GIT2 represses Crk- and Rac1-regulated cell spreading and Cdc42-mediated focal adhesion turnover (original) (raw)
Abstract
G protein-coupled receptor kinase interactors (GITs) regulate focal adhesion (FA) turnover, cell spreading, and motility through direct interaction with paxillin and the Rac-exchange factor Pak-interacting exchange factor β (_β_PIX). However, it is not clear whether GITs function to activate or repress motility or if the predominant GIT forms, GIT1 and GIT2, serve distinct or redundant roles. Here we demonstrate an obligatory role for endogenous GIT2 in repression of lamellipodial extension and FA turnover by Rac1- and Cdc42-dependent signaling pathways, respectively. Moreover, we show that the SH2–SH3 adaptor protein Crk is an essential target of GIT2 inhibition. Unexpectedly, we find that _β_PIX is dispensable for the effects elicited by knockdown of GIT2. Finally, we show that loss of GIT2 is sufficient to induce migration of the nontransformed epithelial cell line MCF10A. These results suggest that inactivation of GIT2 function is a required step for induction of cell motility and that GIT2 may be a target of oncogenic signaling pathways that regulate cell migration.
Keywords: Cdc42, Crk, GIT, PIX, Rac
Introduction
Upon binding to extracellular matrix, integrins nucleate the assembly of macromolecular protein structures that couple the cell membrane to the actin cytoskeleton. These structures are termed focal adhesions (FAs) or focal complexes (FCs), depending on their size, location, molecular composition, and regulation (Zaidel-Bar et al, 2004). While FCs are small, nascent adhesion sites found at membrane protrusions, FAs represent larger, more stable structures that extend underneath the cell body. FA turnover denotes the constant shuttling of proteins from FAs to FCs that underlies the dynamic adhesive behavior required for cell motility (Webb et al, 2002). Given that FAs consist of at least 50 distinct proteins, it is not surprising that elucidation of their structure as well as mechanisms of assembly and turnover remain daunting challenges. Studies to overcome these challenges are pivotal to the understanding of embryogenesis, wound healing, regeneration, and cancer. Indeed, many oncogenes are either constituents of FAs or regulators of FA dynamics.
The Rho family of GTPases plays key roles in cell motility by providing tight coupling of FA turnover and actin polymerization. Growth factor receptors and integrins act cooperatively to regulate the activity of Rho GTPases, resulting in the generation of distinct actin structures (Burridge and Wennerberg, 2004). Activation of Cdc42 and Rac causes rapid membrane protrusion through the generation of filopodia and lamellipodia, respectively, and is associated with increased FA turnover. In contrast, activated RhoA propagates the formation of actin stress fibers and steady-state FAs that mediate stable cell attachment (Hall, 1998).
A key feature of FAs is the presence of numerous proteins that function as molecular scaffolds. Paxillin is a prototypical example, serving as a binding partner for kinases, like FAK and Src, and structural proteins such as vinculin (Brown and Turner, 2004). Paxillin associates with a family of ADP-ribosylation factor GTPase-activating proteins (ArfGAPs), including GIT1 and GIT2, also called CAT1&2, p95-APP1&2 and PKL (Turner et al, 1999; Di Cesare et al, 2000; Zhao et al, 2000). Evidence suggests that G protein-coupled receptor kinase interactors (GITs) might serve to coordinate FAs dynamics and membrane trafficking. In vitro, GIT1 and GIT2 stimulate hydrolysis of GTP bound to Arf1 and Arf6 (Vitale et al, 2000). In cells, GIT molecules appear to function as GAPs for Arf6, consistent with a role for Arf6 in membrane trafficking and actin remodeling at the cell periphery (Di Cesare et al, 2000; Nishiya et al, 2005). Overexpression of GIT1 leads to increased cell motility that is associated with the loss of paxillin from FAs (Zhao et al, 2000; Manabe et al, 2002). In fibroblasts, overexpression of a paxillin deletion mutant, which blocks GIT2 recruitment to FAs, also results in Rac activation, increased cell motility, and actin polymerization (West et al, 2001).
GIT1 and GIT2 can associate with FAK and phosphorylation of GIT2 by FAK appears to promote its interaction with paxillin, indicating that GIT2 may be a key intermediary in FAK-dependent FA turnover (Zhao et al, 2000; Brown et al, 2005). Consistent with a role for GITs in FA dynamics is the observation that phosphorylated GIT2 associates with the SH2 domains of the adaptor molecules Nck, Src, Grb2, and Crk (Brown et al, 2005). A more extensively characterized interaction is that of GITs with the Rac-exchange factor Pak-interacting exchange factor (_β_PIX)/Cloned-out of library-1 (Cool-1), a binding partner of p21-activated kinase (PAK) (Bagrodia et al, 1998, 1999; Manser et al, 1998; Turner et al, 1999). GITs appear to be responsible for the recruitment of _β_PIX and PAK to FAs and the recruitment of PAK may underlie some of the effects of GITs on actin and FA structure (Turner et al, 1999; Zhao et al, 2000; Brown et al, 2002; Loo et al, 2004). However, it is still unclear whether GITs play a stimulatory or inhibitory role in actin polymerization and FA turnover. Moreover, it is not known whether GIT1 and GIT2 possess distinct or redundant functions, which is complicated by the fact that GIT2 is encoded by multiple distinct splice variants of unknown function (Premont et al, 2000).
To understand the functions of GIT family members, we have employed RNA interference (RNAi) to target GIT1 and GIT2. We have found that GIT1 and GIT2 possess nonredundant functions. Specifically, we find that GIT2, but not GIT1, is an essential inhibitor of cell spreading and FA turnover, regulated by Rac1 and Cdc42, respectively. Moreover, induction of cell spreading upon GIT2 knockdown is entirely dependent on the presence of adaptor protein Crk, indicating that, when localized to FAs, GIT2 functions to inhibit Crk-dependent actin remodeling. All these events occur in the presence of unchanged levels of GIT1. Strikingly, in the nontransformed mammary epithelial cell line MCF10A, GIT2 knockdown alone results in dramatically increased cell motility. Taken together, our results show that GIT2 is a resident component of FAs required to repress Crk-, Rac1-, and Cdc42-dependent motile responses.
Results
Specific knockdown of GIT1 and GIT2 by siRNA-mediated silencing
To target GIT1 and GIT2 by RNAi, we expressed short hairpin RNAs (shRNA) using modified retroviral vectors that allowed for selection and detection of transfected cells due to the presence of additional expression cassettes that encode puromycin resistance and GFP, respectively. We find that HeLa cells express predominantly GIT1 and the longest form of GIT2 (GIT2-long), which contrasts with previous reports indicating that HeLa cells also express GIT2-short (Supplementary Figure S1) (Mazaki et al, 2001). To determine the effectiveness of GIT1 and GIT2 shRNAs, HeLa cells were transfected with shRNA expression vectors and 24 h later replated on culture plates or fibronectin-coated coverslips for an additional 72 h in the presence of puromycin to select for transfected cells. Using this protocol, several shRNAs were identified that caused substantial knockdown of GIT1 or GIT2 (data not shown). Comparison of protein levels in cell lysates from knockdowns to serial dilutions of control lysates revealed that the most efficient shRNAs reduced expression by 16–32-fold, that is, to 4–7% of endogenous levels (Figure 1A). This was consistent with real-time PCR analyses of mRNA levels (data not shown). Importantly, GIT1 and GIT2 shRNAs functioned selectively, with no effect of GIT1 shRNA on GIT2 protein levels and vice versa (Figure 1B). Expression of GIT-specific shRNAs also did not result in reduced expression of ERK, vinculin, _β_-catenin, and other proteins (Figure 1B and data not shown). Moreover, by cotransfection of GIT1 and GIT2 shRNA, we could effectively knockdown both proteins simultaneously. This was confirmed in Western blots using a monoclonal antibody generated against p95PKL (chicken GIT2) that recognizes both human GIT1 and GIT2 (Figure 1B). For a full characterization of the antibodies used in this study, see Supplementary Figure S1. We also determined the effects of GIT knockdown on both _β_PIX and PAK levels, as _β_PIX associates directly with GIT1 and GIT2. Interestingly, GIT1&2 double-knockdown cells consistently showed a moderate reduction in _β_PIX protein levels, suggesting that association with GITs might stabilize _β_PIX. In contrast, no reduction in PAK levels was observed (Figure 1B).
Figure 1.
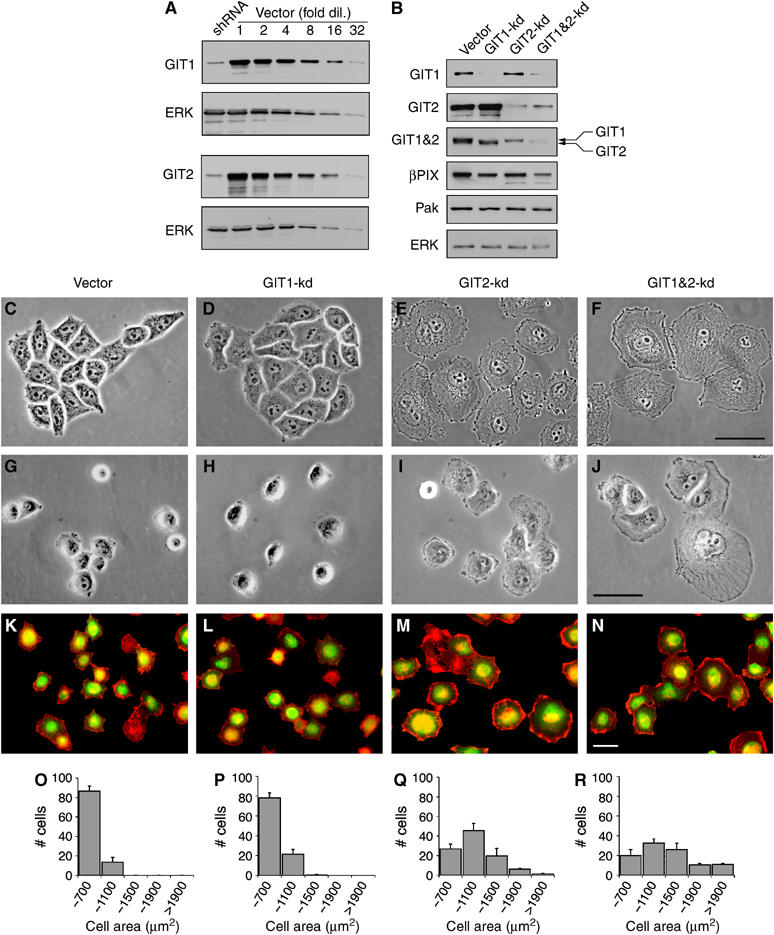
GIT2, but not GIT1, represses membrane ruffling and cell spreading in HeLa cells. (A) Control cell lysates were diluted serially and blotted alongside GIT1-kd or GIT2-kd samples. (B) GIT1, GIT2, and GIT1&2-kd cells were blotted with mouse anti-GIT1, rabbit anti-GIT2, mouse anti-PKL (which recognizes GIT1&2), rabbit anti-_β_-PIX, rabbit anti-PAK, and rabbit anti-ERK antibodies. Phase-contrast images of control (C), GIT1 (D), GIT2 (E), and GIT1&2 (F) knockdown cells cultured on fibronectin coverslips. Spreading of control (G), and GIT1 (H), GIT2 (I), and GIT1&2 (J) knockdown cells on fibronectin for 1 h. HeLa cells were transfected with control (K), GIT1 (L), GIT2 (M), and GIT1&2 (N) shRNA vectors that contain an expression cassette encoding GFP. Knockdown cells were plated on fibronectin for 1 h and stained for F-actin with Alexa594-phalloidin (red). shRNA-expressing cells can be distinguished by green fluorescence. Histograms show the distributions in the cell area of control (O), GIT1 (P), GIT2 (Q), and GIT1&2 (R) knockdown cells plated on fibronectin for 1 h. Values are means±s.d. (_n_=3 experiments). Scale bars: 20 _μ_M.
GIT2 plays a nonredundant role in repression of membrane ruffling and cell spreading
The morphology of HeLa cells cultured on fibronectin following knockdown of GITs was examined by phase-contrast microscopy. Comparison of control, GIT1, and GIT2 knockdown cells revealed that GIT2-deficient cells exhibited a substantial increase in area. GIT2 knockdown cells produced extensive lamellae terminating in phase-dark membrane ruffles and tended to scatter rather than form islets characteristic of this strain of HeLa cells (Figure 1C–F). We next examined the effects of GIT1 and GIT2 knockdown on spreading of HeLa cells on fibronectin for 1 h, a process that requires coordinate regulation of actin membrane dynamics with FA turnover. GIT2 knockdown cells showed enhanced cell spreading and were characterized by broad lamellar extensions and membrane ruffles (Figure 1G–J). In a complementary approach, we transfected HeLa cells with GIT1 and GIT2 shRNA vectors that coexpressed GFP, eliminating the need for selection. When replated on fibronectin for 1 h, GFP-positive GIT2 knockdown cells exhibited a marked enhancement of cell spreading with extensive actin-enriched membrane ruffles frequently circumscribing the entire cell (Figure 1K–N). Quantification of cell area confirmed that GIT2, but not GIT1, knockdown elicited increased cell spreading (Figure 1O–R). Interestingly, when compared to cells with GIT2 knockdown alone, GIT1&2 double-knockdown cells displayed an even larger increase in cell spreading. This suggests that, in the absence of GIT2, GIT1 functions to weakly repress membrane ruffling and cell spreading. However, our results clearly demonstrate that GIT2 is not only necessary but also sufficient to inhibit membrane ruffling and cell spreading.
GIT2 knockdown results in increased FA turnover
GIT1 and GIT2 associate with paxillin and, based on studies using overexpression, GIT1 has been suggested to promote FA disassembly, paxillin redistribution, and increased motility (Di Cesare et al, 2000; Zhao et al, 2000; Manabe et al, 2002). We therefore examined FA structure, specifically the distribution of paxillin, following GIT1 and/or GIT2 knockdown. Control and GIT1 knockdown cells possessed a characteristic pattern of actin stress fibers that terminated in robust FAs (Figure 2A–F). This contrasted with GIT2 knockdown cells that showed extensive lamellipodial extensions and highly diminished paxillin staining (Figure 2G–I). We next determined whether GIT1 was required for the redistribution of paxillin upon GIT2 knockdown, as it seemed possible that GIT2 might serve to negatively regulate the function of GIT1. GIT1&2 double-knockdown cells showed a redistribution of paxillin comparable to GIT2 knockdown alone, indicating that there is no requirement for GIT1 in this process (Figure 2J–L). Closer examination of cells with GIT2, or GIT1&2 double knockdown revealed that paxillin was found in small punctate adhesions located at the cell periphery (Figure 2J′–L′). These resemble FCs seen at the leading edge of motile cells and are thought to represent rapidly cycling adhesions. To confirm that GIT2 knockdown induces FA turnover rather than a selective redistribution of paxillin, we stained the cells with vinculin, another resident FA component. Consistent with a role for GIT2 in inhibiting FA turnover, GIT2 knockdown caused a reduction in the size of vinculin-positive FAs (Figure 2M–N and N′). Finally, the effects of GIT2 shRNA were specific, as they could be reversed by introduction of a GIT2 cDNA in which the shRNA target sequence had been eliminated by silent mutagenesis (Supplementary Figure S2A–D). Moreover, while our principal GIT2 shRNA recognized a sequence common to all predicted GIT2 forms, including GIT2-short, additional shRNAs that target the longest form specifically showed substantial knockdown that correlated with enhanced membrane ruffling and FA turnover (Supplementary Figure S2E–F).
Figure 2.
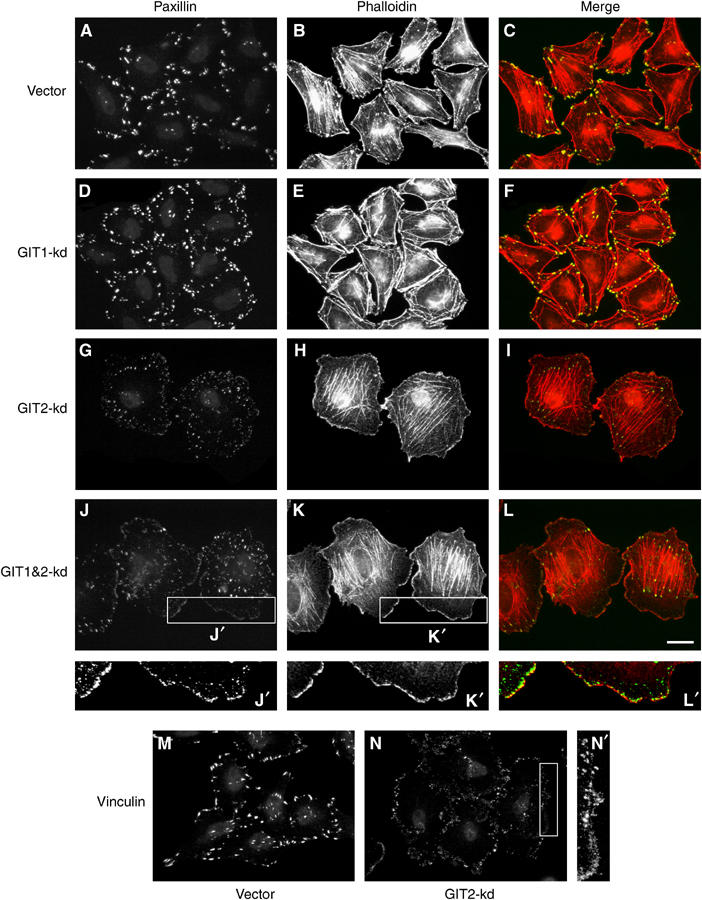
GIT2 knockdown induces FA turnover in HeLa cells. (A–L) Control, GIT1, GIT2, and GIT1&2 knockdown cells were stained for paxillin (green) and F-actin (red). GIT2-kd and GIT1&2-kd cells show a dramatic reduction in FA size that is associated with the redistribution of paxillin to peripheral focal FCs (enlargement J′, K′, L′). (M, N) Control and GIT2 knockdown cells were stained for vinculin. The enlarged image (N′) shows that vinculin also redistributes to peripheral FCs upon GIT2 knockdown. Scale bar: 20 _μ_M.
GIT2 loss does not affect the association of GIT1 with paxillin or the _β_PIX–PAK complex
The observation that GIT1 and GIT2 form hetero-oligomers that assemble into a larger complex that includes _β_PIX, and most likely PAK, suggests that GIT2 may be required for the assembly of a GIT1–GIT2–_β_PIX–PAK complex or its targeting to FAs (Kim et al, 2003; Paris et al, 2003; Premont et al, 2004). We thus analyzed the localization of endogenous GIT1 and GIT2 in HeLa cells. As none of several commercially available antibodies against GIT2 were suitable for immunofluorescence analysis, we generated antibodies against a central region of human GIT2, which shows the most divergence from GIT1. Immunostaining of HeLa cells with GIT1- and GIT2-specific antibodies showed that both these proteins are associated with FAs (Figure 3A–F). This conclusion was further supported by the localization of endogenous GIT2 to FAs in cells transfected with constitutively active RhoA(V14), which leads to the stabilization of FAs (Supplementary Figure S3). Knockdown of GIT1 did not affect the localization of GIT2 (Figure 3E), demonstrating that GIT2 does not require GIT1 to associate with FAs. In contrast, GIT2 knockdown resulted in a redistribution of GIT1 to small peripheral FCs, similar to our observations for paxillin and vinculin (Figure 3C).
Figure 3.
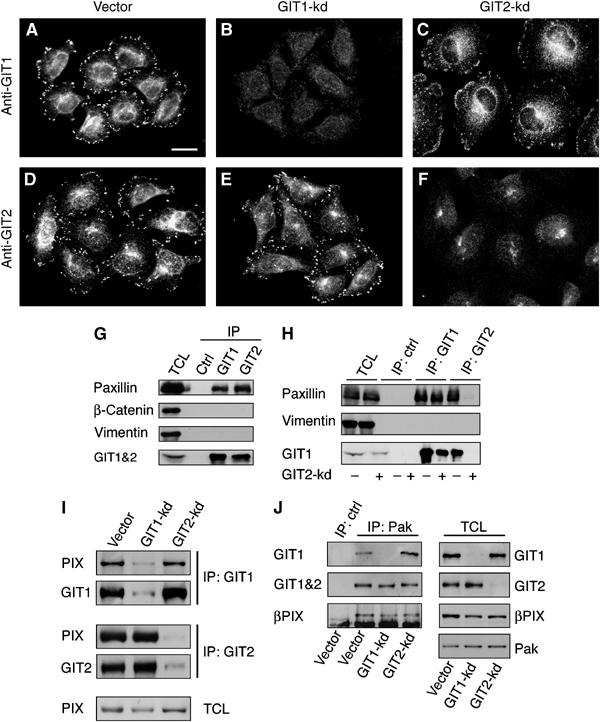
GIT1–_β_PIX–PAK complexes are preserved and GIT1 remains associated with FCs in GIT2 knockdown cells. (A–F) Control, GIT1-kd, and GIT2-kd cells were stained for GIT1 or GIT2. (G) HeLa cells were formaldehyde crosslinked and lysates were prepared as described in Materials and methods. Lysates were immunoprecipitated with GIT1- and GIT2-specific polyclonal antibodies. Following reversal of crosslinks, immunoprecipitates were blotted for paxillin, _β_-catenin, vimentin, and GIT1&2. (H) Crosslinked lysates from control or GIT2-kd cells were immunoprecipitated with GIT1 and GIT2 antibodies and blotted for paxillin, vimentin, and GIT1. (I, J) Control, GIT1-kd and GIT2-kd cells were lysed without formaldehyde crosslinking. (I) Lysates were immunoprecipitated with GIT1- and GIT2-specific polyclonal antibodies and blotted for _β_PIX, GIT1, and GIT2. (J) Lysates from knockdown cells were immunoprecipitated with anti-PAK antibodies and blotted for GIT1, GIT1&2, or _β_PIX. Scale bar: 20 _μ_M.
We next examined the association of endogenous GIT proteins with paxillin in cells, as previous studies have relied on overexpression of tagged constructs. Initial attempts to co-immunoprecipitate paxillin with either endogenous GIT1 or GIT2 were unsuccessful. We hypothesized that this resulted from the association of paxillin with a Triton-insoluble fraction that was not extracted under our lysis conditions. To facilitate cytoskeletal solubilization, we employed reversible formaldehyde crosslinking combined with denaturing lysis and sonication. This technique has been used successfully to study protein–protein interactions (Hall and Struhl, 2002). As shown in Figure 3G, under these conditions the association of both GIT1 and GIT2 with paxillin can be readily detected. Importantly, the interaction of GIT1 with paxillin is preserved in GIT2 knockdown cells (Figure 3H). No association is seen with the abundant proteins _β_-catenin or vinculin, indicating that the associations of GIT1 and GIT2 with paxillin are specific. While we cannot entirely exclude that crosslinking has preserved larger FC assemblies in which GIT1 and paxillin interact indirectly, immunofluorescence staining clearly confirms that GIT1 remains associated with peripheral FCs in the absence of GIT2 (Figure 3C).
Our results indicated that GIT1 remained associated with paxillin and FCs in the absence of GIT2. GIT1, like GIT2, can bind _β_PIX directly and _β_PIX in turn binds directly to PAK (Bagrodia et al, 1998, 1999; Manser et al, 1998). We speculated that GIT2 might regulate the assembly of the GIT1–_β_PIX–PAK complex. We examined the association of endogenous GIT1 and GIT2 with _β_PIX and PAK (without crosslinking). Figure 3I demonstrates that _β_PIX immunoprecipitates with both GIT1 and GIT2. Importantly, GIT1–_β_PIX and GIT2–_β_PIX complexes are not affected by GIT2 and GIT1 knockdown, respectively. This shows that GIT1 and GIT2 can bind independently of one another to _β_PIX. We expanded this observation to include immunoprecipitation of endogenous PAK. As expected, in control cells, PAK associates with _β_PIX, GIT1, and GIT2 (Figure 3J). Importantly, GIT1–_β_PIX–PAK and GIT2–_β_PIX–PAK complexes were found in cells depleted of GIT2 and GIT1, respectively. The presence of an intact GIT1–_β_PIX–PAK complex in GIT2 knockdown cells suggests that this complex is not sufficient to mediate inhibition of FA turnover and lamellipodial extensions. These findings support a model whereby GIT2 plays a nonredundant role at FAs to mediate repression of FA turnover and membrane ruffling.
Rac1 and Cdc42 are required for GIT2-regulated ruffling and turnover, respectively
The effects of GIT2 knockdown are reminiscent of those observed in cells expressing activated Cdc42 and Rac1. To address whether Cdc42 and Rac participated in the response to GIT2 knockdown, we transfected cells depleted of GIT1 or GIT2 with dominant-negative Cdc42(N17) or Rac1(N17). While expression of Cdc42(N17) only weakly inhibited membrane ruffling elicited by knockdown of GIT2 (data not shown), it substantially diminished the redistribution of paxillin from FAs to FCs (Figure 4A–D). In contrast, expression of Rac1(N17) abrogated lamellipodial extensions in cells with GIT2 knockdown (Figure 4I–L), but did not prevent paxillin redistribution (Figure 4E–H), indicating that Rac activation is required for membrane protrusion but not FA turnover. These results show that GIT2 functions to inhibit membrane protrusion and FA turnover by distinct Rac1- and Cdc42-dependent signaling pathways.
Figure 4.
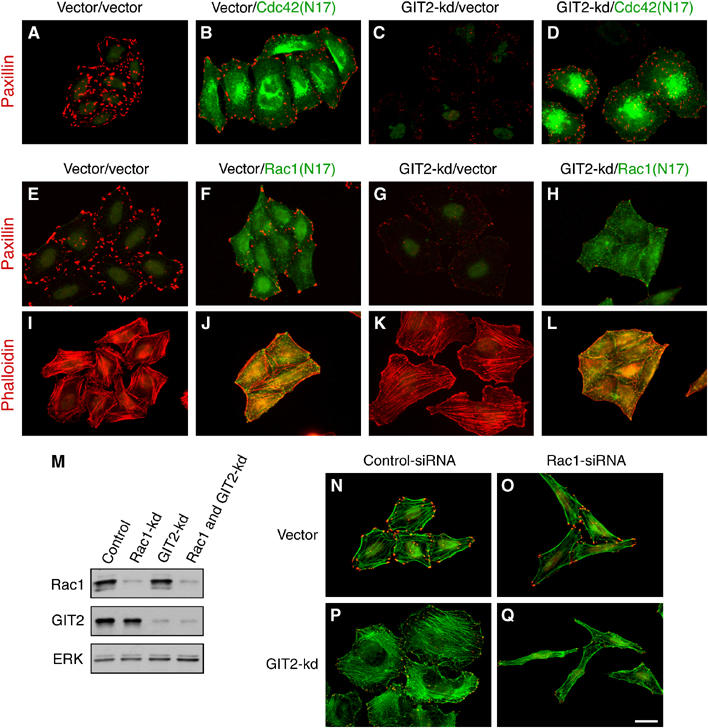
GIT2 represses Rac1 and Cdc42-dependent membrane ruffling and FA turnover, respectively. (A–D) HeLa cells were transfected with GIT2 shRNA with or without myc-tagged Cdc42(N17). Cells were stained for paxillin (red) and myc (green). (E–L) HeLa cells were transfected with GIT2 shRNA with or without myc-tagged Rac1(N17). Cells were stained for paxillin (red) and myc (green) (E–H), or for F-actin (red) and myc (green) (I–L). (M–Q) HeLa cells were transfected with control or Rac1 siRNA together with control or GIT2 shRNA. Cells were lysed and blotted for Rac1, GIT2, and ERK (M), or stained with phalloidin (green) and paxillin (red) (N–Q). Scale bar: 20 _μ_M.
To test conclusively the requirement for Rac1 in effects elicited by knockdown of GIT2, we devised a strategy that allowed us to simultaneously knockdown Rac1 and GIT2. HeLa cells were first transfected with a validated Rac1 siRNA duplex (Noritake et al, 2004) and 6 h later transfected with GIT2 shRNA. Following incubation overnight, cells were selected with puromycin for an additional 72 h and processed either for Western analysis or for immunofluorescence. This protocol leads to a simultaneous reduction in Rac1 and GIT2 levels (Figure 4M). As shown in Figure 4N–Q, introduction of Rac1 siRNA blocked lamellipodial extension induced by GIT2 knockdown. Interestingly, assessment of the bulk content of GTP-bound Rac1 revealed little change in Rac-GTP levels following GIT2 knockdown alone (data not shown), indicating that GIT2 may act locally to control Rac1 GTP-loading or recruitment, as has been suggested for the regulation of Rac1 by paxillin (Yano et al, 2000). However, we cannot exclude that GIT2 may function to inhibit a signaling pathway that acts in concert with Rac1 to regulate actin polymerization. Nevertheless, our results unambiguously identify Rac1-dependent actin remodeling as a target of GIT2-mediated repression.
_β_PIX is dispensable for the effects of GIT2 knockdown on cell motility
_β_PIX is a member of the Dbl family of Rho GEFs and can mediate nucleotide exchange on Rac1 (Manser et al, 1998; Feng et al, 2002). Since _β_PIX is the only detectable PIX family member expressed in HeLa cells (Manser et al, 1998), we speculated that GIT2 might normally function to inhibit the activation of _β_PIX. To test this possibility, we reduced _β_PIX expression by RNAi (Figure 5A and B). Interestingly, GIT2 protein levels were significantly reduced following _β_PIX knockdown, indicating that GIT2 is stabilized through association with _β_PIX. However, neither the enhanced lamellipodia formation nor FA turnover seen following GIT2 knockdown required _β_PIX. In fact, cells lacking _β_PIX showed an increased sensitivity to GIT2 loss, with these cells displaying even more exaggerated membrane extensions than cells with GIT1&2 knockdown alone (Figure 5C–F). Knockdown of _β_PIX was also not required for enhanced cell spreading on fibronectin induced by GIT1&2 knockdown (Figure 5G–J). Interestingly, knockdown of _β_PIX weakly mimicked the effects of GIT2 knockdown, with _β_PIX-deficient cells showing reduced paxillin staining and enhanced spreading (Figure 5E and I). This may result from the reduction in GIT2 levels observed in _β_PIX-deficient cells. These findings demonstrate that _β_PIX is dispensable for the changes in actin and FA structure induced by GIT2 knockdown. Moreover, _β_PIX indirectly appears to contribute to GIT2-mediated suppression of ruffling and FA turnover by stabilizing GIT2.
Figure 5.
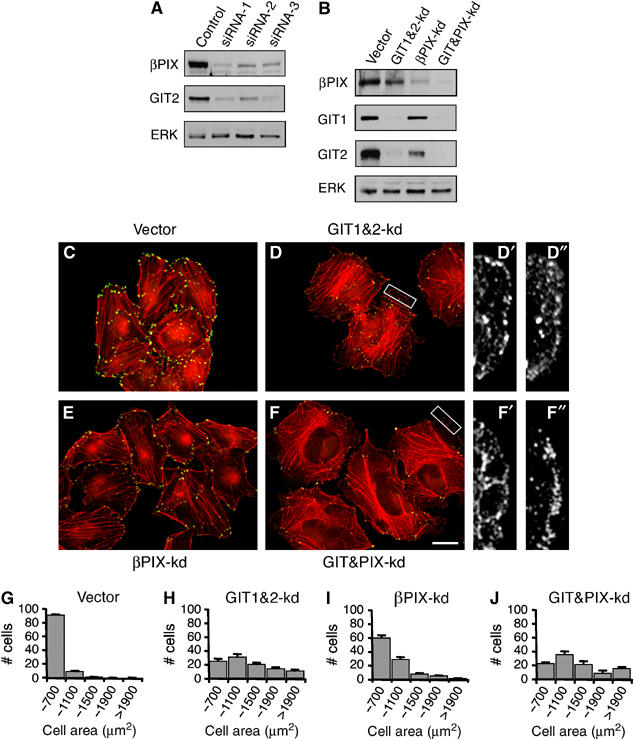
The Rac GEF _β_PIX is required for stabilization of GIT2, but not for membrane ruffling induced by GIT2 knockdown. (A) HeLa cells were transfected with control or _β_PIX siRNA duplexes and blotted for _β_PIX, GIT2, and ERK. (B–F) HeLa cells were transfected with control siRNA or _β_PIX siRNA-1 together with vector or GIT1&2 shRNA. Cells were probed for _β_PIX, GIT1, GIT2, and ERK (B), or stained for paxillin (green) and F-actin (red) (C–F). Enlarged images shows that lamellipodial extensions visualized by actin staining (D′, F′) contain paxillin localized to FCs (D″, F″). (G–J) Quantification of cell size in control, GIT1&2-kd, _β_PIX-kd, and GIT&PIX-kd cells following 1 h replating on fibronectin. Scale bar: 20 _μ_M.
Effects of GIT2 knockdown are dependent on the SH2–SH3 adaptor molecule Crk
Our results indicate that GIT2 functions at FAs in a _β_PIX-independent manner to inhibit membrane ruffling and cell spreading. The SH2–SH3 adaptor molecule Crk is a paxillin-binding partner that functions as part of a signaling pathway that promotes cell motility (Chodniewicz and Klemke, 2004). Moreover, a recent study has demonstrated the existence of a Crk–paxillin–GIT2–_β_PIX complex involved in Rac-dependent epithelial cell motility (Lamorte et al, 2003). To address whether GITs function to repress Crk-dependent actin polymerization and cell spreading, we inhibited CrkII function by RNAi as HeLa cells express readily detectable amounts of CrkII. To knockdown GIT1&2 together with CrkII, HeLa cells were cotransfected with CrkII and GIT1&2 shRNA expression plasmids that encoded for puromycin and GFP, respectively. This resulted in a nearly pure population of cotransfected cells 4 days following transfection (puromycin resistant and >95% GFP positive). This protocol results in the simultaneous reduction of GIT1, GIT2, and CrkII expression (Figure 6A). Inhibition of CrkII substantially reduced lamellipodial extensions (Figure 6B–E) and cell spreading (Figure 6F–I) in GIT1&2 knockdown cells. Interestingly, CrkII knockdown had little effect on paxillin redistribution (Figure 6B–E), suggesting that CrkII induces Rac1-dependent membrane protrusion, but not Cdc42-mediated FA turnover, in response to GIT2 loss.
Figure 6.
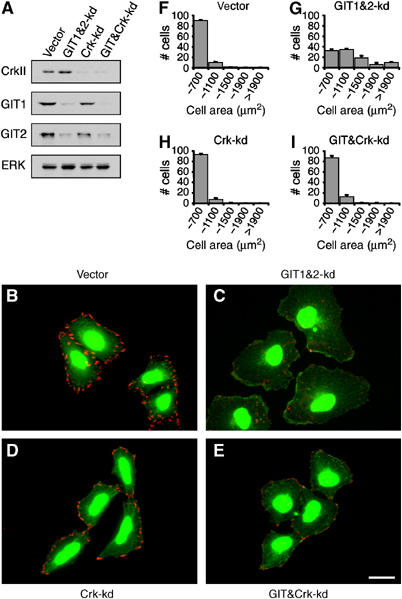
CrkII is required for cell spreading induced by GIT2 knockdown. (A) HeLa cells were transfected with CrkII and GIT1&2 shRNA expression plasmids, which encoded for puromycin resistance and GFP, respectively, or appropriate empty control plasmids. Transfected cells were transiently selected with puromycin and then blotted for CrkII, GIT1, GIT2, and ERK. (B–E) Control, Crk-kd, GIT1&2-kd, and GIT&Crk-kd cells were stained for paxillin (red). GIT1&2-kd cells could be easily identified by expression of GFP. (F–I) Quantification of cell size following 1 h replating on fibronectin. Scale bar: 20 _μ_M.
GIT2 suppresses migration and invasion in nontransformed mammary epithelial MCF10A cells
We examined the effect of GIT2 loss on the motility of the nontransformed mammary epithelial cell line MCF10A. These cells retain many characteristics of normal epithelial cells, including the capacity to growth arrest and polarize (Debnath et al, 2003). We employed retroviral-mediated gene transfer to generate MCF10A cell pools expressing GIT2 shRNA (Figure 7A). Rather than growing as organized islets, GIT2 knockdown cells exhibited a scattered and disorganized morphology (Figure 7B and C). Like HeLa cells, MCF10A cells displayed a robust increase in lamellipodial extensions upon knockdown of GIT2. Moreover, _β_-catenin, which normally localizes to adherens junctions in MCF10A cells, accumulated intracellularly, likely the result of removal of adhesion molecules from membranes no longer engaged in formation of cell–cell contacts. Importantly, no gross morphological effects were observed upon GIT1 knockdown (data not shown). To quantify the effects of GIT2 knockdown on migratory behavior of MCF10A cells, we examined the extent to which control or GIT2 knockdown cells transgressed uncoated and MATRIGELTM-coated transwell filters. Cells with GIT2 knockdown exhibited a ∼2.5-fold increase over control cells in migration across uncoated filters (Figure 7D), while showing a >8-fold increase in migration across matrix-coated filters (Figure 7E). These results demonstrate that loss of GIT2 alone is sufficient to induce migration in nontransformed epithelial cells.
Figure 7.
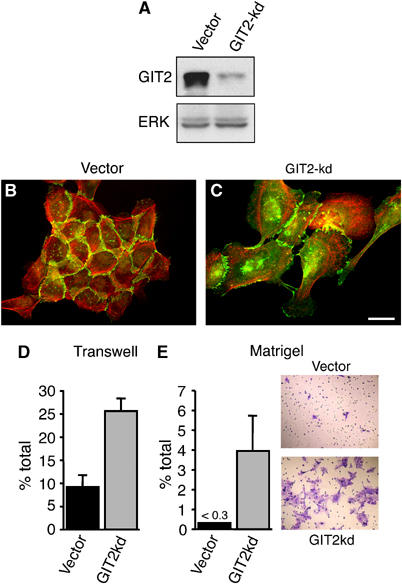
GIT2 represses migration of MCF10A cells. MCF10A cells stably expressing GIT2 shRNA were blotted for GIT2 (A) and stained for _β_-catenin in green and F-actin in red (B, C). (D, E) To quantify changes in motility, control and GIT2 knockdown cells were harvested and added to the top chamber of either uncoated or MATRIGELTM-coated transwell filters. The number of cells migrated to the bottom chamber is expressed as a percentage of the total cells plated. Color panels in panel E show staining of migrated cells with crystal violet. Values are means±s.d. (_n_=5 filters) from a representative experiment. Scale bar: 20 _μ_M.
Discussion
GIT proteins have been implicated in Rac-dependent FA turnover, membrane protrusion, and cell migration, but the literature is conflicting with respect to whether GIT molecules function as positive or negative regulators of these processes. The bulk of the published work relied on overexpression studies to assign a stimulatory role for GITs (Di Cesare et al, 2000; Zhao et al, 2000; West et al, 2001; Manabe et al, 2002; Lamorte et al, 2003; Yin et al, 2005). Moreover, it was not clear if GIT proteins repress or activate Rac, nor was it known whether GIT1 and GIT2 play distinct or redundant roles. An added complexity is that GIT2 mRNA undergoes alternative splicing (Premont et al, 2000).
Using RNAi, we have specifically targeted the expression of endogenous GIT1 and GIT2. We have discovered that endogenous GIT2 functions as a repressor of FA turnover and membrane protrusion. We have unambiguously identified the longest form of GIT2 as the mediator of these functions. First, we have determined that knockdown of endogenous GIT2 does not affect the levels of GIT1. Second, we have targeted GIT2 using multiple siRNAs, demonstrating that the longest from of GIT2 is required to repress membrane ruffling and FA turnover. Finally, we have restored GIT2 function in knockdown cells by introduction of a GIT2-long cDNA lacking the siRNA recognition sequence and showed that they are resistant to knockdown of endogenous GIT2. Taken together, these results demonstrate that GIT2-long plays a nonredundant role in the inhibition of membrane ruffling and FA turnover.
Staining of HeLa cells with GIT2-specific antibodies shows that endogenous GIT2 localizes to FAs, including FAs stabilized by the expression of constitutively active RhoA. This finding differs from previous observations suggesting that GIT2 localized only to Cdc42- and Rac-dependent FCs, but not mature FAs (Brown et al, 2002). This discrepancy likely results from the different antibody and fixation methods used in this study. Nevertheless, we clearly identify both GIT1 and GIT2 as resident FA proteins. This finding does not contradict the observation that GIT2 is also found in FCs. In fact, we find that GIT2 is rapidly targeted to nacent FCs (i.e. upon wounding or cell spreading), where it likely plays a requisite role in their maturation to FAs (data not shown).
Given the effects of GIT2 knockdown on actin and FA structure, we postulated that GIT2 might regulate the association of GIT1 with FAs. This would explain why GIT1 could not substitute for GIT2 in mediating repression. However, we found that GIT1 remained associated with paxillin and FCs in the absence of GIT2. Moreover, we showed that GIT1–_β_PIX–PAK and GIT2–_β_PIX–PAK complexes exist in cells lacking GIT2 and GIT1, respectively. These observations demonstrate that an intact paxillin-GIT1–_β_PIX–PAK complex is unable to functionally compensate for the loss of GIT2 in the repression of FA turnover and cell spreading. We also tested the possibility that GIT2 might inhibit GIT1 function, consistent with previous suggestions that GIT1 may induce FA turnover and membrane protrusion (Di Cesare et al, 2000; Zhao et al, 2000; Manabe et al, 2002). However, knockdown of GIT1 exaggerates membrane ruffling and cell spreading in GIT2-deficient cells. Collectively, these results suggest that both GIT1 and GIT2 act as inhibitors of ruffling and FA turnover, with GIT2 playing both a necessary and sufficient role.
We show that GIT2 inhibits membrane ruffling and FA turnover via distinct pathways. While inhibition of Rac1 signaling by expression of dominant inhibitory Rac1(N17) or by suppression of Rac1 by RNAi attenuates membrane protrusion and cell spreading, it has no effect on the redistribution of paxillin induced by GIT2 knockdown. This contrasts with the effects of Cdc42(N17) expression, which selectively antagonizes the increased FA turnover induced by GIT2 knockdown. The coordinated increase in FA turnover and Rac-dependent actin polymerization is a hallmark of the leading edge of motile cells and our data indicate that GIT2 is a key integrator of these processes. One mechanism for regulating GIT2 functions is through phosphorylation by FAK and Src, which recently has been reported as an obligate step in recruitment of GIT2 to FAs (Brown et al, 2005). As both FAK and Src have been traditionally associated with FA turnover, their promotion of GIT2 recruitment may at first glance seem paradoxical. However, our data suggest that GIT2 may provide a critical negative feedback mechanism to facilitate FA maturation and dampen excessive actin protrusiveness.
How can our findings be reconciled with previous observations suggesting that GIT1 acted as positive regulator of motility and FA turnover? Previous reports have found that the effects of GIT1 expression on actin and FA structure are recapitulated by expression of its C-terminal paxillin-binding domain, in one report consisting of as little as the C-terminal 125 amino acids (Di Cesare et al, 2000; Zhao et al, 2000; Manabe et al, 2002). In light of our findings, these results are most consistent with a scenario in which the effects elicited by overexpression of GIT1 result from displacement of GIT2. Moreover, the role of GIT2 as a repressor is supported by the reported effects of overexpression of paxillinΔLD4, a paxillin mutant that fails to recruit GIT2 to FAs. CHO.K1 cells expressing paxillinΔLD4 show increased Rac1 activation and membrane protrusion similar to those observed following GIT2 knockdown (West et al, 2001; Brown et al, 2005). What remained unclear from these studies was whether the increased Rac activation seen in paxillinΔLD4 cells resulted from a loss of GIT2-mediated repression or a liberation of active GIT2. Our data clarify these observations to show that it is likely a lack of GIT2 recruitment that underlies the effects of paxillinΔLD4 expression.
While our data indicate that GIT1 is largely dispensable for the inhibition of Rac1 at FAs, a recent work by Nishiya et al (2005) suggests that GIT1 may serve such a role when associated with _α_4_β_1 integrin. While not localized to FAs in most cell types, _α_4_β_1 plays a fundamental role in cell migration, being expressed in a subset of migratory cell types, including leukocytes, neural crest cells, and smooth muscle cells. Specifically, recruitment of GIT1 to _α_4 integrins via its association with paxillin acts to locally inhibit activation of Rac1. GIT1 may also be recruited to the lateral membranes of polarized epithelial cells through its association with _β_PIX (Zegers et al, 2003; Audebert et al, 2004). As Rac plays a central role in the formation of epithelial cell contacts (Fukata and Kaibuchi, 2001), it will be important to determine whether GIT1 functions at lateral membranes to inhibit activation of Rac.
_β_PIX binds directly to GIT1 and GIT2 and is targeted to FAs by a GIT-dependent mechanism (Turner et al, 1999; Zhao et al, 2000; Brown et al, 2002). _β_PIX can mediate changes in actin and FA structure in a manner dependent on its catalytic activity (Manser et al, 1998; Obermeier et al, 1998; Koh et al, 2001; Lee et al, 2001). However, we found that _β_PIX is not required for the increase in membrane ruffling or FA turnover induced by GIT2 knockdown, suggesting that _β_PIX is not an activator of Rac1 in this context. In contrast, knockdown of _β_PIX functions synergistically with knockdown of GIT2 to enhance membrane ruffling and cell spreading. This effect may be explained by our observation that _β_PIX regulates GIT2 stability, as we show that knockdown of _β_PIX causes a significant reduction in GIT2 protein levels. _β_PIX has also been reported to facilitate entry of GIT2 into FCs (Brown et al, 2002). Both scenarios are consistent with our observation that _β_PIX knockdown on its own elicits effects that weakly mimic those observed following GIT2 knockdown.
The apparent GEF-independent function of _β_PIX in GIT2 knockdown cells indicates more complex roles for _β_PIX in Rho signaling than first thought. The DH domain of _β_PIX is conformationally constrained, and as a result full-length _β_PIX displays only modest GEF activity against Rac1 (Manser et al, 1998; Feng et al, 2002). Recent work has even suggested that under some circumstances PIX proteins may function as an effector rather than activator of Rho signaling pathways. For example, both α and _β_PIX have been found to interact directly with activated (GTP-bound) Cdc42 (Wu et al, 2003; Baird et al, 2005). _α_PIX has also been shown to bind directly to the p85 regulatory subunit of PI3-kinase and interact indirectly with membrane receptors via its association with PAK (Obermeier et al, 1998; Yoshii et al, 1999; Li et al, 2003). These findings raise the possibility that PIX proteins function upstream of GIT2 to mediate its recruitment to membrane receptors and/or activated Cdc42. In this manner, PIX could facilitate the localized and polarized recruitment of GIT2 to sites of cytokine or growth factor stimulation, that is, the leading edge of migrating cells. Once there, in association with paxillin, GIT2 would function as a break to further actin polymerization and FA turnover.
Our data demonstrate that GIT2 inhibits CrkII-dependent actin polymerization and cell spreading. Originally identified as the oncogene fusion product of the CT10 chicken retrovirus, it is now known that Crk functions as an essential adaptor in evolutionarily conserved signaling pathways that couple integrins to signals that control cell proliferation, survival, and motility (Chodniewicz and Klemke, 2004). Crk binding partners include numerous proteins involved in cell migration, including FA proteins such as paxillin and 130Cas, and motility-promoting signaling molecules such as Sos, PI3 K, Gab2, and the Rac1 exchange factor Dock180. Interestingly, the CrkII SH2 domain has recently been shown to interact directly with phosphorylated GIT2 and a CrkII–paxillin–GIT2–_β_PIX complex has been shown to participate in induction of epithelial motility (Lamorte et al, 2003; Brown et al, 2005). GITs have been suggested to function as Arf6 GAPs at the cell periphery (Di Cesare et al, 2000; Nishiya et al, 2005). Arf6 is a potent inducer of epithelial scattering and migration and a recent report indicates that this is mediated by the Dock180-dependent activation of Rac (Santy et al, 2005). However, expression of dominant-negative Arf6(T27N) did not affect the induction of spreading by GIT2 knockdown (our unpublished observations). While this suggests that GIT2 is not functioning as an Arf6GAP in this context, the possibility that Dock180 is regulating Rac1 activation by GIT2 remains intriguing. We are currently examining the role of CrkII effector pathways in GIT2-dependent motile responses.
We find that GIT2 acts as an obligate repressor of motility and Matrigel invasion in nontransformed mammary epithelial cells. We have previously invoked a role for _β_PIX and activated PAK in contact inhibition of growth and motility in epithelial cells (Zegers et al, 2003). Taken together with our observations here, these data suggest that the PAK–_β_PIX–GIT2 complex plays a fundamental role in epithelial morphogenesis and may be affected during oncogenic transformation of epithelial cells. This intriguing possibility is further supported by the finding that the _β_PIX–GIT complex directly binds the mammalian orthologue of the Drosophila tumor suppressor Scribble (Audebert et al, 2004). GIT2 message is widely expressed in human tissues and a broad variety of cell types (NCBI) and GIT2 protein is readily detected in both transformed (HeLa and 293) and nontransformed (MCF10A, EPH4, and MDCK) cell lines (our unpublished observations). These findings suggest that GIT2 does not function as a classic tumor suppressor, although we cannot exclude the possibility that some tumor types may show loss of GIT2. Instead, we suggest that GIT2 is a candidate target of oncogenic signaling pathways that induce motility and/or invasion. In this scenario, oncogenic signals would inhibit the association of GIT2 with paxillin, leading to the induction of motility. We are currently investigating the role of GIT2 in oncogenic transformation of epithelial cells.
Materials and methods
Plasmids and antibodies
Antibodies rabbit anti-GIT1, goat anti-GIT2, rabbit anti-ERK, rabbit anti-Myc, rabbit anti-PAK, and mouse anti-vimentin were purchased from Santa Cruz Biotechnology. Mouse anti-GIT1, mouse anti-_β_-catenin, mouse anti-paxillin, mouse anti-PKL, and mouse anti-Crk were from BD Biosciences. Secondary antibodies and phalloidin were from Molecular Probes. Additional reagents used were mouse anti-vinculin (Sigma), rabbit anti-_β_PIX (Chemicon), and fibronectin-coated coverslips (BD Biosciences). Rat GIT1 and human GIT2 expression constructs were obtained from Dr Richard Premont. A detailed description of the generation and purification of the GIT2-specific antibodies is provided in Supplementary Methods.
Cell culture and transfection
MCF10A cells were cultured as described previously (Debnath et al, 2003). For transient shRNA-mediated knockdown, HeLa cells were transfected using FuGENE 6 (Roche). At 24 h after transfection, cells were passaged 1:5 into growth medium containing 2.5 _μ_g/ml puromycin and cultured for an additional 72 h. For immunofluorescence analysis, 3 × 104 cells were passaged onto fibronectin-coated coverslips (BD Biosciences). For knockdown of _β_PIX and Rac1, HeLa cells were transfected with 50 nM of siRNA duplexes using LipofectAmine 2000 (Invitrogen). HeLa cells were incubated with siRNA–lipid complexes for 6 h in growth medium. siRNA-transfected cells were either incubated in growth medium for an additional 72 h or transfected with GIT1 and GIT2 shRNA vectors and selected with puromycin as described above. shRNA sequence information and cloning is described in Supplementary Methods.
Immunofluorescence microscopy
Cells were fixed in 2% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 15 min at RT. Processing of samples was generally carried out as described previously (Zegers et al, 2003). Mouse anti-paxillin mAb (cl #165) was used for detection of paxillin. For labeling with GIT1 and GIT2 rabbit polyclonal antibodies, cells were fixed for 2 min at RT in methanol/acetone (1:1).
Immunoprecipitation
For analysis of protein levels following knockdown, cells were lysed in sample buffer containing 0.5% SDS. For protein coimmunoprecipitation, HeLa cells were lysed in Gold-lysis buffer (1% Triton X-100, 20 mM Tris (pH 8.0), 137 mM NaCl, 10 mM MgCl2, 15% glycerol) containing protease inhibitors. Protein concentrations were determined using a BCATM Kit (Pierce). Aliquots of 1 ml, containing approximately 2 mg of total protein, were incubated overnight at 4°C with 1 _μ_g rabbit anti-GIT1, 2 _μ_l rabbit anti-GIT2 serum, or 0.5 _μ_g rabbit anti-PAK antibodies (sc #882). Immunoprecipitates were captured on Protein-G beads. Lysates and immunoprecipitates were subjected to SDS–PAGE and transfered to PVDF. A detailed protocol for reversible formaldehyde crosslinking is provided in Supplementary Methods.
HeLa cell spreading
HeLa cells at 96 h post-transfection were harvested by incubation at 37°C in PBS (without Ca2+ and Mg2+) supplemented with 4 mM EDTA and 1 mM EGTA. Cells were pelleted and resuspended in growth medium at 37°C and plated onto fibronectin coverslips by centrifugation 5 min at 200 g and allowed to spread for 60 min. Cells were either processed for immunofluorescence or examined by phase-contrast microscopy. To quantify cell size, phase-contrast images were taken and individual cells were outlined in Adobe Photoshop. The number of pixels per cell was determined and used to calculate the area of each cell. In all, 100 cells per condition were counted in each of three independent experiments.
MCF10A migration and invasion
To assay for changes in motility, puromycin (2 _μ_g/ml) selected MCF10A stable pools were trypsinized and 1 × 105 cells were added to the top chambers of 24-well transwell plates (BD Biosciences; 8 _μ_M pore size) either uncoated or coated with growth factor reduced MATRIGELTM. Assays were conducted according to the manufacturer's protocol using MCF10A growth medium as a chemoattractant. After overnight incubation, top (nonmigrated) cells were removed, and bottom (migrated) cells quantified by fluorescent labeling using a CyQuant ® cell proliferation assay kit (Molecular Probes). The number of migrated cells was expressed as a fraction of the total cells plated.
Supplementary Material
Supplementary Methods
Acknowledgments
Dr Richard Premont generously provided reagents for this work. We are thankful to Drs Tiziana Parisi and Jeffrey Settleman for critical reading of the manuscript. The salary for SRF was defrayed in part by a Pilot Fund Award from BBRI. This work was supported by R01 CA092354 to SHH.
References
- Audebert S, Navarro C, Nourry C, Chasserot-Golaz S, Lecine P, Bellaiche Y, Dupont JL, Premont RT, Sempere C, Strub JM, Van Dorsselaer A, Vitale N, Borg JP (2004) Mammalian Scribble forms a tight complex with the betaPIX exchange factor. Curr Biol 14: 987–995 [DOI] [PubMed] [Google Scholar]
- Bagrodia S, Bailey D, Lenard Z, Hart M, Guan JL, Premont RT, Taylor SJ, Cerione RA (1999) A tyrosine-phosphorylated protein that binds to an important regulatory region on the cool family of p21-activated kinase-binding proteins. J Biol Chem 274: 22393–22400 [DOI] [PubMed] [Google Scholar]
- Bagrodia S, Taylor SJ, Jordon KA, Van Aelst L, Cerione RA (1998) A novel regulator of p21-activated kinases. J Biol Chem 273: 23633–23636 [DOI] [PubMed] [Google Scholar]
- Baird D, Feng Q, Cerione RA (2005) The Cool-2/alpha-Pix protein mediates a Cdc42-Rac signaling cascade. Curr Biol 15: 1–10 [DOI] [PubMed] [Google Scholar]
- Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE (2005) Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol Biol Cell 16: 4316–4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Turner CE (2004) Paxillin: adapting to change. Physiol Rev 84: 1315–1339 [DOI] [PubMed] [Google Scholar]
- Brown MC, West KA, Turner CE (2002) Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol Biol Cell 13: 1550–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K (2004) Rho and Rac take center stage. Cell 116: 167–179 [DOI] [PubMed] [Google Scholar]
- Chodniewicz D, Klemke RL (2004) Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta 1692: 63–76 [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30: 256–268 [DOI] [PubMed] [Google Scholar]
- Di Cesare A, Paris S, Albertinazzi C, Dariozzi S, Andersen J, Mann M, Longhi R, de Curtis I (2000) p95-APP1 links membrane transport to Rac-mediated reorganization of actin. Nat Cell Biol 2: 521–530 [DOI] [PubMed] [Google Scholar]
- Feng Q, Albeck JG, Cerione RA, Yang W (2002) Regulation of the Cool/Pix proteins: key binding partners of the Cdc42/Rac targets, the p21-activated kinases. J Biol Chem 277: 5644–5650 [DOI] [PubMed] [Google Scholar]
- Fukata M, Kaibuchi K (2001) Rho-family GTPases in cadherin-mediated cell–cell adhesion. Nat Rev Mol Cell Biol 2: 887–897 [DOI] [PubMed] [Google Scholar]
- Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279: 509–514 [DOI] [PubMed] [Google Scholar]
- Hall DB, Struhl K (2002) The VP16 activation domain interacts with multiple transcriptional components as determined by protein–protein cross-linking in vivo. J Biol Chem 277: 46043–46050 [DOI] [PubMed] [Google Scholar]
- Kim S, Ko J, Shin H, Lee JR, Lim C, Han JH, Altrock WD, Garner CC, Gundelfinger ED, Premont RT, Kaang BK, Kim E (2003) The GIT family of proteins forms multimers and associates with the presynaptic cytomatrix protein Piccolo. J Biol Chem 278: 6291–6300 [DOI] [PubMed] [Google Scholar]
- Koh CG, Manser E, Zhao ZS, Ng CP, Lim L (2001) Beta1PIX, the PAK-interacting exchange factor, requires localization via a coiled-coil region to promote microvillus-like structures and membrane ruffles. J Cell Sci 114: 4239–4251 [DOI] [PubMed] [Google Scholar]
- Lamorte L, Rodrigues S, Sangwan V, Turner CE, Park M (2003) Crk associates with a multimolecular Paxillin/GIT2/beta-PIX complex and promotes Rac-dependent relocalization of Paxillin to focal contacts. Mol Biol Cell 14: 2818–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Eom M, Lee SJ, Kim S, Park HJ, Park D (2001) BetaPix-enhanced p38 activation by Cdc42/Rac/PAK/MKK3/6-mediated pathway. Implication in the regulation of membrane ruffling. J Biol Chem 276: 25066–25072 [DOI] [PubMed] [Google Scholar]
- Li Z, Hannigan M, Mo Z, Liu B, Lu W, Wu Y, Smrcka AV, Wu G, Li L, Liu M, Huang CK, Wu D (2003) Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell 114: 215–227 [DOI] [PubMed] [Google Scholar]
- Loo TH, Ng YW, Lim L, Manser E (2004) GIT1 activates p21-activated kinase through a mechanism independent of p21 binding. Mol Cell Biol 24: 3849–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe R, Kovalenko M, Webb DJ, Horwitz AR (2002) GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J Cell Sci 115: 1497–1510 [DOI] [PubMed] [Google Scholar]
- Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L (1998) PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol Cell 1: 183–192 [DOI] [PubMed] [Google Scholar]
- Mazaki Y, Hashimoto S, Okawa K, Tsubouchi A, Nakamura K, Yagi R, Yano H, Kondo A, Iwamatsu A, Mizoguchi A, Sabe H (2001) An ADP-ribosylation factor GTPase-activating protein Git2-short/KIAA0148 is involved in subcellular localization of paxillin and actin cytoskeletal organization. Mol Biol Cell 12: 645–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiya N, Kiosses WB, Han J, Ginsberg MH (2005) An alpha4 integrin–paxillin–Arf–GAP complex restricts Rac activation to the leading edge of migrating cells. Nat Cell Biol 7: 343–352 [DOI] [PubMed] [Google Scholar]
- Noritake J, Fukata M, Sato K, Nakagawa M, Watanabe T, Izumi N, Wang S, Fukata Y, Kaibuchi K (2004) Positive role of IQGAP1, an effector of Rac1, in actin-meshwork formation at sites of cell–cell contact. Mol Biol Cell 15: 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier A, Ahmed S, Manser E, Yen SC, Hall C, Lim L (1998) PAK promotes morphological changes by acting upstream of Rac. EMBO J 17: 4328–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris S, Longhi R, Santambrogio P, de Curtis I (2003) Leucine-zipper-mediated homo- and hetero-dimerization of GIT family p95-ARF GTPase-activating protein, PIX-, paxillin-interacting proteins 1 and 2. Biochem J 372: 391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Claing A, Vitale N, Perry SJ, Lefkowitz RJ (2000) The GIT family of ADP-ribosylation factor GTPase-activating proteins. Functional diversity of GIT2 through alternative splicing. J Biol Chem 275: 22373–22380 [DOI] [PubMed] [Google Scholar]
- Premont RT, Perry SJ, Schmalzigaug R, Roseman JT, Xing Y, Claing A (2004) The GIT/PIX complex: an oligomeric assembly of GIT family ARF GTPase-activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell Signal 16: 1001–1011 [DOI] [PubMed] [Google Scholar]
- Santy LC, Ravichandran KS, Casanova JE (2005) The DOCK180/Elmo complex couples ARNO-mediated Arf6 activation to the downstream activation of Rac1. Curr Biol 15: 1749–1754 [DOI] [PubMed] [Google Scholar]
- Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS (1999) Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J Cell Biol 145: 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N, Patton WA, Moss J, Vaughan M, Lefkowitz RJ, Premont RT (2000) GIT proteins, a novel family of phosphatidylinositol 3,4,5-trisphosphate-stimulated GTPase-activating proteins for ARF6. J Biol Chem 275: 13901–13906 [DOI] [PubMed] [Google Scholar]
- Webb DJ, Parsons JT, Horwitz AF (2002) Adhesion assembly, disassembly and turnover in migrating cells—over and over and over again. Nat Cell Biol 4: E97–E100 [DOI] [PubMed] [Google Scholar]
- West KA, Zhang H, Brown MC, Nikolopoulos SN, Riedy MC, Horwitz AF, Turner CE (2001) The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J Cell Biol 154: 161–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WJ, Tu S, Cerione RA (2003) Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell 114: 715–725 [DOI] [PubMed] [Google Scholar]
- Yano H, Uchida H, Iwasaki T, Mukai M, Akedo H, Nakamura K, Hashimoto S, Sabe H (2000) Paxillin alpha and Crk-associated substrate exert opposing effects on cell migration and contact inhibition of growth through tyrosine phosphorylation. Proc Natl Acad Sci USA 97: 9076–9081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G, Zheng Q, Yan C, Berk BC (2005) GIT1 is a scaffold for ERK1/2 activation in focal adhesions. J Biol Chem 280: 27705–27712 [DOI] [PubMed] [Google Scholar]
- Yoshii S, Tanaka M, Otsuki Y, Wang DY, Guo RJ, Zhu Y, Takeda R, Hanai H, Kaneko E, Sugimura H (1999) alphaPIX nucleotide exchange factor is activated by interaction with phosphatidylinositol 3-kinase. Oncogene 18: 5680–5690 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Cohen M, Addadi L, Geiger B (2004) Hierarchical assembly of cell–matrix adhesion complexes. Biochem Soc Trans 32: 416–420 [DOI] [PubMed] [Google Scholar]
- Zegers MM, Forget MA, Chernoff J, Mostov KE, Ter Beest MB, Hansen SH (2003) Pak1 and PIX regulate contact inhibition during epithelial wound healing. EMBO J 22: 4155–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZS, Manser E, Loo TH, Lim L (2000) Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol Cell Biol 20: 6354–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods