start of the second thymidine treatment, cells were infected with either Ad-Tax or Ad-tTa (negative control) at an m.o.i. of 5. After release, cells were collected at 0, 4, 8, 12, 16, and 20 h, and immunoblotted using Tax, p27_KIP1_, p21_CIP1/WAF1_, pRB, and β-actin antibodies. (B) Relative p27_KIP1_ and p21_CIP1/WAF1_ mRNA levels in Ad-Tax- versus Ad-tTa-transduced HeLa cells. Total cellular RNAs from samples at 0, 8, and 20 h post-release were prepared as in Materials and methods. RT–PCR was carried out as in Figure 3 and normalized against the p27_KIP1_ or p21_CIP1/WAF1_ mRNA level of Ad-tTa-transduced cells at 0 h post-release.
Premature APC activation by Tax is accompanied by p21CIP1/WAF1 and p27KIP1 increase
Temporally, the rise in p21_CIP1/WAF1_ and p27_KIP1_ tax_-expressing cells occurred in S/G2 phase (Figure 4). It paralleled premature APC activation by Tax (Liu et al, 2003, 2005). This prompted us to examine the connection between premature APC activation and the increase of p21_CIP1/WAF1 and p27_KIP1_. To this end, HeLa cells were again arrested and infected with Ad-Tax and Ad-tTa as above, released from the arrest, and analyzed. As reported (Liu et al, 2003) and indicated by flow cytometry, expression of tax during the G1/S block did not result in cell cycle arrest immediately (Figure 5A). Rather, Ad-Tax cells transited through S/G2/M with a 4–5 h delay (compare Ad-Tax and Ad-tTa at 12 h post-release) and then came to a complete stop at the next G1 (Figure 5A top panel, Ad-Tax, 16–28 h post-release). By contrast, Ad-tTa cells progressed normally through S and G2/M after release, and readily initiated another cell cycle (Figure 5A bottom panels, 16–20 h post-release). In control Ad-tTa cells, the levels of p21_CIP1/WAF1_ and p27_KIP1_ are cyclical—increased moderately during G1 (Figure 5B, Ad-tTa, 12 h post-release, see Figure 4 also), and became undetectable during S/G2/M (Figure 5B, 0–8 h post-release, and 16 and 20 h post-release). As expected, levels of cyclin A and B1, and securin in Ad-tTa cells cycled normally—rose sharply during S and declined abruptly after mitosis (Figure 5B right panels, compare 8 and 12 h post-release). In sharp contrast and as previously reported, the levels of cyclins A and B1, and securin in Ad-Tax cells increased but with a significant delay (Figure 5B left panels, compare 8 and 12 h post-release), and never achieved the levels seen in Ad-tTa cells. The reduction in cyclin A, cyclin B1, and securin during the S phase (0–7 h post-release) was concurrent with or was followed immediately by a rise in levels of p21_CIP1/WAF1_ and p27_KIP1_ (8–20 h post-release) during mid to late S, through G2/M and up to the subsequent G1 (Figure 5, also see Figure 4). Most likely as a result of the great surge in p21_CIP1/WAF1_ and p27_KIP1_, cell cycle progression, and cell proliferation ceased abruptly (see Figure 5A top four panels on the right and Figure 5B, Ad-Tax 12–20 h post-release). These temporal correlations raise the possibility that the unscheduled APC activation by Tax and the increase in p21_CIP1/WAF1_ and p27_KIP1_ may be causally linked or may have the same cause.
Unscheduled activation of APC by Tax correlates with a loss of Skp2 and a surge in the levels of p21CIP1/WAF1 and p27KIP1
The levels of p21_CIP1/WAF1_ and p27_KIP1_ are regulated through phosphorylation (by cyclinE/Cdk2), ubiquitination, and proteasome-mediated degradation (Swanson et al, 2000; Hara et al, 2001; Bornstein et al, 2003). The E3 ubiquitin ligase, SCF, together with its substrate-recognition subunit, Skp2, is responsible for the ubiquitination and degradation of p21_CIP1/WAF1_ and p27_KIP1_ (Ganoth et al, 2001; Bashir et al, 2004; Wei et al, 2004). Degradation of p27_KIP1_ also requires its presentation by the cyclin A–Cdk2 complex to SCFSkp2 (Zhu et al, 2004). The level of Skp2 oscillates in a cell cycle-dependent manner (see Figure 5B, Ad-tTa set, compare 8 and 12 or 16 h after release). Two recent reports (Bashir et al, 2004; Wei et al, 2004) showed that Skp2 and another SCF subunit, Cks1, are substrates of the Cdh1-associated APC (APCCdh1). Both become ubiquitinated and degraded in late M and early G1 when APCCdh1 is highly active. This renders SCF inactive and allows p21_CIP1/WAF1_ and p27_KIP1_ to accumulate transiently in G1 (also see Figure 5B Ad-tTa, 12 h post-release). The links between APCCdh1, SCFSKP2, and the levels of p21_CIP1/WAF1_ and p27_KIP1_, together with the data showing that Tax activates APC ahead of schedule (Liu et al, 2003, 2005) led us to examine the status of Skp2 in response to Tax. Consistent with the results previously reported (Bashir et al, 2004; Wei et al, 2004), in the Ad-tTa control, Skp2 accumulated during S phase and became degraded during G1, which was accompanied by a moderate and transient accumulation of p21_CIP1/WAF1_ and p27_KIP1_ (Figure 5B, Ad-tTa, 12 h post-release). By contrast, in parallel with the decrease in cyclin A, cyclin B1, and securin, the level of Skp2 was also reduced in Ad-Tax cells (Figure 5B, Ad-Tax, 4–12 h post-release). The Skp2 mRNA levels in Ad-Tax cells were approximately 80% of the Ad-tTA controls as quantitated by RT–PCR (not shown), indicating that the Tax-induced decrease in Skp2 protein is not due to transcriptional repression. Importantly, the highest level of Skp2 under tax_-positive condition (Figure 5B, Ad-Tax, S phase, 0–7 h post-release) is comparable to the lowest levels of Skp2 under tax_-negative condition (Figure 5B, Ad-tTa, G1, 12 and 16 h post-release). Finally, Skp2 declined further when the Ad-Tax cells eventually exited mitosis and entered into permanent G1 arrest (Figure 5B, Ad-Tax, G1, 20 h post-release and Figure 5A 20, 24, and 28 h post-release). These results strongly suggest that the loss of Skp2 and inactivation of SCFSKP2 occur as a consequence of APC activation by Tax. This in turn causes stabilization of p21_CIP1/WAF1 and p27_KIP1, committing cells to irreversible G1 arrest/senescence.
Tax activates polyubiquitination of Skp2 during S phase
To demonstrate that Tax directly cause polyubiquitination and degradation of Skp2, asynchronously growing HeLa cells were transfected with HA-tagged ubiquitin, CMV-HA-Ub, together with CMV-Flag-Skp2. On the second day, transfected cells were infected with Ad-Tax or Ad-tTa for 16 h and treated with the proteasome inhibitor, MG132, for 4 h. Cell lysates were then immunoprecipitated for Skp2 using the Flag-epitope antibody and immunoblotted with the HA antibody to detect the polyubiquitinated Skp2. Consistent with the idea that unscheduled activation of APC by Tax leads to Skp2 degradation, an increase in the level of polyubiquitinated Skp2 was detected in Ad-Tax cells (Figure 6B, left lane) versus Ad-tTa cells (Figure 6B, right lane). We next transfected HeLa cells with both CMV-HA-Ub and CMV-Flag-Skp2, and then synchronized them at G1/S border. During the second thymidine block, cells were infected with Ad-Tax or Ad-tTa, and then released into thymidine-free medium containing MG-132. Cell lysates were prepared at multiple time points after the release, immunoprecipitated with the Flag antibody, and immunoblotted with the HA antibody for polyubiquitinated Skp2. As shown in Figure 6A, at 4, 6, and 8 h after release when most cells were transiting through S phase, polyubiquitinated Skp2 was readily detected in the Ad-Tax cells (Ad-Tax, ‘+' lanes). This contrasts with the Ad-tTa control, wherein polyubiquitinated Skp2 became apparent only at 8 h post-release when cells started to transit mitosis (Ad-tTa, ‘−' lanes).
Figure 6.
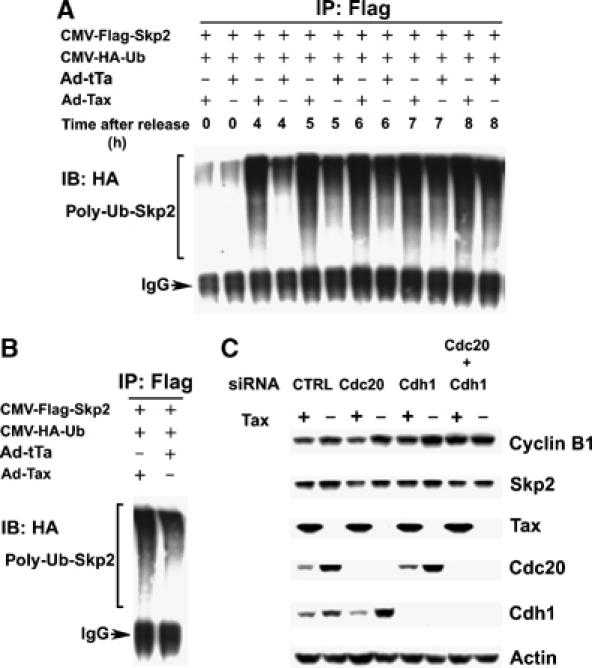
Tax induces polyubiquitination of Skp2 during S phase. (A) Plasmids encoding the HA-tagged ubiquitin (CMV-HA-Ub) and Flag-tagged Skp2 (CMV-Flag-Skp2) were tranfected into HeLa cells using the FuGENE 6 Transfection Reagent. The transfected cells were then synchronized with DTB. At the start of the second thymidine treatment, cells were also infected (at an m.o.i. of 5) with Ad-Tax or Ad-tTa as in Figure 4. The cells were then released (for 0, 4, 5, 6, 7, and 8 h) in complete DMEM containing 10 μM MG132. Immunoprecipitation (IP) of Flag-Skp2 was carried out for lysates collected at each time point (0, 4, 5, 6, 7, and 8 h) after release using the Flag epitope antibody, immunoprecipitates were resolved in an SDS/4–12% PAGE, and immunoblotted (IB) for the polyubiquitinated Skp2 (bracketed) using the HA antibody. (B) Experiments were similar as in (A) except that cells were not synchronized and were infected by Ad-Tax or Ad-tTa for 16 h, then transferred to DMEM containing 10 μM MG132 for 4 h. (C) APC activation by Tax is inhibited by siRNAs against Cdc20 and Cdh1. siRNA transfection was performed as in Materials and methods. Cells were harvest for immunoblots for cyclin B1, Skp2, Tax, Cdc20, Cdh1, and actin at 8 h post-release from the G1/S arrest. The siRNA species used are as indicated. Ad-Tax- and Ad-tTa-transduced cells are labeled as Tax + and −, respectively.
Previous results have shown that Tax binds to both APCCdc20 and APCCdh1 (Liu et al, 2005). Further, APCCdc20 appears to be sufficient for Tax-induced Clb2 degradation in S. cerevisiae (Liu et al, 2003). To determine if Tax targets APCCdc20 or APCCdh1 in HeLa cells, siRNA knockdown experiments were performed. HeLa cells were synchronized by DTB, and transfected with a control siRNA, a Cdc20 siRNA, a Cdh1 siRNA, or a combination of both Cdc20 and Cdh1 siRNAs 2 h before the second thymidine block, followed by infection with Ad-Tax or Ad-tTA. As shown in Figure 6C, the delay in cell cycle progression caused by Tax was reflected by a slower rise in levels of Cdc20 and Cdh1 after release from G1/S arrest. This notwithstanding, siRNA's directed against Cdc20 and Cdh1 were effective in reducing their expression respectively. Importantly, Tax-induced cyclin B reduction was inhibited partially by siRNA against Cdc20, and to a greater extent, by siRNA against Cdh1. The greatest inhibition was achieved when both Cdc20 and Cdh1 siRNA's were used, suggesting that both APCCdc20 and APCCdh1 were affected by Tax (Figure 6C). Finally, in agreement with the role of APCCdh1 in Skp2 degradation, siRNA knockdown of Cdh1 efficiently blocked the loss of Skp2 induced by Tax (Figure 6C).
HTLV-1 transformed T cells express greatly elevated levels of p21CIP1/WAF1 but low levels of p27KIP1
The rapid senescence of HeLa cells induced by Tax raises the question how HTLV-1-transformed T-cell lines manage to express tax abundantly without undergoing cell cycle arrest. We have shown recently that Tax prematurely activates APC in MT4, C8166, and C91PL cells (Liu et al, 2005). The APC activation and reduction of cyclin A, securin, and cyclin B1 in these cells were associated with a delay in S/G2/M progression, but did not lead to G1 arrest (Liu et al, 2005). Transactivation of p21_CIP1/WAF1_ and premature activation of APC by Tax are expected to increase both p21_CIP1/WAF1_ and p27_KIP1_ protein levels in HTLV-1-transformed T cells. Indeed, five HTLV-1-transformed T-cell lines examined all expressed abundant p21_CIP1/WAF1_ mRNA (20–100 fold) and protein when compared to three HTLV-1-unrelated T-cell lines, Jurkat, CEM, and SupT1 (Figure 7 and Table I). Skp2 mRNA in all transformed lines was approximately 20% that of Jurkat. This is likely a result of the overexpression of Skp2 in Jurkat as previously reported (Lim et al, 2002). Compared to Jurkat, CEM cells expressed elevated mRNA levels of both Skp2 and p27_KIP1_ (Table I), while the levels of Skp2, p21_CIP1/WAF1_, and p27_KIP1_ in control SupT1 were similar to that of Jurkat. As expected, increased polyubiquitination of Skp2 in S phase was readily detected in C8166 cells (not shown). Most interestingly, in contrast to HeLa cells transduced with LV-Tax or Ad-Tax (see Figure 3 for comparison) and in spite of lower Skp2 levels (expected to lead to p27_KIP1_ stabilization), all HTLV-I-transformed cells expressed low to undetectable levels of p27_KIP1_ protein and significantly lower levels of p27_KIP1_ mRNA (13–53%) compared to HTLV-1-unrelated controls (Figure 7 and Table I). Based on these results, we speculate that tax_-IRS may be principally mediated by p27_KIP1 and all five independently isolated HTLV-1-transformed T-cell lines examined here have evaded tax_-IRS through a loss of p27_KIP1 expression and/or functions (Figure 9).
Figure 7.
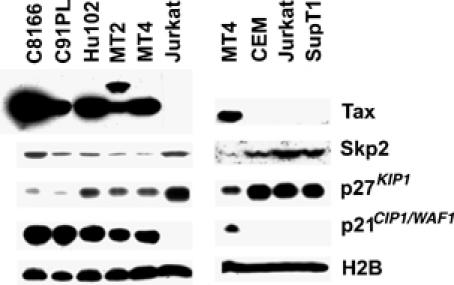
HTLV-I-transformed T cells express high levels of p21_CIP1/WAF1_ but low levels of p27_KIP1_. (A) Tax, Skp2, p21_CIP1/WAF1_, p27_KIP1_, histone 2B (H2B) immunoblots of HTLV-transformed T-cell lines: C8166, C91PL, Hut102, MT2, and MT4 versus HTLV-I-unrelated Jurkat T, CEM, and SupT1 cell lines. (B) Table I. Ratios of Skp2, p21_CIP1/WAF1_, and p27_KIP1_ mRNAs in HTLV-I-transformed cells, and in CEM and SupT1 cells versus Jurkat T cells. The mRNA ratios were determined as described in Materials and methods.
Table 1.
mRNA ratios (HTLV-1-transformed versus HTLV-1-unrelated T-cell lines)
| | P27_KIP1_ | p21_CIP1/WAF1_ | Skp2 | | | ------------ | -------------- | --------- | --------- | | C8166 | 0.13±0.02 | 55±5 | 0.20±0.04 | | C91PL | 0.13±0.03 | 104±16 | 0.17±0.02 | | HuT102 | 0.53±0.08 | 74±4 | 0.23±0.07 | | MT2 | 0.41±0.04 | 32±11 | 0.21±0.02 | | MT4 | 0.55±0.03 | 20±3 | 0.24±0.00 | | Jurkat | 1 | 1 | 1 | | SupT1 | 1.5±0.52 | 2.6±1.0 | 1.1±0.40 | | CEM | 4.1±1.1 | 0.78±0.15 | 4.9±1.2 |
Figure 9.
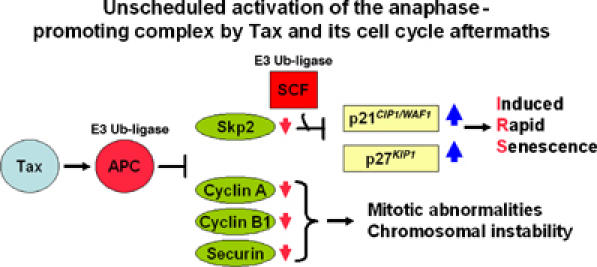
Unscheduled activation of the APC by HTLV-1 Tax and its cell cycle aftermaths. Unscheduled activation of the APC during S phase by HTLV-1 Tax leads to degradation of cyclin A, cyclin B1, and securin, causing mitotic abnormalities/chromosome instability. Concurrently, the degradation of Skp2 results in inhibition/inactivation of SCFSkp2, which, in concert with the loss of cyclin A, leads to the stabilization of p21_CIP1/WAF1_ and especially p27_KIP1_, committing cells to rapid senescence.
Tax can be stably expressed in p27KIP1-null NIH3T3 cells
To determine if a loss of p27_KIP1_ can prevent tax-IRS, a lentivirus vector containing the tax gene and the SV40 promoter-driven puromycin-resistance gene was used to transduce, respectively, a wild-type NIH3T3 cell line, an NIH3T3 cell line with a homozygous deletion of p21_CIP1/WAF1_ gene (p21_CIP1/WAF1−/−_), and importantly, an NIH3T3 cell line with a homozygous deletion of p27_KIP1_ gene (p27_KIP1−/−_). While no cell lines that stably express tax could be established in wild-type or p21_CIP1/WAF1−/−_ NIH3T3 background (unpublished results), several tax_-expressing p27_KIP1−/− (tax+/p27_KIP1_−/−) cell clones were readily obtained (Figure 8). These clones have grown continuously for more than 6 months with stable tax expression. Interestingly, the doubling time of tax+/p27_KIP1−/−_ cells became lengthened compared to that of the parental p27_KIP1−/−_ cells, consistent with the cell cycle slow-down of HTLV-1-transformed T cells previously reported (Liu et al, 2005). Finally, flow cytometry of asynchronously growing p27_KIP1−/−_ and tax+/p27_KIP1−/−_ (clone 2) cells indicated that Tax significantly reduced S (from 53 to 32%) and increased G1 and G2/M cell populations (33–45% and 13–23%, respectively, Figure 8) likely as a result of the premature activation of APC and the transactivation and stabilization of p21_CIP1/WAF1−/−_ (Figure 8). In aggregate, these data support the notion that loss of p27_KIP1_ functions circumvents the G1 arrest/senescence induced by Tax and is necessary for stable tax expression and cell transformation.
Figure 8.
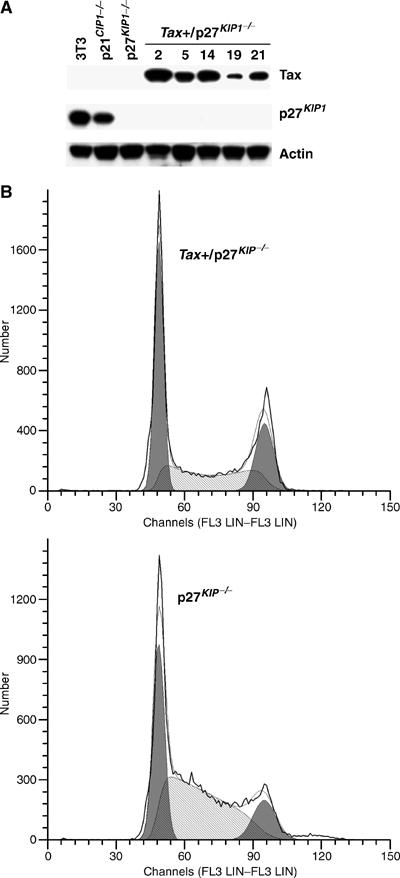
HTLV-I Tax can be stably expressed in p27_KIP1_-null NIH3T3 cells. (A) A lentivirus vector LV-Tax-Puro, containing tax and the SV40 promoter-driven puromycin-resistance gene, was used to transduce a wild-type NIH3T3 cell line, an NIH3T3 cell line with a homozygous deletion of p21_CIP1/WAF1_ gene (p21_CIP1/WAF1−/−_), and a similar cell line with a homozygous deletion of p27_KIP1_ gene (p27_KIP1−/−_). No tax-expressing stable cell lines were obtained in NIH3T3 or p21_CIP1/WAF1−/−_ background. Tax and p27_KIP1_ Immunoblots for five independently isolated puromycin-resistant clones in p27_KIP1−/−_ background are shown. (B) Flow cytometric analyses (G1 (left peak, dark shade), S (light shade), G2/M: (right peak, dark shade)) of asynchronously growing parental p27_KIP1−/−_ cells (G1: 33%, S: 53%, G2/M: 13%) and cells of tax+/p27_KIP1−/−_ clone #2 (G1: 45%, S: 32%, G2/M: 23%).
Discussion
Tax-IRS is caused by a p53- and pRB-independent mechanism
The cellular mechanisms that trigger the senescence pathway are not fully resolved at present. Prevailing evidence indicates that tumor suppressors such as pRb, p53, and members of the INK and CIP/KIP families of CDK inhibitors play significant roles in this process (Chin et al, 1999; Wells et al, 2000, 2003; Ferbeyre et al, 2002). Present data suggest that the permanent proliferative arrest brought about by tax differs mechanistically from that caused by oncogenic ras. Unlike oncogenic ras, tax readily commits immortalized and transformed cells such as HeLa to senescence, and apparently does so without functional p53 and pRB. The _tax_-IRS is accompanied by overt mitotic aberrations, and occurs rapidly after one cell division cycle upon tax expression. Whether oncogenic _ras_-induced senescence occurs within or immediately after a single cell division cycle has not been described. Finally, mitotic abnormalities have not been reported to accompany _ras_-mediated senescence.
Activation of APC by Tax ahead of schedule is responsible for the stabilization of p21CIP1/WAF1 and p27KIP1
The great increase of p21_CIP1/WAF1_ and p27_KIP1_ in tax_-transduced cells is likely to be the cause for tax_-IRS. Both quantitative real-time RT–PCR and pulse-chase experiments have demonstrated that in tax_-expressing cells, protein stabilization is the primary cause for p27_KIP1 increase; while both transactivation and protein stabilization are responsible for p21_CIP1/WAF1 increase. The persistent rise of p21_CIP1/WAF1 and p27_KIP1_ through S/G2/M in tax_-transduced cells also supports the notion that p21_CIP1/WAF1 and p27_KIP1_ are stabilized.
The stabilization of p21_CIP1/WAF1_ and p27_KIP1_ caused by Tax can be best explained by the premature APC activation during S phase, which sets in motion the early inactivation of SCFSkp2. In support of this notion, the polyubiquitination and loss of Skp2 in tax_-transduced cells parallels the unscheduled degradation of cyclin A, cyclin B1, and securin temporally, and precedes or is concurrent with the surge of p21_CIP1/WAF1 and p27_KIP1_ (Figure 6). Skp2 is a natural substrate of APCCdh1 during late M/early G1 (Bashir et al, 2004; Wei et al, 2004). Present data support the idea that the target of Tax activation is APC rather than Cdc20 or Cdh1. In Tax-expressing HeLa cells, both APCCdh1 and APCCdc20 contribute to the reduction in cyclin B1 level (Figure 6C). The most dramatic reduction in Skp2 and increase in p21_CIP1/WAF1_ and p27_KIP1_ occurred when Tax+ cells entered G1. This is consistent with the notion that at the late stage of mitotic progression, Tax-activated APCCdh1 plays a major role in stabilizing p21_CIP1/WAF1_ and p27_KIP1_. Indeed, siRNA knockdown of Cdh1 effectively dampened the loss of Skp2 induced by Tax (Figure 6C). Previous analyses of S. cerevisiae mutants have suggested that Cdh1 is not required for Tax-induced loss of Clb2 and cell cycle arrest (Liu et al, 2003). It is not clear whether this is due to a difference in the manner by which Cdh1 is regulated in S. cerevisiae versus HeLa cells, or alternatively, S. cerevisiae APCCdc20 may contribute to Tax-induced Skp2 degradation. In conclusion, the loss of cyclin A, cyclin B1, and securin as caused by Tax leads to mitotic aberrations/chromosomal instability (Figures 1 and 3); while the loss of Skp2 and the rise in p21_CIP1/WAF1_ and p27_KIP1_ independently commits cells to _tax_-IRS (Figures 2, 3 and 8). These results are summarized in a model depicted in Figure 9.
APC activation by Tax requires cellular transit through S phase. During the G1/S arrest, no accumulation of p21_CIP1/WAF1_ and p27_KIP1_ occurred in spite of Tax expression (Figures 4 and 5). Only after transit to the mid to late S phase did the tax_-transduced cells begin to accumulate p21_CIP1/WAF1 and p27_KIP1_. We think this is because the cellular factors needed for APC activation, such as the polo-like kinase and Cdc20 are destroyed during mitotic exit and synthesized during S phase. Thus, only when these factors are restored during S phase is Tax able to act.
The downregulation of p27_KIP1_ in S and G2 is impaired in Skp2_−/− cells (Hara et al, 2001). This has led to the suggestion that the role of Skp2 may be to control p27_KIP1 during S and G2 (Hara et al, 2001). That the Tax-induced loss of Skp2 and rise of p21_CIP1/WAF1_ and p27_KIP1_ occur during S and G2 is consistent with this conclusion. The loss of Skp2 alone, however, cannot be the sole cause for the build-up of p21_CIP1/WAF1_ and p27_KIP1_ and _tax_-IRS. Skp2_−/− mice are viable. Although Skp2_−/− cells accumulate p27_KIP1, they do not become G1 arrested and commit senescence (Nakayama et al, 2004). If a loss of Skp2 cannot account fully for the tax_-IRS, then what can? Koff and co-workers have shown recently that the degradation of p27_KIP1 requires noncatalytic involvement of cyclin A–Cdk2 (Zhu et al, 2004). Therefore, the premature loss of cyclin A induced by Tax likely also contributes to p27_KIP1 stabilization. Cks1, another subunit of SCFSkp2, is also a target of APCCdh1 (Ganoth et al, 2001; Bashir et al, 2004; Wei et al, 2004). It is possible that Tax also promotes the loss of Cks1 through unscheduled APC activation, thereby add to the severity of SCFSkp2 inhibition (Figure 9). Based on these considerations, we speculate that unscheduled activation of APC in S phase (by Tax or other compounds) may be exploited as a means to arrest the proliferation of cancer cells whose p53 and pRB pathways are no longer functional.
Tax-IRS and the development of ATL
HTLV-1 causes ATL in a small percentage (2–6%) of infected individuals after a long latency period of up to 20–40 years. How HTLV-1 infection progresses from clinical latency to full-blown T-cell malignancy is not well understood. The long incubation period and the low frequency of clinical progression to ATL suggest that complex and interdependent viral and cellular events are involved. ATL patients show evidence of a monoclonal integration of HTLV-1 proviral DNA in CD4+CD25+ leukemia cells. Leukemia/lymphoma apparently emerges from a pool of HTLV-1-positive CD4+ T cells that persist for decades in infected individuals, possibly through oligoclonal expansion. How might tax affect ATL development? The analyses of HTLV-1-transformed T-cell lines and the stable expression of tax in p27_KIP1−/−_ cells (Figure 7) indicate that abrogation and/or inactivation of p27_KIP1_ (complete loss, haplo-insufficiency, transcriptional repression, and/or inactivating phosphorylation of p27_KIP1_) may allow HTLV-1-infected cells to evade tax_-IRS and allow the mitogenic activity of tax and the chromosomal instability it induces to take effect, leading to cell transformation and development of ATL. Finally, chromosomal instability induced by Tax and host cytotoxic T-lymphocyte response against Tax may then select for a complete loss of tax expression from ATL. Examination of HTLV-1-transformed and ATL cells for the status of p27_KIP1 gene and/or pathways that inactivate p27_KIP1_ is likely to provide useful clues for the emergence, progression, and treatment of ATL.
Materials and methods
Cell lines
HeLa, 293T, NIH3T3, and NIH3T3 with a homozygous deletion of p21_CIP1/WAF1_ (p21_CIP1/WAF1−/−_) or p27_KIP1_ gene (p27_KIP1−/−_) (provided by Dr Andrew Koff, Memorial Solan-Kettering Cancer center, New York) were grown in Dulbecco's modified Eagle's medium (DMEM). HTLV-transformed T cells, C8166, C91PL, Hut102, MT2, and MT4, and HTLV-unrelated T-cell lines, Jurkat, CEM, and SupT1, were grown in RPMI medium. All media were supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 100 U/ml each of penicillin and streptomycin.
Plasmids and antibodies
The lentivirus vector for tax, LV-Tax, was derived by inserting a _Bam_HI–_Sal_I fragment containing the coding sequence of the HTLV-1 tax into the _Bam_HI and _Xho_I sites of the pHR'CMV-LacZ vector (Naldini et al, 1996) in place of the lacZ gene. The control LV-GFP vector was similarly constructed by replacing the lacZ gene with the EGFP gene on a _Bam_HI–_Kpn_I fragment. A lentivirus vector, LV-Tax/puromycin, containing tax and the puromycin-resistance gene was derived by inserting an _Eco_R1–_Kpn_1 fragment containing the SV40 promoter and the puromycin-resistance gene downstream of the Tax cDNA.
The antibody for Tax (4C5) was made in this lab; others include cyclin A (SC-751), cyclin B1 (SC-752), p21_CIP1/WAF1_ (SC-397), p27_KIP1_ (SC-1641), p16INK4a (SC-759), p53 (SC-6243), actin (SC-1616), HA tag (SC-7392), and Cdc20 (SC-5269) antibodies from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; the Cdh1 antibody from Oncogene, Inc. (CC43), the Flag antibody from Sigma (F3165); the Skp2 antibody (32–3400) from Zymed; and the pRB antibody from BD Pharmingen (#554136). The antibody against human securin was generously provided by Hui Zou of the UT Southwestern Medical School.
Production of lentivirus vectors
Lentiviral vectors were produced by transient transfection of HEK293T cells using a calcium phosphate transfection kit (Invitrogen Life Technologies, Carlsbad, CA). The vector plasmid (10 μg) HR'CMV-Tax together with two packaging plasmids, pCMVD8.2ΔVpr (vpr-null, env-null, 10 μg) and pCMV-VSV-G (vesticular stomatitis virus G-glycoprotein, 2 μg), were used to transfect 5 × 106 293T cells that had been plated in a T75 flask 1 day before. The calcium phosphate/DNA precipitate was added to cells in complete DMEM medium. After 16 h, cells were washed with 5 ml PBS to remove the DNA, and incubated in 5 ml complete DMEM. Vector supernatants were collected at 24 and 48 h post-transfection. The supernatant was centrifuged at low speed to remove cellular debris and concentrated by ultracentrifugation in a Beckman SW28 rotor at 24 000 r.p.m. for 2 h at 4°C. The vector-enriched pellet was resuspended in DMEM and aliquoted for storage at −80°C. Vector stocks were normalized by p24-Gag content measured with an enzyme-linked immunosorbent assay (Beckman-Coulter) and tittered by immunofluorescence or visualization of GFP after gene transduction.
Cell cycle synchronization and infection by lentivirus or adenovirus vectors
Cell cycle synchronization and infection of synchronized HeLa cells with adenovirus vectors were as reported previously (Liu et al, 2003). Infection with lentiviral vectors was carried out 1 h after release from the G1/S arrest.
Senescence-associated _β_-galactosidae (SA _β_-Gal) staining
HeLa cells (2.5 × 104/well) in monolayer were grown in a six-well plate, transduced with LV-Tax or LV-GFP at an m.o.i. of 5, grown for 3 days, washed in 2 ml PBS, fixed for 3–5 min at room temperature in 2 ml of a solution containing 2% formaldehyde, 0.2% glutaraldehyde (or 3% formaldehyde), washed, and incubated for 24 h at 37°C with a solution (3 ml) containing 1 mg/ml of 5-bromo-4-chloro-3-indolyl B-D-galactoside (X-Gal), 40 mM citric acid/sodium phosphate at pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2.
Detection of Skp2 polyubiquitination in asynchronous HeLa cells
Two hundreds thousand HeLa cells were grown in a 10 cm Petri dish and transfected with 10 μg each of an expression plasmid for the FLAG-tagged human Skp2 and that for the HA-tagged human ubiquitin (HA-Ub) using the FuGENE 6 transfection reagents (Roche Applied Science). The second day, cells were infected with Ad-Tax or Ad-tTa at an m.o.i. of 5 for 16 h, and then treated with 10 μM MG132 for 4 h. Cells were lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.5% DOC, 1% NP-40, 0.1% SDS) containing 1 × protease inhibition cocktail (Roche Applied Science) and immunoprecipitated with a mouse monoclonal Flag antibody (F3165, Sigma, Inc. USA) at 4°C overnight. The immunoprecipitates were washed three times with PBS and immunoblotted with a mouse anti-HA antibody (SC-7392, Santa Cruz).
Detection of Skp2 polyubiquitination in synchronized HeLa cells
HeLa cells were transfected similarly as above. At 4 h after transfection, cells were subjected synchronized by DTB. Throughout the duration of the second thymidine treatment, cells were infected with Ad-Tax or Ad-tTa at an m.o.i of 5. The cells were then released into complete medium containing 10 μM MG132, and harvested at the indicated times (0, 4, 5, 6, 7, and 8 h) post-release for immunoprecipitation and immunoblots.
Quantitative real-time PCR
HeLa cells were transduced with LV-Tax or LV-GFP for 3 days. Cells were harvested and total cellular RNA was isolated using the Trizol reagent (Invitrogen). The amount of total RNA in each sample was estimated based on the levels of 28S, 18S, and 5S rRNAs resolved in native 1% agarose gels. The first-strand cDNA synthesis was carried out with a kit from Amersham Biosciences according to the manufacturer's protocol. The cDNA samples were then stored frozen at −80°C. The real-time PCR for p21_CIP1/WAF1_, p27_KIP1_, and the internal β-actin control were performed using the universal master mix and kits from the Applied Biosystems (part number 4331182), which consist of gene-specific TagMan MGB probes (6-FAM™ dye-labeled) and unlabeled PCR primer pairs for p21_CIP1/WAF1_ (Hs003555782_m1, CDKN1A), p27_KIP1_ (Hs00153277_m1, CDKN1B), and β-actin (Hs99999903_m1, ActB), respectively. Each real-time PCR reaction contained the first-strand cDNA, the respective primer pair, and the gene-specific TagMan MGB probe, and was carried out in an ABI Prism 7000 Sequence Detection System. The PCR condition was 95°C for 10 min, followed by 95°C for 15 s, and 60°C for 1 min in each cycle, for 40 cycles. Four concentrations (successive four-fold dilution) of the first-strand cDNA were analyzed to establish linearity of the real-time PCR. The levels of p21_CIP1/WAF1_ and p27_KIP1_ mRNA in either LV-Tax/HeLa or LV-GFP/HeLa sample were normalized against that of the β-actin. The fold increases of p21_CIP1/WAF1_ and p27_KIP1_ mRNA in LV-Tax/HeLa versus LV-GFP/HeLa were calculated and plotted. Similarly, RT–PCR was used to measure p21_CIP1/WAF1_, p27_KIP1_, and Skp2 mRNA levels using the level of 18S ribosomal RNA as an internal control. The PCR primer pairs for Skp2 and 18S RNA were (Hs00180634_m1, Skp2) and (Hs99999901_ s1, 18S), respectively. The levels of p21_CIP1/WAF1_, p27_KIP1_ and Skp2 mRNA in HTLV-transformed T cells and HTLV-1-unrelated CEM, Jurkat, and SupT1T cells were normalized against that of the 18S RNA first. The ratios of p21_CIP1/WAF1_, p27_KIP1_, and Skp2 mRNA of Jurkat versus the other T cells were then computed. The RT–PCR for Ad-Tax- and Ad-tTa-transduced cells was carried out similarly.
Pulse chase for p27KIP1
HeLa cells were transduced with LV-Tax or LV-GFP for 3 days. One hundred thousand cells from each transduction were washed with 10 ml warm PBS twice to remove methionine and cysteine. The cells were then trypsinized and resuspended in 10 ml warm PBS, spun down, and resuspended in 1 ml methionine-free, cysteine-free DME complete medium containing 2% dialyzed FBS. The cells were starved for methionine and cysteine for 20 min at 37°C, thereupon, 560 μCi of promix L-S35 (14.3 μCi/μl, AGQ0080, Amersham Biosciences, Piscataway, NJ, USA) containing L-S35-methionine and L-S35-cysteine was added and the cells were pulse-labeled for 1 h. Radiolabeled cells were spun down and the radioactive media removed. The cells were then washed with 5 ml of warm PBS, transferred to 4 ml of a chase medium composed of DME complete medium supplemented with 10% FBS and 2 mM each of methionine and cysteine. Aliquot of cells (1 ml) was then taken at the indicated time points (0, 1, 3, and 6 h), harvested, lysed, and immunoprecipitated using a p27_KIP1_ antibody (SC-528, Santa Cruz).
siRNA knockdown of Cdc20 and Cdh1
Two hundred thousand HeLa cells were seeded in each well of six-well plates and synchronized by double thymidine treatment as described above. At 2 h before the second thymidine treatment, cells were transfected with 120 pmol of a control siRNA, a Cdc20 siRNA, a Cdh1 siRNA, or a combination of both Cdc20 and Cdh1siRNA's. Transfections were carried out using oligofectamine (Invitrogen, Inc.) as prescribed by the manufacturer. After 4 h into the second thymidine treatment, cells were infected with either Ad-Tax or Ad-tTa (m.o.i.=5). The infection was carried out for the duration of the remainder of the second thymidine treatment for 12 h. Cells were then released from the arrest and collected at 8 h after release for immunoblots. The siRNA oligonucleotides, control nontargeting siRNA (Cat No: D-001210-01-20), Cdc20 siRNA (p55CDC, human, Cat No: M-003225-03), and Cdh1 siRNA (CDH1, human, Cat No: M-015377-01), were purchased from Dharmacon Inc.
Acknowledgments
We thank M Pagano for the Flag-Skp2 plasmid, Dr S Hatakeyama for the HA-ubiquitin expression plasmid, ISY Chen, PM Cannon, and I Christodoulopoulos for lentiviral vectors, Andrew Koff for wild-type, p21-null, and p27-null NIH3T3 cells, H Zou for the human securin antibody, G Franchini, H Yu, K Lee, T Dunn, and O Cohen-Fix for helpful discussions, and X Xiang and P Grimley for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health to C-ZG.
References
- Akagi T, Ono H, Shimotohno K (1996) Expression of cell-cycle regulatory genes in HTLV-I infected T-cell lines: possible involvement of Tax1 in the altered expression of cyclin D2, p18Ink4 and p21Waf1/Cip1/Sdi1. Oncogene 12: 1645–1652 [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M (2004) Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428: 190–193 [DOI] [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A (2003) Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 278: 25752–25757 [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1: 193–199 [DOI] [PubMed] [Google Scholar]
- Cereseto A, Washington PR, Rivadeneira E, Franchini G (1999) Limiting amounts of p27Kip1 correlates with constitutive activation of cyclin E–CDK2 complex in HTLV-I-transformed T-cells. Oncogene 18: 2441–2450 [DOI] [PubMed] [Google Scholar]
- Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB (1999) A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res 59: 3761–3767 [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97: 527–538 [DOI] [PubMed] [Google Scholar]
- Chowdhury IH, Farhadi A, Wang XF, Robb ML, Birx DL, Kim JH (2003) Human T-cell leukemia virus type 1 Tax activates cyclin-dependent kinase inhibitor p21/Waf1/Cip1 expression through a p53-independent mechanism: inhibition of cdk2. Int J Cancer 107: 603–611 [DOI] [PubMed] [Google Scholar]
- Chu ZL, Di Donato JA, Hawiger J, Ballard DW (1998) The tax oncoprotein of human T-cell leukemia virus type 1 associates with and persistently activates IkappaB kinases containing IKKalpha and IKKbeta. J Biol Chem 273: 15891–15894 [DOI] [PubMed] [Google Scholar]
- de la Fuente C, Wang L, Wang D, Deng L, Wu K, Li H, Stein LD, Denny T, Coffman F, Kehn K, Baylor S, Maddukuri A, Pumfery A, Kashanchi F (2003) Paradoxical effects of a stress signal on pro- and anti-apoptotic machinery in HTLV-1 Tax expressing cells. Mol Cell Biochem 245: 99–113 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW (2002) Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 22: 3497–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini G, Wong-Staal F, Gallo RC (1984) Human T-cell leukemia virus (HTLV-I) transcripts in fresh and cultured cells of patients with adult T-cell leukemia. Proc Natl Acad Sci USA 81: 6207–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu DX, Kuo YL, Liu B, Jeang KT, Giam CZ (2003) Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J Biol Chem 278: 1487–1493 [DOI] [PubMed] [Google Scholar]
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A (2001) The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol 3: 321–324 [DOI] [PubMed] [Google Scholar]
- Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S, Nakayama K (2001) Degradation of p27(Kip1) at the G(0)-G(1) transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem 276: 48937–48943 [DOI] [PubMed] [Google Scholar]
- Helt AM, Galloway DA (2003) Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24: 159–169 [DOI] [PubMed] [Google Scholar]
- Jeang KT, Giam CZ, Majone F, Aboud M (2004) Life, death, and tax: role of HTLV-I oncoprotein in genetic instability and cellular transformation. J Biol Chem 279: 31991–31994 [DOI] [PubMed] [Google Scholar]
- Jin DY, Giordano V, Kibler KV, Nakano H, Jeang KT (1999) Role of adapter function in oncoprotein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I Tax interacts directly with IkappaB kinase gamma. J Biol Chem 274: 17402–17405 [DOI] [PubMed] [Google Scholar]
- Kawata S, Ariumi Y, Shimotohno K (2003) p21(Waf1/Cip1/Sdi1) prevents apoptosis as well as stimulates growth in cells transformed or immortalized by human T-cell leukemia virus type 1-encoded tax. J Virol 77: 7291–7299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B, Okayama A, Donnelly R, Tachibana N, Essex M (1991) Polymerase chain reaction analysis of defective human T-cell leukemia virus type I proviral genomes in leukemic cells of patients with adult T-cell leukemia. J Virol 65: 5471–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang MH, Geisbert T, Yao Y, Hinrichs SH, Giam CZ (2002) Human T-lymphotropic virus type 1 oncoprotein tax promotes S-phase entry but blocks mitosis. J Virol 76: 4022–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MS, Adamson A, Lin Z, Perez-Ordonez B, Jordan RC, Tripp S, Perkins SL, Elenitoba-Johnson KS (2002) Expression of Skp2, a p27(Kip1) ubiquitin ligase, in malignant lymphoma: correlation with p27(Kip1) and proliferation index. Blood 100: 2950–2956 [DOI] [PubMed] [Google Scholar]
- Liu B, Hong S, Tang Z, Yu H, Giam CZ (2005) HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc Natl Acad Sci USA 102: 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liang MH, Kuo YL, Liao W, Boros I, Kleinberger T, Blancato J, Giam CZ (2003) Human T-lymphotropic virus type 1 oncoprotein tax promotes unscheduled degradation of Pds1p/securin and Clb2p/cyclin B1 and causes chromosomal instability. Mol Cell Biol 23: 5269–5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, Kitagawa M, Nakayama K, Hatakeyama S (2000) Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 19: 2069–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI (2004) Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 6: 661–672 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63: 1129–1136 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Sun SC, Ballard DW (1999) Persistent activation of NF-kappaB by the tax transforming protein of HTLV-1: hijacking cellular IkappaB kinases. Oncogene 18: 6948–6958 [DOI] [PubMed] [Google Scholar]
- Swanson C, Ross J, Jackson PK (2000) Nuclear accumulation of cyclin E/Cdk2 triggers a concentration-dependent switch for the destruction of p27Xic1. Proc Natl Acad Sci USA 97: 7796–7801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG Jr (2004) Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428: 194–198 [DOI] [PubMed] [Google Scholar]
- Wells SI, Aronow BJ, Wise TM, Williams SS, Couget JA, Howley PM (2003) Transcriptome signature of irreversible senescence in human papillomavirus-positive cervical cancer cells. Proc Natl Acad Sci USA 100: 7093–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, Howley PM (2000) Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J 19: 5762–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Sun SC (2000) Activation of IKKalpha and IKKbeta through their fusion with HTLV-I tax protein. Oncogene 19: 5198–5203 [DOI] [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A (1998) Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 93: 1231–1240 [DOI] [PubMed] [Google Scholar]
- Zhu XH, Nguyen H, Halicka HD, Traganos F, Koff A (2004) Noncatalytic requirement for cyclin A-cdk2 in p27 turnover. Mol Cell Biol 24: 6058–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]