The TT8 Gene Encodes a Basic Helix-Loop-Helix Domain Protein Required for Expression of DFR and BAN Genes in Arabidopsis Siliques (original) (raw)
Abstract
The TRANSPARENT TESTA8 (TT8) locus is involved in the regulation of flavonoid biosynthesis in Arabidopsis. The tt8-3 allele was isolated from a T-DNA–mutagenized Arabidopsis collection and found to be tagged by an integrative molecule, thus permitting the cloning and sequencing of the TT8 gene. TT8 identity was confirmed by complementation of tt8-3 and sequence analysis of an additional allele. The TT8 gene encodes a protein that displays a basic helix-loop-helix at its C terminus and represents an Arabidopsis ortholog of the maize R transcription factors. The TT8 transcript is present in developing siliques and in young seedlings. The TT8 protein is required for normal expression of two flavonoid late biosynthetic genes, namely, DIHYDROFLAVONOL 4-REDUCTASE (DFR) and BANYULS (BAN), in Arabidopsis siliques. Interestingly, TRANSPARENT TESTA GLABRA1 (TTG1) and TT2 genes also control the expression of DFR and BAN genes. Our results suggest that the TT8, TTG1, and TT2 proteins may interact to control flavonoid metabolism in the Arabidopsis seed coat.
INTRODUCTION
Flavonoids are derived from phenylalanine and malonyl-CoA and constitute one of the largest groups of secondary metabolites in plants. They are based on a 15-carbon skeleton, which can be modified to yield different subclasses, including flavonols, anthocyanins, and proanthocyanidins (Figure 1). In plants, flavonoid derivatives are responsible for the pigmentation pattern of vegetative parts and seeds and are involved in a wide range of biological functions. For example, they protect against UV radiation, serve as signal molecules in plant–microbe interactions, and participate in plant defense responses (reviewed in Dooner et al., 1991; Koes et al., 1994; Dixon and Paiva, 1995; Shirley, 1996). Recent studies have also stressed the involvement of flavonoids in seed coat–imposed dormancy as well as in seed storability (Winkel-Shirley, 1998; Debeaujon et al., 2000). Moreover, flavonoids are receiving increasing interest as health-promoting components of animal and human diets (Lairon and Amiot, 1999). These diverse roles can be correlated, at least in part, with the well-documented antioxidant properties of phenylpropanoid derivatives, especially flavonoids (Rice-Evans et al., 1997), and with their inhibitory effect on enzymatic activities (Castelluccio et al., 1995).
Figure 1.

Outline of the Flavonoid Metabolism in Arabidopsis.
Flavonoids are secondary metabolites derived from products of the phenylpropanoid biosynthetic pathway and the Krebs cycle. The final compounds include flavonols (colorless pigments), anthocyanins (pink and red pigments), and condensed tannins (brown pigments). The characterized enzymes are shown in boldface. The various mutants identified are noted adjacent to the step they affect, with putative regulatory loci given in parentheses. ban, banyuls; CHI, chalcone isomerase; CHS, chalcone synthase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumarate:CoA ligase; DFR, dihydroflavanol 4-reductase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; FLS, flavonol synthase; icx1, increased chalcone synthase expression1; LAR, leucoanthocyanidin reductase; LDOX, leucoanthocyanidin dioxygenase; PAL, phenylalanine ammonia-lyase; tt, transparent testa; ttg, transparent testa glabra.
Investigating the structure and regulation of the flavonoid biosynthetic pathway in plants may thus help us to better understand and monitor flavonoid metabolism with regard to properties of the end products (Weisshaar and Jenkins, 1998). Flavonoid biosynthesis has been studied extensively by several methods, from protein purification to screening libraries with heterologous probes (reviewed in Holton and Cornish, 1995). The ubiquitous and nonessential nature of pigments for plant viability has made it possible to identify many flavonoid mutants, which has facilitated the genetic and molecular dissection of the pathway. To date, most of the enzymes involved in flavonoid biosynthesis have been characterized in several plant species, including maize, snapdragon, petunia (Holton and Cornish, 1995), and Arabidopsis (Shirley et al., 1995; Bharti and Khurana, 1997). The first three steps are catalyzed successively by chalcone synthase (CHS), chalcone isomerase (CHI), and flavanone 3-hydroxylase (F3H). Dihydrokaempferol can be subsequently hydroxylated by flavonoid 3′-hydroxylase (F3′H), giving rise to dihydroquercetin, or converted by the dihydroflavonol 4-reductase (DFR), resulting in anthocyanin-type end products (Figure 1).
The accumulation of flavonoids within plants or seeds is subject to fine temporal and spatial control involving several levels of regulation (e.g., transcriptional or post-translational regulation) and diverse developmental stimuli or environmental factors (Procissi et al., 1997; Burbulis and Winkel-Shirley, 1999; Pelletier et al., 1999). Despite numerous studies, the molecular mechanisms involved in the regulation of flavonoid biosynthesis remain to be identified. The bulk of our information concerning the regulatory network comes from genetic and molecular analyses of maize mutations. Two major classes of transcription factors have been described: the R/B family, which shows sequence homology to the basic helix-loop-helix (bHLH) DNA binding/dimerization region found in animal MYC oncogene products (Ludwig et al., 1989), and the C1/Pl family, which encodes proteins with similarity to the DNA binding domain of the mammalian MYB proto-oncogene proteins (Cone et al., 1986; Paz-Ares et al., 1987). Both R- and C1-like proteins directly interact and bind as heterodimers to the promoter sequences of target genes (Goff et al., 1992). Genes encoding bHLH- and MYB-related proteins have also been found in dicots. For example, the DELILA gene of Antirrhinum specifies an R orthologous product that regulates floral anthocyanin pigmentation (Martin et al., 1991; Goodrich et al., 1992). Similarly, the Jaf13 and ANTHOCYANIN2 (AN2) genes control pigmentation of the petunia floral limb and encode bHLH and MYB proteins, respectively (Quattrocchio et al., 1998, 1999). Moreover, some of the regulatory genes have been shown to function when ectopically expressed in heterologous plant cells, demonstrating their functional homology. For instance, several maize _R_-like regulatory loci increase the amount of pigmentation as well as the expression of flavonoid genes in Arabidopsis, tobacco, and petunia (Lloyd et al., 1992; Bradley et al., 1998). In recent studies with petunia, a novel WD40-type regulatory protein, which is encoded by the AN11 gene, has been characterized. This protein is thought to control anthocyanin gene expression through post-translational regulation of transcription factors (de Vetten et al., 1997).
However, despite the structural and functional similarities of flavonoid genes, plant pigmentation has a wide range of distribution patterns and biological functions, which suggests that fine-tuning regulation of target gene expression by a plant depends on its species (Mol et al., 1998). In some species, structural or regulatory genes belong to multigene families in which each of the paralogous members shows a specific spatial and temporal expression pattern. For example, the different CHS genes of morning glory have highly variable expression patterns (Durbin et al., 2000). Similarly, the different R and C1 alleles control organ, tissue, and cell specificity of anthocyanin deposition in maize (Ludwig and Wessler, 1990). On the other hand, variations in plant pigmentation patterns can be explained by the divergent evolution of the promoter sequences from target structural genes (Quattrocchio et al., 1998).
Unraveling flavonoid metabolism in the model plant Arabidopsis may provide new and invaluable information that will improve our understanding of the regulatory network. First, the Arabidopsis genome displays less redundancy and smaller multigene families than does maize or petunia. Indeed, in Arabidopsis, all but one of the characterized flavonoid enzymes, flavonol synthase (FLS), are encoded by single-copy genes. Second, biochemical analyses have been used to characterize Arabidopsis flavonoid end products. Flavonol and anthocyanin derivatives accumulate in vegetative parts, whereas flavonols and proanthocyanidins, essentially derived from dihydroquercetin, accumulate in the endothelium layer of the seed coat (Chapple et al., 1994) (Figure 1). Third, many Arabidopsis mutants impaired in flavonoid metabolism are available. Most of these mutants have been isolated on the basis of modified seed pigmentation and are therefore known as tt (for transparent testa) mutants (Koornneef, 1981, 1990). To date, 21 TT loci have been identified (N. Nesi, I. Debeaujon, C. Jond, M. Caboche, and L. Lepiniec, unpublished results), several of which have been characterized. The TT4 gene encodes the CHS enzyme, TT5 encodes CHI, TT6 encodes F3H, TT7 encodes F3′H (B. Winkel-Shirley, personal communication), and TT3 encodes DFR (Figure 1). Recently, a new gene, called BANYULS (BAN), was cloned and shown to encode a putative leucoanthocyanidin reductase (LAR) specifically involved in proanthocyanidin accumulation in the seed endothelium layer (Devic et al., 1999).
Concerning the regulation of flavonoid metabolism, Kubo et al. (1999) reported the cloning of a homeobox gene, ANTHOCYANINLESS2, that is involved in anthocyanin accumulation in leaves. In addition, TRANSPARENT TESTA GLABRA1 (TTG1) encodes a WD40 repeat protein (Walker et al., 1999) and may therefore represent an orthologous gene at the petunia AN11 locus. The TTG2 gene was shown to specify a transcription factor of the WRKY type (C. Johnson and D.R. Smyth, personal communication). Finally, additional analyses also suggest a regulatory role for the TT8 protein. Indeed, a slight reduction in pigmentation correlated with a decrease in DFR mRNA and an increase in C4H (cinnamate 4-hydroxylase) mRNA accumulation has been reported in young seedlings of the tt8-1 mutant (Shirley et al., 1995; Bell-Lelong et al., 1997).
In our laboratory, the screening of the Versailles T-DNA–mutagenized Arabidopsis collection (Bechtold et al., 1993) led to the identification of 22 independent insertional lines showing a modification of seed coat pigmentation (N. Nesi, I. Debeaujon, C. Jond, M. Caboche, and L. Lepiniec, unpublished results). In this study, we report the genetic and molecular characterization of a new T-DNA–tagged tt8 allele. The cloning of the TT8 gene revealed that it encodes a bHLH domain protein with strong similarity to _R_-related maize genes. Additional experiments also demonstrated that TT8 modulates the expression of two flavonoid late structural genes, namely, DFR and BAN, supporting a major role of the TT8 protein in the flavonoid regulatory network. Finally, we show that two other TT genes, TTG1 and TT2, are also required for normal expression of DFR and BAN genes in Arabidopsis siliques.
RESULTS
Isolation and Genetic Characterization of a New tt8 Allele
As part of our strategy to identify genetic loci controlling flavonoid metabolism in the seed coat, we visually examined mutagenized Arabidopsis populations for individuals with modified seed pigmentation. The deb122 mutant was identified among the T3 progeny of one of 15,000 T-DNA lines from the Versailles T-DNA mutant collection. Seeds produced by deb122 plants were yellowish and easily distinguishable from brown wild-type seeds (Figure 2A). Reciprocal crosses between deb122 and wild-type plants revealed that 100% of the F1 seeds exhibited the phenotype conferred by the maternal plant genotype. Furthermore, all F2 seeds displayed the wild-type phenotype, and the F3 seeds segregated into deb122 and wild-type seeds with a ratio of ∼1:3 (47 plants produced deb122 seeds among 193 plants; 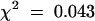 , meaning that the result is significant with a risk of 5%). Together, these results are consistent with the inheritance of a recessive, monogenic, nuclear, and maternal trait conferring the deb122 mutation, which therefore resembles tt mutations. To test allelism with known TT loci, we monitored crosses between deb122 plants and several tt mutants. The deb122 line did not complement the tt8-1 mutation previously described by Bürger (1971). Given that an additional tt8 allele, tt8-2, had been identified among the Kranz and Röbbelen Arabidopsis Information Service mutant collection (Koornneef, 1990), the new allele in deb122 line was thus named tt8-3.
, meaning that the result is significant with a risk of 5%). Together, these results are consistent with the inheritance of a recessive, monogenic, nuclear, and maternal trait conferring the deb122 mutation, which therefore resembles tt mutations. To test allelism with known TT loci, we monitored crosses between deb122 plants and several tt mutants. The deb122 line did not complement the tt8-1 mutation previously described by Bürger (1971). Given that an additional tt8 allele, tt8-2, had been identified among the Kranz and Röbbelen Arabidopsis Information Service mutant collection (Koornneef, 1990), the new allele in deb122 line was thus named tt8-3.
Figure 2.

Phenotype of tt8 and Complemented Seeds.
(A) Seeds of the deb122 mutant (right) of Arabidopsis compared with those of the wild-type genotype (left). Mutant and wild-type genotypes are both from the Wassilewskija-2 (Ws-2) ecotype.
(B) T2 progeny of a tt8-3 (deb122) homozygous plant transformed with the pBIB-Hyg T-DNA binary vector carrying the DEB122 genomic region.
 .
.
The tt8-3 Mutation Cosegregates with a Single T-DNA Insertion
Because the tt8-3 line was obtained from a T-DNA–mutagenized population, we first checked whether the mutant phenotype cosegregated with a single T-DNA copy, which would permit subsequent molecular isolation of the TT8 gene. The T-DNA used to generate the Versailles collection carries a kanamycin resistance marker for the selection of transgenic plants (Bouchez et al., 1993). The analysis of F2 plantlets from a cross between plants with homozygous tt8-3 and wild-type genotypes revealed that the T-DNA insertion segregated as a single locus: 3502 kanamycin-resistant plantlets and 1117 kanamycin-sensitive plantlets represent a segregation ratio of 3:1 (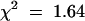 , meaning that the result is significant with a risk of 5%). In addition, all the homozygous tt8-3/tt8-3 plants carried a kanamycin resistance marker, whereas none of the homozygous TT8/TT8 plants was kanamycin resistant, suggesting that the tt8-3 mutation was tightly linked to the T-DNA inserted at the DEB122 locus. Furthermore, probing genomic DNA from homozygous tt8-3 plants by DNA gel blot hybridization with different T-DNA–derived probes indicated that a single full-length T-DNA copy was present in the genome of the mutant (data not shown). From the above genetic and molecular analyses, we assumed that the tt8-3 allele carries a single T-DNA copy. Thus, we isolated genomic DNA fragments flanking the T-DNA insert in line tt8-3 by using a polymerase chain reaction (PCR) walking approach (Devic et al., 1997).
, meaning that the result is significant with a risk of 5%). In addition, all the homozygous tt8-3/tt8-3 plants carried a kanamycin resistance marker, whereas none of the homozygous TT8/TT8 plants was kanamycin resistant, suggesting that the tt8-3 mutation was tightly linked to the T-DNA inserted at the DEB122 locus. Furthermore, probing genomic DNA from homozygous tt8-3 plants by DNA gel blot hybridization with different T-DNA–derived probes indicated that a single full-length T-DNA copy was present in the genome of the mutant (data not shown). From the above genetic and molecular analyses, we assumed that the tt8-3 allele carries a single T-DNA copy. Thus, we isolated genomic DNA fragments flanking the T-DNA insert in line tt8-3 by using a polymerase chain reaction (PCR) walking approach (Devic et al., 1997).
Using PCR, we amplified 600 bp and 1.8 kb of tt8-3 plant genomic DNA flanking the right and left T-DNA borders, respectively. Primers deb122RB1 and deb122LB2 were then designed from the right and left genomic T-DNA borders and used to amplify a 954-bp fragment (called the deb122 probe) from wild-type genomic DNA. Sequence comparison of this PCR product with both genomic T-DNA borders showed that T-DNA integration into the tt8-3 genome resulted in a deletion of 29 bp of plant genomic DNA. The deb122 probe was then used to screen the Institut für Genbiologische Forschung Arabidopsis bacterial artificial chromosome (BAC) library (Mozo et al., 1998). Thus, we identified six overlapping BAC clones (F17A8, F21A7, F23D16, F14E11, F13G23, and F22O14) assigned to chromosome 4, which is consistent with the mapping of the TT8 locus (Shirley et al., 1995). The positive BAC F17A8 (GenBank accession number AL049482) was chosen for the following studies because its full sequence was established and annotated during the progress of this work (Bevan et al., 1998). Database searches revealed that ∼4 kb of the F17A8 sequence (positions 80,383 to 84,577; see Figure 3A), including the deb122 probe, encodes a putative protein (CAB39649) that is similar to known bHLH transcription factors.
Figure 3.

Molecular Analysis of the TT8 Locus.
(A) Diagram of the DEB122 region on BAC F17A8. The arrow indicates the orientation of the putative DEB122 protein. The primers deb122RB1 and deb122LB2, used to amplify the deb122 probe, are indicated. The two HindIII restriction sites were used to generate an 8-kb genomic fragment (pBIB-Hyg-8) for complementation assays.
(B) Genomic organization of the TT8 gene (Ws-2 ecotype). The positions and relative sizes of the exons and introns of the genomic clone are indicated by black and white boxes, respectively. The respective positions of the translation start (ATG) and the translation stop (TAG) codons are shown. The solid bar represents the conserved bHLH region. The T-DNA (7 kb) is inserted into the second intron of the tt8-3 allele and causes a 29-bp deletion indicated by the striped box, whereas ethyl methanesulfonate mutagenesis causes a nucleotide transition in the sixth intron of the tt8-1 allele (shown by an arrow). The position of a transposon-like element (1.5 kb) in the Col-0 wild-type genome is shown by the dashed arrow. The primers used for molecular analyses are noted below the diagram. RB, right border; LB, left border.
Complementation of the tt8-3 Insertional Mutation with a Genomic Clone
To demonstrate the complementation of the phenotype of the tt8 mutant by ectopic expression of the wild-type DEB122 gene, we transformed homozygous tt8-3 plants with a genomic construct derived from the F17A8 clone. For this purpose, an 8-kb HindIII genomic fragment containing the DEB122 region was inserted into the binary vector pBIB-Hyg to form the pBIB-Hyg-8 construct (see Figure 3A). This construct was introduced into the tt8-3 mutant by way of Agrobacterium. Fifty-one hygromycin-resistant transformants were recovered, and for all of them, T2 progeny produced brown-colored seeds (Figure 2B), indicating that the seed phenotype of tt8-3 had reverted to that of the wild type. All transformants were also kanamycin resistant, which confirmed that they carried the original T-DNA copy. In addition, the 51 transformants exhibited different segregation patterns on hygromycin selection medium (data not shown), indicating that they were derived from independent transformation events. These results provide strong evidence that the genomic insert from pBIB-Hyg-8 contained the TT8 gene.
Structural Analysis of the TT8 Locus
The sequence of the genomic insert from pBIB-Hyg-8 was submitted to the Eukaryotic GeneMark.hmm program available at http://dixie.biology.gatech.edu/GeneMark/eukhmm.cgi to predict the putative gene structure. The algorithm sug-gested the presence of a single gene containing five introns. These predictions allowed us to design specific oligonucleotide primers for the cloning of the full-length TT8 cDNA by PCR amplification.
We first conducted a primer extension analysis to map in fine detail the transcription start site of the TT8 gene and to confirm the likely translation initiation codon (ATG). The major reverse transcription product (data not shown) allowed us to localize the transcription start site 44 bp upstream of the putative translation start codon, as shown in Figure 4. The 5′ untranslated region (UTR) contains no other ATG codon, which confirms the position of the first Met residue. A putative TATA box was found 29 bp upstream of the transcription start site. The 3′ UTR was amplified from an Arabidopsis silique cDNA library (Giraudat et al., 1992), as described in Methods. Sequencing six independent clones yielded two types of polyadenylation sites (Figure 4). Primers deb122-5′ and deb122-3′ (Figure 3B) were then designed to amplify by PCR the longer of the two TT8 transcripts. A 1833-bp PCR fragment was obtained, corresponding to a 1557-bp coding region surrounded by 44 and 232 bp of 5′ and 3′ UTRs, respectively. The full-length cDNA sequence has DDBJ/EMBL/GenBank accession number AJ277509. Alignment of the genomic and cDNA sequences showed that exon 4 was missed by the gene structure prediction algorithm previously used, probably because this exon is very short (15 bp). Therefore, the TT8 gene actually contains six introns (Figure 3B). All the splice junctions conformed to the GT-AG consensus (Brown et al., 1996).
Figure 4.

Nucleotide and Deduced Amino Acid Sequences of the TT8 cDNA.
Uppercase letters indicate the extent of the full-length TT8 transcript (GenBank accession number AJ277509); the untranscribed 5′ region is in lowercase letters. The amino acid residues derived from the translation of this sequence are shown (one-letter code) below the corresponding codons. The functional translation stop site (TAG) is marked with an asterisk. Position 307 shows an amino acid polymorphism related to the Arabidopsis ecotype. A putative nuclear localization signal is shown in boldface above the bHLH domain, and the bHLH domain is boxed in black. The presumed TATA box is underlined, and the intron positions are shown by arrowheads. The arrows indicate the two polyadenylation sites found, and the corresponding putative polyadenylation signals are underlined by the dashed lines.
Homozygous tt8-3 plants were transformed with the TT8 coding region under the control of the cauliflower mosaic virus (CaMV) 70S promoter. Analysis of 95 hygromycin-resistant primary transformants showed that 82 of them had brown seeds. This result is in agreement with the previous complementation assay using a genomic construct and provides additional evidence that the cloned cDNA corresponds to the TT8 locus.
Analysis of the TT8 gene sequence was conducted with three ecotypes: Wassilewskija-2 (Ws-2), Enkheim-2 (En-2), and Columbia-0 (Col-0). Although several nucleotide changes were observed in the coding sequence between ecotypes, only one led to amino acid modification: an S residue at position 307 in the Col-0 and En-2 sequences was replaced by a Y residue in Ws-2 ecotype (Figure 4). A more surprising difference was the length of the fifth intron, which was 1532 bp longer in Col-0 than in Ws-2 and En-2 (Figure 3B). This additional sequence showed characteristic traits of transposable elements, including 16-bp terminal inverted repeats (CACTACAAAAAAAAGG) resembling those of the CACTA transposon family (reviewed in Gierl and Saedler, 1992) and 3 bp (ATG) surrounding both sides of the terminal repeats that may have resulted from duplication of the target genomic site. However, no typical open reading frame was found within the transposon-like sequence.
The TT8 Gene Encodes a bHLH Domain Protein
The open reading frame of the TT8 cDNA specifies a 518–amino acid protein with a calculated molecular mass of 59.2 kD. A putative nuclear localization signal (amino acids 355 to 371) was recognized by using the PSORT algorithm (Nakai and Kanehisa, 1992). Using the BLAST algorithm (Altschul et al., 1990) to compare the TT8 amino acid sequence with translations of DNA sequences from various organisms revealed that the TT8 protein belongs to the broad range of MYC-related proteins from animals and plants. Indeed, a typical bHLH signature was found near the C terminus of the TT8 sequence. The motif generally consists of a basic region (14 amino acids) and two α helices separated by a loop of variable length. The bHLH structure and most of its invariant amino acid residues are conserved in TT8 (E-368, R-369, R-371, L-375, N-376, F-379, L-382, R-383, V-386, P-387, K-391, K-394, I-397, L-398, Y-404, V-405, and L-408), except for two R/K residues that are substituted in all plant bHLH proteins (Figure 5A). Similarities between the TT8 protein and animal MYC-type sequences are limited to the bHLH motif, as described previously for other plant bHLH proteins (Ludwig et al., 1989; Goodrich et al., 1992).
Figure 5.

Deduced TT8 Amino Acid Sequence and Comparison with bHLH-Related Protein Sequences.
(A) Amino acid sequence comparison of the bHLH regions encoded by TT8 and several MYC-related proteins. Asterisks indicate amino acid residues found in MYC-related protein sequences from all eukaryotic organisms, and conserved amino acids are boxed in black. Dashes indicate gaps inserted to improve the alignment. Sequences shown are those from human c-MYC (GenBank accession number X00198), newt MRF-4 (X82836), Xenopus NeuroD (U28067), yeast CBF1 (M33620), maize IN1 (U57899), Antirrhinum DELILA (M84913), Arabidopsis RD22BP1 (AB000875), and Arabidopsis TT8 (this study).
(B) Distance analysis of several plant bHLH-related factor sequences recovered by using a BLAST algorithm on the GenBank and EMBL databases. The following bHLH protein sequences were used to build the tree (accession numbers are in parentheses): maize Lc (M26227), maize R-S (X15806), maize B-Peru (X57276), maize IN1 (U57899), maize MYC7E (AF061107), rice Ra (U39860), pea PG1 (U18348), Gerbera MYC1 (AJ007709), bryophyte MYC-RP (AB024050), Antirrhinum DELILA (M84913), petunia Jaf13 (AF020545), and Arabidopsis AtMYC1 (D83511), Arabidopsis AtMYC146 (AF013465), Arabidopsis RD22BP1 (AB000875), and Arabidopsis TT8 (this study). Sequences were aligned using ClustalX and manually adjusted. For tree construction, only the N terminus region and the bHLH domain were used, as marked in (C). The consensus tree presented was obtained by neighbor-joining analysis, bootstrapped with 1000 iterations by ClustalX, and drawn with the TreeView program. Bootstrap values are indicated at each branchpoint; branches with a bootstrap score <850 were eliminated. Relative branch length (0.1) is indicated below the tree. Proteins in boldface have been reported to be involved in plant flavonoid metabolism.
(C) Sequence comparison between TT8 and three other bHLH proteins involved in plant flavonoid pigmentation. Identical amino acids are boxed in black, and similar amino acids are boxed in gray. Brackets delimit the bHLH region. Sequences used are from maize IN1 (GenBank accession number U57899), maize B-Peru (X57276), Antirrhinum DELILA (M84913) and Arabidopsis TT8 (this study). Asterisks above the sequences indicate amino acid residues used to build the distance tree shown in (B). Dashes were introduced to optimize alignment.
The sequence relationships among plant bHLH proteins are illustrated in Figure 5B. Parsimonious analyses performed by using PAUP software and the PROTPARS matrix (see Methods) gave a very similar relationship (data not shown). This suggests that the tree also represents phylogenetic relationships. TT8 appears to be closely related to the maize INTENSIFIER1 (IN1) protein, which had been described as a putative repressor of the anthocyanin biosynthetic pathway in the maize aleurone (Burr et al., 1996). TT8 and IN1 form a subclass that seems to be distantly related to the major phylogenetic group, including all other known bHLH proteins involved in flavonoid metabolism. This latter group also includes Arabidopsis AtMYC1 and AtMYC146, whose biological functions in plants have not been reported (Urao et al., 1996; Bate and Rothstein, 1997).
The strong similarity between TT8 and IN1 is particularly obvious when full sequences are compared (Figure 5C). Apart from the bHLH domain, the N-terminal region shows high similarities among plant bHLH proteins. In this region (amino acids 20 to 190 in the TT8 sequence), TT8 exhibits 59% amino acid identity with IN1, 49% with B-Peru, and 52% with DELILA. In maize, the N terminus of the B-Peru protein is assumed to be involved in the protein–protein interactions with the C1 MYB-related factors (Goff et al., 1992).
Analysis of tt8 Alleles
To gain insight into the nature of tt8 mutations, we determined the accumulation of TT8 mRNA in siliques of tt8-3 and tt8-1 in comparison with the parental ecotypes (Ws-2 and En-2, respectively). Because TT8 transcripts were not detected by RNA gel blot analyses (data not shown), we used a reverse transcription–PCR (RT-PCR) approach. As a positive control, we also monitored the accumulation of EF1_α_A4 transcripts, encoding a translation elongation factor of Arabidopsis (Liboz et al., 1990). No TT8 mRNA was detected in siliques of tt8-3 (Figure 6A, lane 2), demonstrating that the mutation in the T-DNA–tagged allele caused a complete loss of function. The T-DNA copy was found to be inserted into the second intron in tt8-3 genomic DNA (Figure 3B). On the other hand, TT8 transcripts were produced in tt8-1, which probably resulted from ethyl methanesulfonate mutagenesis (Koornneef, 1990). Therefore, we sequenced the RT-PCR products from wild-type En-2 and tt8-1 plants (Figure 6A, lanes 3 and 4, respectively). Apparently the sixth intron was not spliced in the tt8-1 transcript because of a G-to-A nucleotide change at position +1874 of the gene (Figure 3B). This base transition led to the loss of the GT splicing consensus site, thus preventing the production of correct transcripts in tt8-1. Because no stop codon occurs within the translated intron, the resulting protein should be 28 amino acids longer than the wild-type protein (Figure 6B).
Figure 6.

Analysis of tt8 Mutations.
(A) Detection of TT8 transcripts in siliques of two tt8 mutants and their corresponding parental ecotype by RT-PCR. The full-length TT8 transcript was detected by ethidium bromide staining after 35 cycles of amplification. The expression of the EF1_α_A4 gene was used as a control.
(B) Characterization of the donor and acceptor splicing sites of intron 6 in the wild type (En-2) and tt8-1 mutant. Brackets indicate correctly spliced sites; dots indicate an internal intron. Amino acid residues are shown (one-letter code) below the sequence. The asterisk indicates the mutated nucleotide in the _tt8-1_–derived sequence.
The TT8 Gene Is Expressed throughout Seed Development and in Young Seedlings
The expression pattern of the TT8 gene was investigated in different wild-type tissues and in siliques during seed development by using a quantitative RT-PCR strategy. Primers deb122RB1 and deb122LB3 (Figure 3B) were used to amplify a 780-bp fragment from TT8 transcripts. As shown in Figure 7A, the TT8 transcripts were detected in 4-day-old seedlings, buds, flowers, and developing siliques. No signal was observed in rosette leaves, stems, and roots, even when PCR amplification was extended to 40 cycles (data not shown). In reproductive organs, TT8 was faintly expressed in buds and flowers, whereas the accumulation of the transcript rapidly increased during the very early stages of seed development. The expression reached a maximum at the globular embryo stage, which roughly corresponded to the third day after pollination and was maintained fairly uniformly throughout seed formation.
Figure 7.

Analysis of TT8 Expression during Plant Development.
(A) TT8 transcripts were detected by quantitative RT-PCR in vegetative parts that included 4-day-old seedlings, rosette leaves, stems, and roots from 10-day-old plantlets and in reproductive organs that included buds, flowers, and developing seeds. Accumulation of the EF1_α_A4 transcript was used as an internal control. The PCR products were detected by DNA gel blot analysis after 21 amplification cycles. The blots were hybridized with the respective probes.
(B) Comparison of TT8 expression pattern in reproductive organs with those of C4H, CHS, and DFR genes. PCR and hybridization assays were conducted exactly as those in (A).
The accumulation of TT8 transcripts in siliques was then compared with the expression pattern of several genes involved in phenylpropanoid and flavonoid biosynthetic pathways during seed development. Figure 7B shows that no mRNA variation was observed for the C4H gene. Similar results were obtained for three other phenylpropanoid genes that encode phenylalanine ammonia-lyase1 (PAL1) and two 4-coumarate:CoA ligases (4CL1 and 4CL3) (data not shown). Moreover, the flavonoid biosynthetic genes apparently can be divided into two groups with respect to their expression pattern. On one hand, the CHS gene, the expression of which was high from the bud stage onward, can be classified within the flavonoid “early” biosynthetic genes (EBGs), as defined for seedlings (Kubasek et al., 1992). Similar results were observed for CHI, F3H, FLS1, and F3_′_H transcripts (data not shown). On the other hand, the DFR transcripts were detected in flowers but not in buds and peaked at the globular stage (Figure 7B). The DFR gene was therefore designated as a flavonoid “late” biosynthetic gene (LBG), as were the BAN and LDOX (for leucoanthocyanidin dioxygenase) transcripts (data not shown). The accumulation of TT8 mRNA during seed development just preceded and overlapped those of flavonoid LBGs but did not appear to be correlated with expression of phenylpropanoid genes and flavonoid EBGs (Figures 7A and 7B).
TT8 Is Required for Normal Expression of DFR and BAN, Two Flavonoid LBGs
We systematically tested the expression of all known flavonoid biosynthetic genes in siliques of the tt8-3 mutant and compared the expression patterns to those of the wild type. Figure 8 presents some results of this analysis conducted on globular embryo-stage siliques. At this stage, all flavonoid structural genes tested were highly expressed in wild-type plants (Figure 7). Under our conditions, siliques of the tt8-3 mutant exhibited wild-type amounts of C4H mRNA, unlike the seedlings (Bell-Lelong et al., 1997). Analysis of other phenylpropanoid genes—PAL1, 4CL1, and _4CL3—_also revealed no significant variation in mRNA content between tt8-3 and the wild type (data not shown). Monitoring the expression of the above-mentioned flavonoid EBGs gave similar results. Concerning flavonoid LBGs, DFR transcripts were severely reduced in tt8-3 siliques, and no BAN transcripts could be detected at all (Figure 8), whereas LDOX mRNA accumulation was identical to that of the wild type (data not shown).
Figure 8.

Expression of Flavonoid Biosynthetic Genes in Globular Embryo Stage Siliques.
Siliques were obtained from wild-type genotype and three tt mutants: tt8-3, ttg1-1, and tt2-3. Expression of the different genes was monitored by quantitative RT-PCR.
Expression of DFR and BAN Genes also Depends on TTG1 and TT2 Genes
Further expression analyses were conducted to compare the effects of tt8 mutations on flavonoid metabolism with those of ttg1 and tt2, both of which modify the pattern of seed pigmentation. TTG1 encodes a WD40 repeat protein (Walker et al., 1999). With respect to flavonoid accumulation in different organs, TT2 was assumed to encode a seed-specific regulatory protein or a member of a differentially expressed structural enzyme family (Shirley et al., 1995). For our purpose, we used the ttg1-1 allele, which harbors a nonsense mutation (Walker et al., 1999), and the tt2-3 allele, a T-DNA null mutant isolated from the Versailles collection (N. Nesi, C. Jond, I. Debeaujon, M. Caboche, and L. Lepiniec, unpublished results). Figure 8 reveals that DFR expression could not be detected in siliques of the ttg1-1 mutant, as shown previously in seedlings (Shirley et al., 1995). DFR transcripts were substantially less in siliques of the tt2-3 allele (Figure 8). This result has not been observed in seedlings (Shirley et al., 1995). As shown for tt8-3, BAN transcripts were not produced in siliques of ttg1-1 and tt2-3 mutants.
DISCUSSION
The TT8 Gene Encodes a New Arabidopsis bHLH Protein
During screening for seed color mutants, we isolated a new tt8 allele, called tt8-3, which is T-DNA tagged. The pigmentation patterns of the tt8 seeds and young seedlings are modified, suggesting that the TT8 gene is involved in flavonoid metabolism. In this study, we report the genetic and molecular analyses of tt8 mutations and the cloning of the TT8 gene. The corresponding protein displays the typical features of a transcription factor with a bHLH signature at its C terminus, a domain found in all animal c-MYC–related proteins (Massari and Murre, 2000). Two tt8 alleles are characterized. In the tt8-3 mutant, insertion of a T-DNA copy into the second intron led to a complete loss of function (i.e., no transcript was produced). One can hypothesize that T-DNA insertion may disturb the structural characteristics of the intron that are essential for correct splicing, such as AT-GC content or a branch site consensus sequence (Brown et al., 1996). Such T-DNA insertions were reported in many instances, including an insertion for one of the ban alleles (Devic et al., 1999). In tt8-1, a single nucleotide mismatch at the sixth exon/intron boundary induced the production of a mRNA 84 bp longer than that of the wild type. The additional 28–amino acid stretch is located just at the N terminus of the bHLH domain. Its presence may impair correct conformation of the whole protein and thus confer the phenotype of the tt8 mutant.
In plants, the first bHLH protein, Lc, has been reported for maize (Ludwig et al., 1989). Among plant bHLH proteins, TT8 belongs to the family of proteins involved in the control of plant flavonoid pigmentation. Indeed, the TT8 amino acid sequence is highly similar to those from maize R, bryophyte MYC-RP, Antirrhinum DELILA, and petunia Jaf13 proteins (Ludwig et al., 1989; Goodrich et al., 1992; Quattrocchio et al., 1998; Gong et al., 1999). This suggests a common evolutionary origin for all of these genes. Moreover, the significant conservation in the N-terminal region between TT8 and maize B-Peru raises the possibility that TT8 may interact with MYB-related proteins in Arabidopsis, as has been described for the maize factor (Goff et al., 1992). All of these findings strongly support the hypothesis that TT8 may act as a transcription factor that interacts with other proteins to modulate the expression of target genes.
Interestingly, distance analysis shows that two Arabidopsis bHLH proteins, namely, AtMYC1 (Urao et al., 1996) and AtMYC146 (Bate and Rothstein, 1997), belong to the flavonoid group, on the basis of their sequence features. However, their biological functions have not yet been determined. AtMYC1 transcripts are detected mainly in the tissues of developing siliques and to a lesser extent in leaves and stems (Urao et al., 1996); AtMYC146 accumulates in those same tissues (data not shown). Experiments will be undertaken to test whether these proteins could represent homologs of TT8 in the flavonoid biosynthetic pathway.
Important information derived from the distance tree is that TT8 and the maize IN1 proteins both belong to the same subclass, which appears to be distantly related to other well-known pigmentation-related bHLH factors (Figure 5B). Remarkably, proteins from both monocots and dicots are found within the two above-mentioned subclasses, suggesting that two different bHLH proteins may have existed in the genome of the common ancestor. Another point worth emphasizing is that IN1 reportedly acts as a negative regulator of anthocyanin biosynthesis in maize aleurone. Indeed, homozygous in1 kernels are black as a result of anthocyanin overaccumulation, in comparison with the deep purple pigmentation of wild-type seeds. The in1 mutation may activate genes involved in anthocyanin biosynthesis, as proposed by Burr et al. (1996). The role of IN1, however, appears to differ from that of TT8, which is assumed to be a positive regulator of flavonoid metabolism based on the phenotype of the tt8 mutant. One possible explanation for this apparent discrepancy is that the black pigmentation observed in in1 mutant kernels may reflect a metabolic rechanneling rather than a direct effect of the mutation on an anthocyanin-negative regulatory gene. This would be similar to Arabidopsis ban mutants, the immature seeds of which are pink as a result of accumulated anthocyanin end product in the endothelium layer of the seed coat. Devic et al. (1999) demonstrated that the BAN gene does not encode a negative regulator of anthocyanin production, as Albert et al. (1997) first suggested, but rather encodes a putative LAR, which leads to proanthocyanidin accumulation, or a positive regulator of such an enzyme. As a consequence, mutations affecting the BAN gene alter LAR activity, thus directing the biosynthetic pathway to the production of purple anthocyanins rather than brown proanthocyanidins.
TT8 Is a Regulator of “Late” Flavonoid Metabolism
Analysis of TT8 gene expression revealed that TT8 transcripts accumulate in 4-day-old seedlings. This accumulation closely matches the transient developmental peak in flavonoids observed in young seedlings (Kubasek et al., 1992). TT8 was also observed in developing siliques, where accumulation of flavonoid pigments has been reported during the early stages of seed formation (Chapple et al., 1994; Devic et al., 1999). In addition, previous works have suggested that the tt8-1 mutation modifies the steady state amount of DFR mRNAs in young seedlings of Arabidopsis, whereas no effect was observed on the expression of CHS, CHI, F3H, FLS, and LDOX genes (Shirley et al., 1995; Pelletier and Shirley, 1996; Pelletier et al., 1997).
To extend these observations to seeds, we systematically analyzed the expression of all known flavonoid biosynthetic genes in siliques of tt8-3. For this, we first investigated the expression pattern of flavonoid biosynthetic genes during seed development and demonstrated that the accumulation of CHS, CHI, F3H, FLS1, and F3_′_H transcripts precedes that of DFR, BAN, and LDOX. According to Kubasek et al. (1992), flavonoid structural genes in the first group were therefore classified as “early” genes and those in the second group as “late” genes. Remarkably, the order of induction nearly follows the order of biosynthetic steps within the flavonoid pathway (Figure 1) and fits with the results obtained from Arabidopsis seedlings (Kubasek et al., 1992; Pelletier and Shirley, 1996; Pelletier et al., 1997).
Considering the tt8-3 mutant, our results clearly demonstrate that TT8 is not necessary for the expression of flavonoid EBGs in siliques. However, the mRNA amounts of two flavonoid late structural genes, DFR and BAN, are markedly affected in tt8-3 siliques. The expression of DFR is reduced in siliques of tt8-3, supporting the previous results obtained with young seedlings (Shirley et al., 1995). Other plant bHLH factors also affect the expression of DFR orthologous genes in snapdragon, petunia, and moss (Martin et al., 1991; Goodrich et al., 1992; Quattrocchio et al., 1998; Gong et al., 1999), which suggests a common, conserved regulatory mechanism among plants. Moreover, no BAN transcript could be detected in tt8-3 siliques. This result is consistent with the fact that TT8 is epistatic to BAN (Albert et al., 1997) and with the finding that no catechins accumulate in tt8 seeds (Debeaujon et al., 2000). In addition, because BAN is specifically expressed in the seed coat endothelium (Devic et al., 1999), we assume that TT8 mRNA is at least localized in the endothelium layer within the developing siliques. Interestingly, TT8 is not required for LDOX expression in siliques, as previously shown in seedlings (Pelletier et al., 1997). Therefore, additional independent regulatory elements may occur in the control of late biosynthetic steps.
Together, our findings strongly highlight the key regulatory role of TT8 in the control of two genes of the late flavonoid pathway in siliques: DFR is the first enzyme committed to the anthocyanin and proanthocyanidin biosynthesis, and BAN is at the branch point leading to the catechin and proanthocyanidin subpathway (Figure 1). These results are in agreement with the fact that TT8 begins to accumulate just before DFR and BAN transcripts during seed formation. However, because the amount of TT8 mRNA remains fairly constant throughout seed development, TT8 should not be the rate-limiting factor involved in the decrease in expression of structural genes observed from the curled cotyledon embryo stage onward (Figure 7B). Additional regulators may thus be involved in this process. Moreover, bHLH-related factors are well known to interact with other transcriptional factors. For instance, interactions between bHLH- and MYB-type plant proteins have been widely documented, especially in maize. Indeed, R has been shown to activate the transcription of several flavonoid structural genes in coordination with the C1 MYB protein (Goff et al., 1992; Lesnick and Chandler, 1998).
Expression of Flavonoid LBGs also Depends on TTG1 and TT2: New Clues into the Puzzle of Flavonoid Gene Regulation
Another important point of this study is provided by the analysis of flavonoid gene expression in two other mutants, ttg1-1 and tt2-3. Our results demonstrate that the TTG1 and TT2 loci are also necessary for the expression of DFR and BAN genes in siliques. These data suggest that the TT2 protein is required for flavonoid regulation in seeds. Perhaps the TT8, TT2, and TTG1 genes might interact to modulate the activity of structural target genes. In preliminary experiments, the TT8 mRNA was decreased only in siliques of the ttg1-1 mutant, but no difference was observed in the tt2-3 mutant (data not shown). This result suggests that TTG1 is at least required for normal expression of TT8 in siliques, unlike TT2.
Several issues remain to be addressed to improve our knowledge of the flavonoid gene regulatory network in Arabidopsis. First, the relationships among the TT8, TT2, and TTG1 factors have to be further investigated. Information about the putative genetic interactions among these three loci will be obtained by analyses of the corresponding double mutant phenotypes as well as by biochemical studies of their flavonoid content. The recent molecular analysis of the TTG1 gene has shown that it encodes a WD40 repeat protein localized in the cytosol (Walker et al., 1999), which suggests the existence of an intermediate factor or factors downstream of TTG1 to control gene expression. The cloning of the TT2 gene will provide invaluable information about its function and its subcellular localization. We also plan to test the occurrence of in vivo molecular interactions among TT8, TT2, and TTG1 proteins. Moreover, examining whether TT8 (and perhaps TT2) can directly bind the DFR and BAN promoters in vivo and therefore promote the transcriptional activation of the corresponding genes will also be essential. Interestingly, within the first 200 bp of the DFR promoter are two putative bHLH recognition sequences (CACGTG) (Shirley et al., 1992); both match the binding consensus site for plant bHLH proteins (CANNTG; Abe et al., 1997). Similarly, the BAN sequence displays one putative binding site for bHLH proteins, a site located 150 bp upstream of the translation start codon (I. Debeaujon, unpublished results).
In conclusion, this study demonstrates that TT8, TTG1, and TT2 are involved in the control of late genes, thus confirming that the entire flavonoid pathway is not coordinately regulated, just as has been found for several species (Martin et al., 1991; Quattrocchio et al., 1993; Pelletier and Shirley, 1996; Pelletier et al., 1997). Further investigations are necessary to understand the regulatory mechanism. Interestingly, in petunia, bHLH, WD40, and MYB proteins have been proposed to act together on late flavonoid metabolism (reviewed in Mol et al., 1998). In addition to TT8, TTG1, and TT2, two other TT genes, TT1 (M. Sagasser, K. Hahlbrock, and B. Weisshaar, unpublished results) and TTG2 (C. Johnson and D.R. Smyth, personal communication) have been shown to encode regulatory factors in Arabidopsis. To date, how these regulatory loci control flavonoid metabolism in Arabidopsis has not yet been demonstrated.
METHODS
Plant Material and Growth Conditions
Arabidopsis thaliana plants of the ecotype Wassilewskija (Ws-2) as well as tt8-3 (deb122) and tt2-3 mutant seeds (both from the Ws-2 ecotype) were obtained from the Station de Génétique et d'Amélioration des Plantes (INRA, Versailles, France). The tt8-1 (F107) and tt8-2 (F17) mutants originated from the Kranz and Röbbelen Arabidopsis Information Service collection. Both the tt8-1 allele and the corresponding Enkheim-2 (En-2) wild-type ecotype used for our studies were kindly provided by the Nottingham Arabidopsis Stock Centre (UK) (Stock Centre seed nos. N111 and N1138, respectively). The ttg1-1 allele used for the expression analyses was a gift from B. Winkel-Shirley (Virginia Polytechnic Institute, Blacksburg, WV).
Plants were routinely grown in a greenhouse (16-hr photoperiod; 10 to 15°C night/20 to 25°C day temperature) on sterilized compost irrigated twice a week with mineral nutrient solution. For crosses and seed production, plants were grown in individual pots. For plant transformation, batches of seeds were directly sown on 10 × 15-cm pots. Plantlets were thinned to 20 to 30 per pot at 2 weeks and then were allowed to grow until the flowering bolts were ∼10 cm high and the first siliques appeared.
For aseptic growth, seeds were surface-sterilized and plated on Murashige and Skoog (1962) medium. Petri dishes were first incubated at 4°C for 48 to 72 hr to break dormancy and homogenize germination and then were kept in a growth chamber (16-hr photoperiod; 15°C night/20°C day temperature). Selection of T-DNA–transformed tt8-3 seeds was performed by germination on Murashige and Skoog medium containing kanamycin (Sigma) at 50 mg/L. To obtain young seedlings and root material for RNA analysis, we grew plantlets in vitro for 4 and 10 days, respectively, on B5 mineral medium (Duchefa, Haarlem, The Netherlands) containing 2% sucrose and 1% agar. In the case of root production, plates were in a nearly vertical position.
Cloning of the T-DNA–Flanking Genomic Sequences in tt8-3
Genomic DNA was isolated from 10-day-old plantlets grown in vitro by extraction with a hexadecyltrimethylammonium bromide buffer, according to Doyle and Doyle (1990), and treated with RNase A (0.1 μg/μL). Genomic regions flanking the T-DNA insertion in the tt8-3 mutant were isolated by using a polymerase chain reaction (PCR) walking approach. The procedure was essentially performed as described by Devic et al. (1997). Genomic DNA (∼500 ng) from tt8-3 plants was digested with ScaI or DraI restriction enzymes and ligated to an adapter duplex to produce two genomic walk libraries. T-DNA–flanking genomic regions were amplified by using a pair of nested primers specific to the T-DNA borders in combination with a pair of nested primers specific to the adapter designed by Devic et al. (1997): AP1, 5′-GGATCCTAATACGACTCACTATAGGGC-3′; and AP2, 5′-CTATAGGGCTCGAGCGGC-3′. A 1.8-kb fragment was amplified from the ScaI library by using nested primers specific to the T-DNA left border: TailB, 5′-CGGCTATTGGTAATAGGACACTGG-3′; and LBBAR1, 5′-CAACCCTCAACTGGAAACGGGCCGGA-3′. At the right border, a 600-bp fragment was obtained from the DraI library by using T-DNA primers: Tail2, 5′-TCGTTAAAACTGCCTGGCACAG-3′; and RBGUS1, 5′-CCAGACTGAATGCCCACAGGCCGTC-3′. The PCR assays were performed with 35 cycles of denaturation at 94°C for 30 sec (3 min for the first cycle), annealing at 60°C for 30 sec, and elongation at 72°C for 2 min and 30 sec (10 min for the last cycle). The amplified fragments were cloned into a pGEM-T plasmid according to the recommendations of the supplier (Promega) and sequenced. Two primers (deb122RB1, 5′-AGGAAGACAACTCAA-CCAGC-3′; and deb122LB2, 5′-TCATCAGAATACAATTCTCAA-ATCT-3′) were designed from plant genomic DNA and used to characterize the T-DNA target site in the wild-type genomic DNA. This primer set amplifies a 954-bp fragment on genomic DNA, which was used as a probe (deb122 probe) for subsequent genomic library screening. To recover a genomic clone containing the wild-type DEB122 gene, we screened the Institut für Genbiologische Forschung bacterial artificial chromosome (BAC) library (Mozo et al., 1998) from the RessourcenZentrum/PrimärDatenbank im Deutschen HumanGenomProjekt (RZPD, Berlin, Germany) (see also http://www.rzpd.de/ for information).
Constructs and Plant Transformation
All constructs used for plant transformation were made in the pBIB-Hyg T-DNA binary vector (Becker, 1990), which carries a hygromycin-resistance marker for in vitro selection of the transformants. Intermediate constructs were introduced into the Escherichia coli DH12S and verified by restriction of plasmid DNA. Final constructs were introduced into the Agrobacterium tumefaciens C58C1Rif(pmp90) strain (Koncz et al., 1984) by electroporation and checked by PCR assays using primers derived from sequences of both pBIB-Hyg and the insert.
For complementation with the genomic fragment, ∼700 ng of BAC F17A8 was digested to completion with HindIII restriction enzyme. Restriction fragments were directly ligated into the HindIII site of the pBIB-Hyg vector and introduced into E. coli. To recover clones carrying the 8-kb fragment corresponding to the DEB122 locus, we screened bacterial clones by colony hybridization with the deb122 probe. A positive clone named pBIB-Hyg-8 was isolated, checked by HindIII restriction, introduced into Agrobacterium, and used for plant transformation assays.
The construct for ectopic expression of the TT8 transcript was obtained as follows. Using primers complementary to the region upstream from the translation start site (deb122ATG, 5′-ATGGAT-GAATCAAGTATTATTCCGG-3′) and downstream from the stop codon (deb122Stop, 5′-CTATAGATTAGTATCATGTATTATG-3′), we amplified the TT8 full-coding region (_TT8_-CR) from a green siliques cDNA library (Giraudat et al., 1992) by PCR. The 1557-bp PCR fragment was directly blunt-end ligated (SmaI) between the double-enhanced cauliflower mosaic virus (CaMV) 35S promoter and the CaMV polyadenylation signal of the pLBR19 vector (Guerineau et al., 1992). A clone carrying the _TT8_-CR fragment in sense orientation was isolated and sequenced to ensure that no mutation was introduced by the DNA polymerase. To raise transgenic plants, we excised the 70S-promoter::_TT8_-CR::Term overexpression cassette from pLBR19 as a KpnI-XhoI fragment and cloned it into the KpnI-SalI–digested pBIB-Hyg vector, thus generating the pBIB-_TT8_-CR plasmid.
Plant transformation was performed as described by Bechtold et al. (1993). T1 seeds were sown on Murashige and Skoog medium containing hygromycin (50 mg/L). Resistant T1 seedlings were transferred to soil to set seeds. The phenotype of T2 seeds was examined for phenotypic complementation of the mutation by the transgene.
RNA Analyses
For expression analyses in developing siliques, individual flowers on the primary inflorescence were tagged on the day of pollination (stigma just extruded from the corolla). Siliques were harvested from 20 to 30 plants grown together. Each sample consisted of four adjacent immature siliques beginning with the first silique below the last flower. To determine precisely the embryo developmental stage prevailing during each stage, we removed seeds from siliques and cleared the seeds for 3 to 5 hr in a chloral hydrate–glycerol–water solution (8:2:1 [v/v/v]) on a microscope slide. Cleared seeds were observed with a Microphot-FXA (Nikon, Tokyo, Japan) microscope equipped with Nomarski differential interference contrast optics. Stems and rosette leaves samples were harvested from 1-month-old plants.
Tissue samples were ground in liquid nitrogen, and total RNA was extracted with the RNeasy plant mini kit (Qiagen, Chatsworth, CA) according to the instructions of the manufacturer. The extracts were treated with 30 units of RNase-free DNase I (Qiagen) and eluted with 35 μL of diethyl pyrocarbonate–treated water. For reverse transcription (RT)–PCR studies, 5 μg of DNA-free RNA extract was converted into first-strand cDNA by using the SuperScript preamplification system for first-strand cDNA synthesis (Gibco BRL) and oligo(dT)12–18. The cDNA samples were diluted 10-fold, and 2 μL of dilution was amplified in a 50-μL PCR mixture containing DNA polymerase buffer (Gibco BRL), 2.5 mM MgCl2, 200 μM of each deoxynucleotide triphosphate, 0.2 μM of each gene-specific primer, and 1 unit of Taq DNA polymerase (Gibco BRL). PCR was conducted for 18, 21, 24, 35, or 40 cycles with the following thermal profiles: 94°C for 30 sec (3 min for the first cycle), 60°C for 30 sec, and 72°C for 2 min 30 sec, with a 10-min terminal extension step at 72°C.
For quantitative RT-PCR assays, reactions were performed with 18, 21, and 24 cycles to ensure that amplifications were within the linear range. In this study, only the results obtained with 21 cycles are presented, because their results were included in the linear zone. The PCR products were size-separated on a 1% (w/v) agarose gel, blotted onto positively charged nylon membrane (GeneScreen Plus; Du Pont), and hybridized with primed 32P-labeled fragments. Probes were labeled with α-32P-dCTP, using the random primers DNA labeling system kit (Gibco BRL). After overnight hybridization in a solution of 7% SDS, 0.25 M Na2HPO4/NaH2PO4, pH 7.2, and 2 mM EDTA at 65°C, blots were washed twice in 2 × SSC (1 × SSC is 0.15 M NaCl and 0.015 M sodium citrate), 0.5% sarkosyl, and 0.2% tetra-Natrium diphosphate for 15 min at 65°C before autoradiography.
To determine whether the same amounts of RNA had been sampled from the different tissues, we also tested two control primers, EF1αA4-UP (5′-ATGCCCCAGGACATCGTGATTTCAT-3′) and EF1αA4-RP (5′-TTGGCGGCACCCTTAGCTGGATCA-3′), designed from exons 1 and 2 of the EF1αA4 gene (GenBank accession number X16432), respectively, under the same conditions. The length of the amplified product from EF1αA4 transcripts was 706 bp. For mRNA detection of the genes of interest, the gene-specific primers used were as follows: for TT8, deb122RB1 (upstream primer) and deb122LB3 (reverse primer, 5′-CTCCACGTGGCAAACGATGATTGG-3′); for C4H (GenBank accession number U71080), C4H-UP (5′-CACTGTTTACGGCGAGCATTGG-3′) and C4H-RP (5′-AGAACCGTGTCGAGTTCGTTCC-3′); for CHS (M20308), CHS-UP (5′-ATGGCTGGTGCTTCTTCTTTGG-3′) and CHS-RP (5′-TCTCTCCGACAGATGTGTCAGG-3′); for DFR (M86359), DFR-UP (5′-ATGGTTAGTCAGAAAGAGACCG-3′) and DFR-RP (5′-GTCTTATGATCGAGTAATGCGC-3′); and for BAN (AF092912), BAN-RT5′ (5′-AACAACTAAATCTCTATCTCTGTA-3′) and BAN-RT3′ (5′-GAATGAGACCAAAGACTCATATAC-3′), which were designed by Devic et al. (1999). For each set of primers, the size of the amplified product is 780 bp for TT8, 670 bp for C4H, 725 bp for CHS, 670 bp for DFR, and 1210 bp for BAN. Each blot was repeated at least in triplicate with RNA from independent experiments.
The transcription start site of the TT8 gene was localized by primer extension analysis with a gene-specific 27-mer oligonucleotide, which was complementary to the sense strand sequence of the TT8 cDNA. Primer deb122EXT (5′-GATTCATCCATCGTTCCCGGA-GATACG-3′), from –16 to +11 relative to the translation start site (ATG), was radiolabeled at its 5′ terminus with T4 polynucleotide kinase and γ-32P-dATP. Approximately 105 counts per minute of the radiolabeled primer was hybridized with 30 μg of total RNA, which was isolated from Arabidopsis green siliques. The hybridization mixture was incubated in a 65°C water bath for 90 min and then allowed to cool overnight to room temperature. After hybridization, first-strand cDNA was synthesized from the annealed primer by adding reverse transcriptase and deoxynucleotide triphosphates, as described above. The reaction was stopped by incubating at 70°C for 15 min. The reaction product was resuspended in sequencing gel loading buffer, denatured at 95°C, resolved by electrophoresis through a polyacrylamide–7 M urea gel, and visualized by autoradiography. To provide length markers, we sequenced parts of the pB-SMB plasmid (Perkin-Elmer) with forward and reverse universal primers.
For the cloning of the 3′ untranslated region (UTR) of TT8 cDNA, two successive PCR amplifications were conducted with the cDNA library described by Giraudat et al. (1992). To this end, two specific nested primers from the TT8 sequence, deb122LB4 (5′-TGATCTTCAGAGTTCCTTTCCTCC-3′) and deb122LB5 (5′- GGAGATAAGGGC-GAAAGTAAGAGGG-3′), were successively used in combination with the T7 primer, derived from the λ Zap II cloning vector (Stratagene). Primers deb122-5′ (5′-ATTTTTAGAGAGAGAGCTACCACG-3′) and deb122-3′ (5′-AGTACTAAATTGGACAACGAACAA-3′) allowed amplification of the full-length TT8 cDNA.
Sequence Analysis
DNA sequences of all isolated clones were done by Genome Express (Evry, France). To manage the sequence data, the DNA Strider 1.3 program (Marck, 1988) was used. Similarity searches of the databases were performed according to Altschul et al. (1990) with the NCBI BLAST server (http://www.ncbi.nih.gov/BLAST/). The protein molecular mass was estimated by using the MWCALC program, and the nuclear localization signal was located by using the PSORT program (Nakai and Kanehisa, 1992), both at Infobiogen (http://www.infobiogen.fr/services). To perform distance analysis among plant basic helix-loop-helix proteins (bHLH), an alignment of selected published bHLH amino acid sequences was generated with the ClustalX program (Thompson et al., 1997) and optimized manually. The final alignment file is available upon request. The matrix of distances was subjected to a cluster analysis by using the neighbor-joining program from the ClustalX package; for statistical analysis, 1000 bootstrap replications were performed. The consensus tree was established by using the TreeView program (version 1.5.3, available at http://taxonomy.zoology.gla.ac.uk/rod/rod.html). The sequences were also used for maximum parsimony analysis with the Phylogenetic Analysis Using Parsimony (PAUP) program, with or without the PROTPARS matrix (PAUP version 3.1.1; developed by D.L. Swofford in 1990 at the Natural History Survey, Champaign, IL).
Acknowledgments
We are especially indebted to Gareth Jenkins, Virginie Guyon, and two anonymous reviewers for critical reading of the manuscript; to Anne-Marie Lescure for her constant support during the progress of this work; and to Maarten Koornneef for helping I.D. to conduct the allelism tests of the Versailles tt collection in his laboratory. We also thank Nicole Bechtold for her invaluable help with T-DNA mutant lines from Versailles, the Nottingham Stock Centre for providing tt8-1 and En-2 seed, and Brenda Winkel-Shirley for the gift of the ttg1-1 allele. Bernd Weisshaar, David R. Smyth, and Brenda Winkel-Shirley are acknowledged for sharing their unpublished results. We thank Jérôme Giraudat for providing an Arabidopsis silique cDNA library and the RZPD for the hybridization filter and individual BAC clones. Akira Suzuki is gratefully acknowledged for excellent technical advice with the primer extension analysis. We also thank Joël Talbotec and Pierre Marie for plant care. N.N. is the recipient of a Ministère de l'Education Nationale, de la Recherche et de la Technologie fellowship.
References
- Abe, H., Yamaguchi-Shinozaki, K., Urao, T., Iwasaki, T., Hosokawa, D., and Shinozaki, K. (1997). Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid–regulated gene expression. Plant Cell 9**,** 1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert, S., Delseny, M., and Devic, M. (1997). BANYULS, a novel negative regulator of flavonoid biosynthesis in the Arabidopsis seed coat_._ Plant J. 11**,** 289–299. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. (1990). Basic local alignment search tool. J. Mol. Biol. 215**,** 403–410. [DOI] [PubMed] [Google Scholar]
- Bate, N.J., and Rothstein, S.J. (1997). An _Arabidopsis Myc_-like gene (MYC-146) with homology to the anthocyanin regulatory gene Delila (accession no. AF013465) (PGR 97–140). Plant Physiol. 115**,** 315. [Google Scholar]
- Bechtold, N., Ellis, J., and Pelletier, G. (1993). _In planta Agrobacterium_-mediated gene transfer by infiltration of adult Arabidopsis thaliana plants. C. R. Acad. Sci. Paris 316**,** 1194–1199. [Google Scholar]
- Becker, D. (1990). Binary vectors which allow the exchange of plant selectable markers and reporter genes. Nucleic Acids Res. 18**,** 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell-Lelong, D.A., Cusumano, J.C., Meyer, K., and Chapple, C. (1997). Cinnamate-4-hydroxylase expression in Arabidopsis. Plant Physiol. 113**,** 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan, M., et al. (1998). Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature 391**,** 485–488. [DOI] [PubMed] [Google Scholar]
- Bharti, A.K., and Khurana, J.P. (1997). Mutants of Arabidopsis as tools to understand the regulation of phenylpropanoid pathway and UVB protection mechanisms. Photochem. Photobiol. 65**,** 765–776. [DOI] [PubMed] [Google Scholar]
- Bouchez, D., Camilleri, C., and Caboche, M. (1993). A binary vector based on Basta resistance for in planta transformation of Arabidopsis thaliana. C. R. Acad. Sci. Paris 316**,** 1188–1193. [Google Scholar]
- Bradley, J.M., Davies, K.M., Deroles, S.C., Bloor, S.J., and Lewis, D.H. (1998). The maize Lc regulatory gene up-regulates the flavonoid biosynthetic pathway of Petunia. Plant J. 13**,** 381–392. [Google Scholar]
- Brown, J.W.S., Smith, P., and Simpson, C.G. (1996). Arabidopsis consensus intron sequences. Plant Mol. Biol. 32**,** 531–535. [DOI] [PubMed] [Google Scholar]
- Burbulis, I.E., and Winkel-Shirley, B. (1999). Interactions among enzymes of the Arabidopsis flavonoid biosynthetic pathway. Proc. Natl. Acad. Sci. USA 96**,** 12929–12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürger, D. (1971). Die morphologischen Mutanten des Göttinger Arabidopsis-Sortiments, einschliesslich der Mutanten mit abweichender Samenfarbe. Arabidopsis Inf. Serv. 8**,** 36–42. [Google Scholar]
- Burr, F.A., Burr, B., Scheffler, B.E., Blewitt, M., Wienand, U., and Matz, E.C. (1996). The maize repressor-like gene intensifier1 shares homology with the r1/b1 multigene family of transcription factors and exhibits missplicing. Plant Cell 8**,** 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelluccio, C., Paganga, G., Melikian, N., Bolwell, G.P., Pridham, J., Sampson, J., and Rice-Evans, C. (1995). Antioxidant potential of intermediates in phenylpropanoid metabolism in higher plants. FEBS Lett. 368**,** 188–192. [DOI] [PubMed] [Google Scholar]
- Chapple, C.C.S., Shirley, B.W., Zook, M., Hammerschmidt, R., and Somerville, S.C. (1994). Secondary metabolism in Arabidopsis. In Arabidopsis, E.M. Meyerowitz and C.R. Sommerville, eds (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), pp. 989–1030.
- Cone, K.C., Burr, F.A., and Burr, B. (1986). Molecular analysis of the maize anthocyanin regulatory locus C1. Proc. Natl. Acad. Sci. USA 83**,** 9631–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debeaujon, I., Léon-Kloosterziel, K.M., and Koornneef, M. (2000). Influence of the testa on seed dormancy, germination and longevity in Arabidopsis. Plant Physiol. 122**,** 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vetten, N., Quattrocchio, F., Mol, J., and Koes, R. (1997). The an11 locus controlling flower pigmentation in petunia encodes a novel WD-repeat protein conserved in yeast, plants, and animals. Genes Dev. 11**,** 1422–1434.9192870 [Google Scholar]
- Devic, M., Albert, S., Delseny, M., and Roscoe, T.J. (1997). Efficient PCR walking on plant genomic DNA. Plant Physiol. Biochem. 35**,** 331–339. [Google Scholar]
- Devic, M., Guilleminot, J., Debeaujon, I., Bechtold, N., Bensaude, E., Koornneef, M., Pelletier, G., and Delseny, M. (1999). The BANYULS gene encodes a DFR-like protein and is a marker of early seed coat development. Plant J. 19**,** 387–398. [DOI] [PubMed] [Google Scholar]
- Dixon, R.A., and Paiva, N.L. (1995). Stress-induced phenylpropanoid metabolism. Plant Cell 7**,** 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooner, H.K., Robbins, T.P., and Jorgensen, R.A. (1991). Genetic and developmental control of anthocyanin biosynthesis. Annu. Rev. Genet. 25**,** 173–199. [DOI] [PubMed] [Google Scholar]
- Doyle, J.J., and Doyle, J.L. (1990). Isolation of plant DNA from fresh tissues. Focus 12**,** 13–15. [Google Scholar]
- Durbin, M.L., McCaig, B., and Clegg, M.T. (2000). Molecular evolution of the chalcone synthase multigene family in the morning glory genome. Plant Mol. Biol. 42**,** 79–92. [PubMed] [Google Scholar]
- Gierl, A., and Saedler, H. (1992). Plant-transposable elements and gene tagging. Plant Mol. Biol. 19**,** 39–49. [DOI] [PubMed] [Google Scholar]
- Giraudat, J., Hauge, B.M., Valon, C., Smalle, J., Parcy, F., and Goodman, H.M. (1992). Isolation of the Arabidopsis ABI3 gene by positional cloning. Plant Cell 4**,** 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff, S.A., Cone, K.C., and Chandler, V.L. (1992). Functional analysis of the transcriptional activator encoded by the maize B gene: Evidence for a direct functional interaction between two classes of regulatory proteins. Genes Dev. 6**,** 864–875. [DOI] [PubMed] [Google Scholar]
- Gong, Z.-Z., Yamagishi, E., Yamazaki, M., and Saito, K. (1999). A constitutively expressed _Myc_-like gene involved in anthocyanin biosynthesis from Perilla frutescens: Molecular characterization, heterologous expression in transgenic plants and transactivation in yeast cells. Plant Mol. Biol. 41**,** 33–44. [DOI] [PubMed] [Google Scholar]
- Goodrich, J., Carpenter, R., and Coen, E.S. (1992). A common gene regulates pigmentation pattern in diverse plant species. Cell 68**,** 955–964. [DOI] [PubMed] [Google Scholar]
- Guerineau, F., Lucy, A., and Mullineaux, P. (1992). Effect of two consensus sequences preceding the translation initiator codon on gene expression in plant protoplasts. Plant Mol. Biol. 18**,** 815–818. [DOI] [PubMed] [Google Scholar]
- Holton, T.A., and Cornish, E.C. (1995). Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell 7**,** 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes, R.E., Quattrocchio, F., and Mol, J.N.M. (1994). The flavonoid biosynthetic pathway in plants: Function and evolution. BioEssays 16**,** 123–132. [Google Scholar]
- Koncz, C., Kreuzaler, F., and Kalman, Z., and Schell, J. (1984). A simple method to transfer, integrate and study expression of foreign genes, such as chicken ovalbumin and α-actin in plant tumors. EMBO J. 3**,** 1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koornneef, M. (1981). The complex syndrome of ttg mutants. Arabidopsis Inf. Serv. 18**,** 45–51. [Google Scholar]
- Koornneef, M. (1990). Mutations affecting the testa colour in Arabidopsis. Arabidopsis Inf. Serv. 27**,** 1–4. [Google Scholar]
- Kubasek, W.L., Shirley, B.W., McKillop, A., Goodman, H.M., Briggs, W., and Ausubel, F.M. (1992). Regulation of flavonoid biosynthetic genes in germinating Arabidopsis seedlings. Plant Cell 4**,** 1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, H., Peeters, A.J.M., Aarts, M.G.M., Pereira, A., and Koornneef, M. (1999). ANTHOCYANINLESS2, a homeobox gene affecting anthocyanin distribution and root development in Arabidopsis. Plant Cell 11**,** 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lairon, D., and Amiot, M.-J. (1999). Flavonoids in food and natural antioxidants in wine. Curr. Opin. Lipidol. 10**,** 23–28. [DOI] [PubMed] [Google Scholar]
- Lesnick, M.L., and Chandler, V.L. (1998). Activation of the maize anthocyanin gene a2 is mediated by an element conserved in many anthocyanin promoters. Plant Physiol. 117**,** 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liboz, T., Bardet, C., Le Van Thai, A., Axelos, M., and Lescure, B. (1990). The four members of the gene family encoding the Arabidopsis thaliana translation elongation factor EF-1α are actively transcribed. Plant Mol. Biol. 14**,** 107–110. [DOI] [PubMed] [Google Scholar]
- Lloyd, A.M., Walbot, V., and Davis, R.W. (1992). Arabidopsis and Nicotiana anthocyanin production activated by maize regulators R and C1. Science 258**,** 1773–1775. [DOI] [PubMed] [Google Scholar]
- Ludwig, S.R., and Wessler, S.R. (1990). Maize R gene family: Tissue-specific helix-loop-helix proteins. Cell 62**,** 849–851. [DOI] [PubMed] [Google Scholar]
- Ludwig, S.R., Habera, L.F., Dellaporta, S.L., and Wessler, S.R. (1989). Lc, a member of the maize R gene family responsible for tissue-specific anthocyanin production, encodes a protein similar to transcriptional activators and contains the _myc_-homology region. Proc. Natl. Acad. Sci. USA 86**,** 7092–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marck, C. (1988). “DNA Strider”: A “C” program for the fast analysis of DNA and protein sequences on the Apple Macintosh family of computers. Nucleic Acids Res. 16**,** 1829–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, C., Prescott, A., Mackay, S., Bartlett, J., and Vrijlandt, E. (1991). Control of anthocyanin biosynthesis in flowers of Antirrhinum majus. Plant J. 1**,** 37–49. [DOI] [PubMed] [Google Scholar]
- Massari, M.E., and Murre, C. (2000). Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20**,** 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol, J., Grotewold, E., and Koes, R. (1998). How genes paint flowers and seeds. Trends Plant Sci. 3**,** 212–217. [Google Scholar]
- Mozo, T., Fischer, S., Shizuya, H., and Altmann, T. (1998). Construction and characterization of the IGF Arabidopsis BAC library. Mol. Gen. Genet. 258**,** 562–570. [DOI] [PubMed] [Google Scholar]
- Murashige, T., and Skoog, F. (1962). A revised medium for rapid growth and bioassays in tobacco tissue culture. Physiol. Plant. 15**,** 473–497. [Google Scholar]
- Nakai, K., and Kanehisa, M. (1992). A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 14**,** 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares, J., Ghosal, D., Wienand, U., Peterson, P.A., and Saedler, H. (1987). The regulatory c1 locus of Zea mays encodes a protein with homology to myb proto-oncogene products and with structural similarities to transcriptional activators. EMBO J. 6**,** 3553–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier, M.K., and Shirley, B.W. (1996). Analysis of flavanone 3-hydroxylase in Arabidopsis seedlings: Coordinate regulation with chalcone synthase and chalcone isomerase. Plant Physiol. 111**,** 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier, M.K., Murrell, J.R., and Shirley, B.W. (1997). Characterization of flavonol synthase and leucoanthocyanidin dioxygenase genes in Arabidopsis: Further evidence for differential regulation of “early” and “late” genes. Plant Physiol. 113**,** 1437–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier, M.K., Burbulis, I.E., and Winkel-Shirley, B. (1999). Disruption of specific flavonoid genes enhances the accumulation of flavonoid enzymes and end-products in Arabidopsis seedlings. Plant Mol. Biol. 40**,** 45–54. [DOI] [PubMed] [Google Scholar]
- Procissi, A., Dolfini, S., Ronchi, A., and Tonelli, C. (1997). Light-dependent spatial and temporal expression of pigment regulatory genes in developing maize seeds. Plant Cell 9**,** 1547–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrocchio, F., Wing, J.F., Leppen, H.T.C., Mol, J.N.M., and Koes, R.E. (1993). Regulatory genes controlling anthocyanin pigmentation are functionally conserved among plant species and have distinct sets of target genes. Plant Cell 5**,** 1497–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrocchio, F., Wing, J.F., van der Woude, K., Mol, J.N.M., and Koes, R. (1998). Analysis of bHLH and MYB domain proteins: Species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. Plant J. 13**,** 475–488. [DOI] [PubMed] [Google Scholar]
- Quattrocchio, F., Wing, J., van der Woude, K., Souer, E., de Vetten, N., Mol, J., and Koes, R. (1999). Molecular analysis of the anthocyanin2 gene of petunia and its role in the evolution of flower color. Plant Cell 11**,** 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice-Evans, C.A., Miller, N.J., and Paganga, G. (1997). Antioxidant properties of phenolic compounds. Trends Plant Sci. 2**,** 152–159. [Google Scholar]
- Shirley, B.W. (1996). Flavonoid biosynthesis: “New” functions for an “old” pathway. Trends Plant Sci. 1**,** 377–382. [Google Scholar]
- Shirley, B.W., Hanley, S., and Goodman, H.M. (1992). Effects of ionizing radiation on a plant genome: Analysis of two Arabidopsis transparent testa mutations. Plant Cell 4**,** 333–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley, B.W., Kubasek, W.L., Storz, G., Bruggemann, E., Koornneef, M., Ausubel, F.M., and Goodman, H.M. (1995). Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. Plant J. 8**,** 659–671. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., and Higgins, D.G. (1997). The CLUSTALX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25**,** 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urao, T., Yamaguchi-Shinozaki, K., Mitsukawa, N., Shibata, D., and Shinozaki, K. (1996). Molecular cloning and characterization of a gene that encodes a MYC-related protein in Arabidopsis. Plant Mol. Biol. 32**,** 571–576. [DOI] [PubMed] [Google Scholar]
- Walker, A.R., Davison, P.A., Bolognesi-Winfield, A.C., James, C.M., Srinivasan, N., Blundell, T.L., Esch, J.J., Marks, M.D., and Gray, J.C. (1999). The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell 11**,** 1337–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisshaar, B., and Jenkins, G.I. (1998). Phenylpropanoid biosynthesis and its regulation. Curr. Opin. Plant Biol. 1**,** 251–257. [DOI] [PubMed] [Google Scholar]
- Winkel-Shirley, B. (1998). Flavonoids in seeds and grains: Physiological function, agronomic importance and the genetics of biosynthesis. Seed Sci. Res. 8**,** 415–422. [Google Scholar]