FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCɛ, B-Raf and S6K2 (original) (raw)
Abstract
Patients with small cell lung cancer (SCLC) die because of chemoresistance. Fibroblast growth factor-2 (FGF-2) increases the expression of antiapoptotic proteins, XIAP and Bcl-XL, and triggers chemoresistance in SCLC cells. Here we show that these effects are mediated through the formation of a specific multiprotein complex comprising B-Raf, PKCɛ and S6K2. S6K1, Raf-1 and other PKC isoforms do not form similar complexes. RNAi-mediated downregulation of B-Raf, PKCɛ or S6K2 abolishes FGF-2-mediated survival. In contrast, overexpression of PKCɛ increases XIAP and Bcl-XL levels and chemoresistance in SCLC cells. In a tetracycline-inducible system, increased S6K2 kinase activity triggers upregulation of XIAP, Bcl-XL and prosurvival effects. However, increased S6K1 kinase activity has no such effect. Thus, S6K2 but not S6K1 mediates prosurvival/chemoresistance signalling.
Keywords: B-Raf, PKCɛ, S6K2, survival, SCLC
Introduction
Small cell lung cancer (SCLC) represents 20% of all lung tumours. Despite initial sensitivity to therapy, relapse with chemoresistant disease is rapid and overall survival is very poor. Therefore, elucidation of the mechanisms underlying SCLC chemoresistance is necessary. Growth factors can provide prosurvival signals, and, in particular, fibroblast growth factor-2 (FGF-2) has been implicated in driving chemoresistance in cancers including SCLC (Pardo et al, 2002; Pardo et al, 2003). Moreover, elevated serum concentrations of FGF-2 is an independent prognostic factor for adverse outcome in SCLC (Ruotsalainen et al, 2002). We previously reported that FGF-2 induced the activation of the extracellular-regulated kinase signalling pathway (MEK/ERK), thereby triggering resistance to etoposide (Pardo et al, 2002; Pardo et al, 2003), a drug commonly used in the treatment of SCLC. The prosurvival effect occurred via increased translation of the antiapoptotic molecules Bcl-2, Bcl-XL, XIAP and cIAP1 (Pardo et al, 2002; Pardo et al, 2003). Consequently, further elucidation of the links between FGF-2-induced MEK/ERK signalling and this translational response are warranted.
Ribosomal S6 kinases S6K1 and S6K2, also known as S6Kα and S6Kβ (Gout et al, 1998; Shima et al, 1998; Lee-Frumen et al, 1999), both regulate the translational machinery (Dufner and Thomas, 1999). Each kinase has a cytoplasmic and nuclear form but most work has focused on the cytoplasmic proteins, which for simplicity we refer to here as S6K1 and S6K2. They were thought to have overlapping functions as they both phosphorylate the S6 protein. However, recent data suggest that their substrates and roles may be distinct although the precise function of S6K2 is still unclear (Valovka et al, 2003; Richardson et al, 2004). Thus, despite high homology, they differ substantially in their N- and C-terminal domains; S6K1 knockout mice are small despite increased expression levels of S6K2 (Shima et al, 1998), whereas S6K2 null mice have no obvious phenotype (Pende et al, 2004); the activation of S6K1 is insensitive to MEK inhibition, but we and others have shown that S6K2 is a novel target of MEK signalling (Martin et al, 2001; Pardo et al, 2001; Wang et al, 2001). The latter findings raise the possibility that S6K2 might mediate MEK/ERK-induced chemoresistance.
S6K2 is also regulated by protein kinase C (PKC) (Valovka et al, 2003), a family of proteins involved in the activation of MEK/ERK in several cell systems including SCLC cells (Kawauchi et al, 1996; Seufferlein and Rozengurt, 1996; Zou et al, 1996). The PKC family comprises classical (cPKCs: PKCα, PKCβ, PKCγ), nonclassical (nPKCs: PKCδ, PKCɛ, PKCη and PKCθ) and atypical (aPKCs: PKCζ and PKCι/λ) classes. While the activation of cPKCs is both Ca2+ and phorbol ester dependent, nPKCs only require phorbol esters and aPKCs are independent of both agents (Way et al, 2000). Depending on the stimulus used, distinct subclasses of PKC lead to different physiological effects (Way et al, 2000). Interestingly, PKCɛ can mediate prosurvival/chemoresistance in lung cancer cells (Ding et al, 2002), but the signalling mechanism underlying this effect was not identified. Other signalling molecules that could potentially be implicated in this process include Raf-1 and/or B-Raf given their known involvement in growth factor receptor coupling to MEK/ERK and PKC (Cheng et al, 2001; Hamilton et al, 2001).
Here we show that PKCɛ, B-Raf and S6K2 form a signalling complex in SCLC and HEK293 cells in response to FGF-2 treatment. S6K1 or Raf-1 failed to associate with PKCɛ. Downregulation of PKCɛ induced, while PKCɛ overexpression protected SCLC cells from drug-induced cell death. This correlated with increased S6K2, but not S6K1, activity and enhanced Bcl-XL and XIAP levels. Increased S6K2, but not S6K1 kinase, activity also enhanced cell survival and upregulated Bcl-XL and XIAP. However, downregulation of S6K2, but not S6K1, prevented FGF-2-mediated antiapoptotic effects. This is the first report of divergent biological activities for S6K2 and S6K1. Thus, S6K2, unlike S6K1, is selectively recruited into a signalling complex containing PKCɛ and B-Raf and likely controls FGF-2-mediated translation of mRNA species involved in the regulation of cell death.
Results
PKC_ɛ_ levels correlate with Bcl-XL and XIAP expression and cell survival
In view of the reported involvement of PKCɛ in lung cancer cell survival (Ding et al, 2002), we initially investigated whether PKCɛ levels correlated with the expression of Bcl-XL and XIAP, known regulators of H510 and H69 SCLC cell survival (Pardo et al, 2002; Pardo et al, 2003). Western blots revealed that H69 cells with low levels of PKCɛ displayed lower Bcl-XL and XIAP expression than H510 cells that contained high levels of PKCɛ (Figure 1A and B). A similar correlation between PKCɛ XIAP and Bcl-XL levels existed in seven additional SCLC cell lines but was not seen for other PKCs, including PKCδ (data not shown). Also, in most cell lines, an inverse correlation between the levels of PKCα and PKCɛ seemed to exist (Figure 1A and Supplementary Figure 1C). These results suggested that PKCɛ might control the expression of Bcl-XL and XIAP in SCLC cells. Indeed, ɛ-H69 cells overexpressing wild-type PKCɛ showed increased levels of both Bcl-XL and XIAP compared to vector-alone (V-H69) cells (Figure 1C). The ɛ-H69 cells also showed increased background phosphorylation of ERK (Figure 1C), enhanced survival in normal culture conditions (Figure 1D) and resistance to etoposide (VP-16)-induced cell death (Figure 1E).
Figure 1.
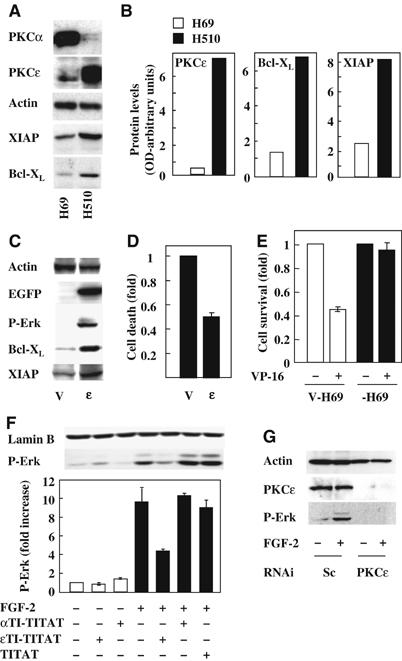
PKCɛ levels correlate with XIAP and Bcl-XL expression and Erk phosphorylation in SCLC cells. (A) H69 and H510 cell lysates were Western blotted for the expression of PKCα, PKCɛ, XIAP, Bcl-XL and actin. (B) Representative blots from (A) were quantified by optical densitometry normalised for actin. (C) H69 cells transfected with empty (V) or a wt-PKCɛ-GFP-expressing vector (ɛ) were analysed for phospho-ERK, XIAP and Bcl-XL levels. (D) Baseline level cell death in V-H69 and ɛ-H69 cells growing in 10% FCS was determined by flow cytometry using Annexin V staining. (E) ɛ-H69 and V-H69 cells in SFM were treated with or without 0.1 μM etoposide (VP-16) and cell numbers determined 96 h later. Conditions were performed in quadruplicates and the average cell number±s.e.m. represented as fold over untreated. (F) H510 cells in SFM were treated with or without 40 μM ɛTI-TITAT, αTI-TITAT or TITAT for 4 h before stimulation for 5 min with or without FGF-2 (0.1 ng/ml). Cell lysates were Western blotted for biphospho-ERK. (F-lower panel) Results from three independent experiments were analysed by optical densitometry and represented as average±s.e.m. fold increase over control. (G) H510 cells transfected with PKCɛ or scrambled (sc) siRNA were stimulated with or without FGF-2. Lysates were analysed for PKCɛ levels and Erk phosphorylation. (A, C, F and G) Lamin B and actin immunodetection were used as loading controls.
FGF-2-induced ERK phosphorylation is mediated by PKC_ɛ_
FGF-2-induced MEK/ERK signalling increases Bcl-2, Bcl-XL, XIAP and cIAP1 expression in SCLC cells (Pardo et al, 2002, 2003). In view of the preceding results, we reasoned that PKCs such as PKCɛ might mediate FGF-2-induced MEK/ERK signalling in H510 cells. To investigate this notion, we tested the effect of the cell-permeable Ca2+ chelator BAPTA and a panel of inhibitors including Gö6976, Hispidin, Rottlerin and GF109203X which target PKCα/β1, PKCβ, nPKCs or is nonselective, respectively. Only GF109203X and Rottlerin inhibited FGF-2-mediated ERK phosphorylation in H510 cells although the compounds were all active as they blocked acute PDBu-induced ERK phosphorylation (Supplementary Figure 1A). Rottlerin inhibits both PKCδ (Gschwendt et al, 1994) and PKCɛ (Davies et al, 2000), but taken together with our previous findings, it is plausible that PKCɛ might be the critical mediator of FGF-2-induced ERK signalling in H510 cells. In agreement with this, comparison of PKC isoform expression levels in seven SCLC cell lines showed that only PKCɛ correlated with FGF-2-induced ERK phosphorylation (Supplementary Figure 1B).
To confirm the involvement of PKCɛ in FGF-2-mediated ERK signalling, we used a cell-permeable translocation inhibitor peptide for PKCɛ (ɛTI-TITAT) and compared this with a PKCα inhibitor (αTI-TITAT) or carrier peptide alone (TITAT) (Vives et al, 1997). Treatment with ɛTI-TITAT led to a 60% inhibition of ERK phosphorylation in response to FGF-2 (Figure 1F). In contrast, neither αTI-TITAT nor TITAT inhibited this response. To verify these findings, we downregulated PKCs in H510 cells using either synthetic short interfering RNA (siRNA) as smart pools (P) or deconvoluted individual siRNA's. Preliminary experiments confirmed the efficacy and selectivity of these pooled or individual siRNA's (Supplementary Figure 3A and data not shown). Figure 1G demonstrates that such downregulation completely prevented FGF-2-induced ERK activation while scrambled siRNA had no effect. Similar results were seen in HEK293 cells using either the same siRNA molecules (data not shown) or pSR vectors encoding short-hairpin RNAi (shRNAi) targeting distinct sequences within PKCɛ (Supplementary Figure 3C). In contrast, parallel experiments targeting other PKC isoforms including PKCδ had no such effect (data not shown). Taken together, these results implicate PKCɛ in FGF-2-mediated ERK signalling in both H510 and HEK293 cells.
PKC_ɛ_, B-Raf and S6K2 form a multiprotein complex following FGF-2 treatment in H510 cells
We have previously demonstrated that MEK/ERK signalling is required for S6K2 activation by FGF-2 in H510 SCLC cells. However, in H69 cells, where the FGF receptors are uncoupled from MEK/ERK, FGF-2 fails to activate S6K2 (Pardo et al, 2001) and also fails to induce chemoresistance (Pardo et al, 2002). Hence, further investigation of these two cell lines could provide a valuable opportunity to elucidate the molecular mechanisms by which PKCɛ integrates signals to both S6K2 and ERK following FGF-2 stimulation. Therefore, lysates from H510 and H69 cells treated with or without FGF-2 were co-immunoprecipitated to identify potential differences in PKCɛ phosphorylation and binding partners. As phosphorylation of T566 and S729 on PKCɛ are known to correlate with activity of this kinase (Parekh et al, 1999; Cenni et al, 2002), phospho-specific antibodies to these sites were employed in the analysis. In H510 cells, FGF-2 increased phosphorylation of both these sites within 5 min (Figure 2A upper panel), a time-course consistent with ERK phosphorylation (Figure 2A lower panel). This correlated with the co-immunoprecipitation of S6K2 and B-Raf but not S6K1 or Raf-1. In contrast, in H69 cells, FGF-2 failed to induce phosphorylation of residues T566 or S729 on PKCɛ (Figure 2A). Moreover, FGF-2 did not trigger coassociation of S6K2, S6K1, B-Raf or Raf-1 with PKCɛ and, as previously described, failed to induce ERK phosphorylation in these cells. However, these proteins were easily detected in total cell lysates from H69 cells (Figure 2A, lower panel). Thus, FGF-2 appears to activate PKCɛ and may induce the formation of a novel signalling complex comprising PKCɛ/B-Raf and S6K2 in H510 cells but not in H69 cells.
Figure 2.
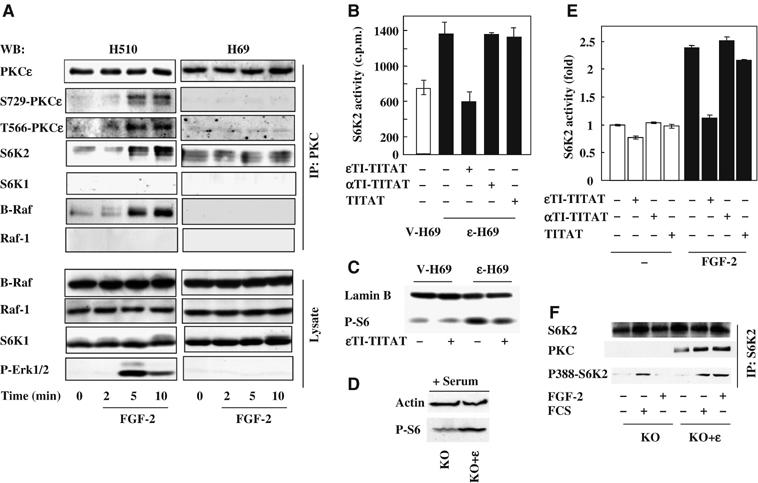
PKCɛ forms a multiprotein complex with B-Raf and S6K2 in H510 cells following FGF-2 and regulates S6K2 activity. (A) H510 and H69 cells in SFM were treated with FGF-2 for the times indicated. Cell lysates were subjected to immunoprecipitation with a PKCɛ antibody before Western blotting (WB) for the molecules indicated. (A, lower panel) Total cell lysate was Western blotted as indicated. (B, E) S6K2 was immunoprecipitated from V-H69 and ɛ-H69 (B) or H510 cells (E) following 4 h treatment with or without ɛTI-TITAT, αTI-TITAT and TITAT. Immunoprecipitates were subjected to in vitro kinase assays with S6-peptide as a substrate and the results shown are average c.p.m.±s.e.m. from triplicates of a representative experiment. (C) The phosphorylation of the endogenous S6 protein from V-H69 and ɛ-H69 cells in SFM treated with or without ɛTI-TITAT was determined using a phospho-S6 antibody. (D, F) PKCɛ KO MEFs re-expressing (KO+ɛ) or not (KO) PKCɛ were (D) grown in 10% FCS and analysed for phospho-S6 levels or (F) stimulated with or without FGF-2 and FCS before S6K2 immunoprecipitation and Western blotted as indicated. (C and D) Lamin B and actin immunodetection were used as a loading control. (A–F) Results shown are representative of at least three independent experiments.
We confirmed the protein identities of this new FGF-2-induced signalling complex in H510 cells, by repeating the co-immunoprecipitation experiments using antibodies directed against S6K2, S6K1, Raf-1 or B-Raf (Supplementary Figure 2A and B). As B-Raf activation has repeatedly been implicated in ERK signalling (Peraldi et al, 1995; Erhardt et al, 1999; Calipel et al, 2003; Dillon et al, 2003; Wan et al, 2004), our results suggest the existence of a signalling module in which both B-Raf and PKCɛ might be required for ERK phosphorylation downstream of FGF-2. In addition, the association of S6K2 to PKCɛ in an FGF-2-dependent manner raised the possibility of this PKC isoform being involved in S6K2 activation.
To investigate this, we compared the basal S6K2 activity in the PKCɛ-overexpressing H69 cells (ɛ-H69) with that in the empty-vector-transfected cells (V-H69). Background S6K2 activity was increased by two-fold in ɛ-H69 as compared to V-H69 cells (Figure 2B). This increase was dependent on PKCɛ basal activity as PKCɛ inhibition with ɛTI-TITAT lowered S6K2 activity in ɛ-H69 cells to a level comparable to that found in V-H69 cells (Figure 2B). In contrast, incubation of ɛ-H69 cells with the PKCα translocation inhibitor (αTI-TITAT) or TITAT alone had no effect on S6K2 activity. This elevated S6K2 activity correlated with increased S6 phosphorylation in vivo, which was prevented by ɛTI-TITAT (Figure 2C). Conversely, in PKCɛ null MEFs (KO), re-expression of PKCɛ (KO+ɛ) (Ivaska et al, 2002) enhanced S6 phosphorylation in response to serum or FGF-2 (Figure 2D and data not shown). We have previously shown that the KO+ɛ MEF cell line shows equivalent physiological responses to wild-type MEF cell lines despite slightly increased PKCɛ expression levels (Ivaska et al, 2002; Kermorgant et al, 2004). Moreover, in H510 cells, FGF-2-induced S6K2 activation was also inhibited by ɛTI-TITAT but not by αTI-TITAT or TITAT (Figure 2E). To further substantiate the role of PKCɛ in S6K2 activation by FGF-2 or serum, we compared PKCɛ null MEFs (KO) with the KO+ɛ cells for coassociation of PKCɛ with S6K2 and phosphorylation of T388 in the C-terminal of S6K2, a site known to correlate with activation of this kinase. Only the KO+ɛ MEFs showed FGF-2-induced association of PKCɛ with S6K2, which paralleled enhanced phosphorylation of S6K2 upon T388 (Figure 2F). Serum also induced coassociation of these two kinases, but unlike FGF-2, stimulated T388 phosphorylation both in the KO and KO+ɛ cells. Taken together, these data demonstrate that PKCɛ, B-Raf and S6K2 are part of a multiprotein complex that forms in response to FGF-2 stimulation and regulates S6K2 activity.
A PKC_ɛ_, B-Raf and S6K2 complex forms in HEK293 cells: PKC_ɛ_ downregulation disrupts B-Raf association with S6K2
To determine whether FGF-2 could induce formation of the B-Raf/PKCɛ/S6K2 complex in additional cell types, we utilized HEK293 cells, KO and KO+ɛ cells. B-Raf could be co-immunoprecipitated with either PKCɛ or S6K2 following FGF-2 stimulation in 293 cells or KO+ɛ but not in the KO cells lacking PKCɛ (Figure 3A, C and 2F and data not shown). Moreover, neither PKCα, Raf-1 nor S6K1 associated with S6K2 (data not shown). Thus, induction of this novel signaling complex by FGF-2 is not restricted to SCLC cells.
Figure 3.
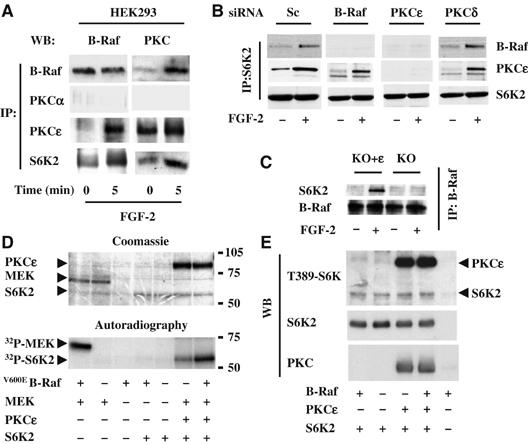
PKCɛ is required for B-Raf association with S6K2. (A) HEK293 cells were stimulated with FGF-2 and immunoprecipitates (IP) for the molecules indicated analysed by Western blotting (WB) for either B-Raf or PKCɛ. (B) HEK293 cells transfected with siRNAi for B-Raf, PKCα, PKCɛ, PKCδ or scramble control (sc) were stimulated with FGF-2 and S6K2 immunoprecipitates analysed by WB for B-Raf and PKCɛ. (C) MEFs from PKCɛ KO mice, re-expressing (KO+ɛ) or not (KO) PKCɛ were stimulated with or without FGF-2. B-Raf immunoprecipitates were analysed by WB for S6K2. (D, E) Recombinant PKCɛ, V600EB-Raf and S6K2 proteins were combined as indicated and subjected to in vitro kinase assay with 32P-γATP (D) or cold ATP (E). Recombinant GST-MEK was used as a positive control for V600EB-Raf activity. Samples were run on SDS–PAGE, Coomassie-stained (D, upper panel) or transferred to nitrocellulose (E), then exposed to an X-Ray film (D, lower panel) or subjected to WB for the molecules indicated (E). Results shown are representative of a minimum of three independent experiments.
To identify the possible sequence of interactions involved in the assembly of this multiprotein complex, we selectively downregulated B-Raf, PKCɛ or as controls PKCα and PKCδ and assessed the effect on complex formation. HEK293 cells were transfected with pooled or individual siRNA or pSR vectors encoding shRNAi. Target selectivity and ability to impair FGF-2-induced ERK phosphorylation was determined (Supplementary Figure 3A–C and data not shown). We then assessed the effect of downregulating these proteins on the associations of B-Raf and PKCɛ with S6K2 in response to FGF-2. Knockdown of PKCδ or -α or use of a scrambled RNAi had no effect on the formation of the complex (Figure 3B and Supplementary Figure 3D). In the absence of B-Raf, PKCɛ still associated with S6K2. However, B-Raf failed to associate with S6K2 in the absence of PKCɛ (Figure 3B and Supplementary Figure 3D). Importantly, identical results were seen with siRNA or shRNAi strategies targeting different sequences, although the former was more efficient at target protein knockdown. This suggests that while PKCɛ association to S6K2 could be direct, B-Raf association to S6K2 requires PKCɛ. In agreement with this, FGF-2 only induced association of B-Raf with S6K2 in the KO+ɛ but not KO cells (Figure 3C).
To further investigate this and examine whether PKCɛ and/or B-Raf could modulate the phosphorylation status of S6K2, purified preparations of these kinases were coincubated in various combinations with 32Pi-ATP. When activated B-Raf (V600EB-Raf) was coincubated with S6K2 no phosphorylation of S6K2 was seen, although in parallel experiments V600EB-Raf could efficiently phosphorylated MEK (Figure 3D lower panel). In contrast, PKCɛ induced a marked phosphorylation of S6K2, which was further enhanced by the addition of V600EB-Raf (Figure 3D, lower panel). Coomassie staining confirmed that these changes were not a consequence of unequal loading of the added kinases (Figure 3D, upper panel). Repetition of this experiment using cold ATP and Western blotting for T388S6K2 (also detects T389S6K1 and an equivalent site on PKCɛ) showed that this was not the phosphorylation site on S6K2 induced by PKCɛ or V600EBRaf (Figure 3E). Collectively, these results indicate that PKCɛ can directly associate and phosphorylate S6K2, whereas B-Raf likely requires the presence of PKCɛ to join the complex.
S6K2, but not S6K1, kinase activity increases survival and upregulates of Bcl-XL and XIAP
As FGF-2-induced cell survival requires PKCɛ, which forms a complex with BRaf and S6K2, but excludes S6K1, it is plausible that the two S6K isoforms differ in their ability to control cell survival. To test this hypothesis, we generated several clones of HEK293Tet cells (Invitrogen) expressing kinase active tetracycline-inducible constructs of the cytoplasmic forms of both S6K1 and S6K2. Tetracycline selectively increased the protein levels of transfected S6K isoforms with no effect on the parental cell line (293Tet) in all clones tested (Figure 4A and data not shown). In vitro kinase assay and Western blotting for S6 phosphorylation confirmed that tetracycline treatment increased the activity of the corresponding kinase (Figure 4B and D) similar to that seen following FGF-2 stimulation ((Pardo et al, 2001) and data not shown). We then assessed the effect of this selective increase in kinase activity on the ability of HEK293Tet cell clones to survive serum starvation. In the absence of tetracycline. the proportion of cell death was similar in the KA-S6K1, KA-S6K2 and vector alone HEK293Tet cells. Following tetracycline exposure. only the KA-S6K2-overxpressing cells had reduced cell death (Figure 4C), seen between 12 and 24 h after serum withdrawal (Supplementary Figure 4A). These results could not be attributed to enhanced cell proliferation as KA-S6K2-expressing cells showed no increase in DNA synthesis compared to V-293Tet cells. However, Bcl-XL and XIAP expression were selectively increased in KA-S6K2 cells. This was not further induced by FGF-2, could not be suppressed by selective MEK inhibition with PD098059 and was not seen in either KA-S6K1- or wild-type S6K2-overexpressing cells (Figure 4D and data not shown). Like the KA-S6K2-overexpressing cells, stimulation of HEK293Tet cells with FGF-2 showed increased survival and Bcl-XL and XIAP expression, but the latter could be blocked by MEK inhibition (Figure 4E, Supplementary Figure 4C). Taken together, these results suggest that S6K2 but not S6K1 regulates Bcl-XL and XIAP expression and that activated S6K2 is sufficient to reproduce FGF-2-induced prosurvival effects.
Figure 4.
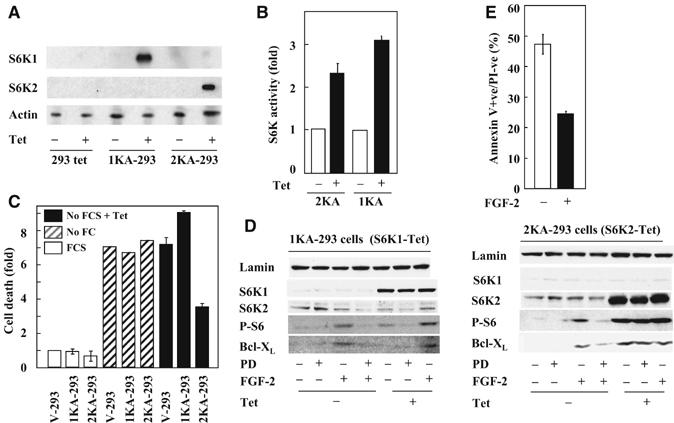
Specific induction of S6K2 kinase activity in HEK293 cells increase cell viability and upregulates Bcl-XL. HEK293-Tet clones transfected with an inducible vector for kinase active S6K1 (1KA), S6K2 (2KA) or empty vector were treated with tetracycline for 6 h before (A) Western blotting (WB) as indicated or (B) S6K1 or 2 kinase assay using an S6 peptide as substrate. (C) V-, 1KA- and 2KA-293 cells were incubated in the absence of tetracycline with (white bars) or without (hatched bars) serum for 18 h or with tetracycline in the absence of serum (black bars) and the proportion of apoptotic cells determined using annexin V staining. (D) Cell lysates from 2KA and 1KA-293 cells treated with or without tetracycline, FGF-2 and PD098059 were analysed for Bcl-XL expression and S6 phosphorylation. (E) HEK293 cells incubated with or without FGF-2 (4 h) before serum deprivation (18 h) were analysed by annexin V staining. (A, D) Actin and lamin B immunodetections were used as a loading control. (B, C, E) Results represent the average of triplicates±s.e.m. (A–E) Results are representative of at least three independent experiments.
S6K2 but not S6K1 downregulation enhances cell death and inhibits clonogenic growth
To further support the notion that S6K2 is a critical mediator of cell survival, we employed RNAi to specifically downregulate S6K isoforms and examined cell death by counting viable cells. First, S6K1 or S6K2 RNAi pSR vectors were transfected into HEK293 cells and selective downregulation of the respective targets was verified by Western blotting (Figure 5A, upper panel). Compared to S6K1 (S6K1pSR), downregulation of S6K2 (S6K2pSR) increased cell death by about two-fold in normal growth conditions (Figure 5A, lower panel). Upon serum withdrawal, background cell death increased in both vector and S6K1 knockdown cells. However, S6K2 knockdown induced more cell death (Figure 5A, lower panel).
Figure 5.
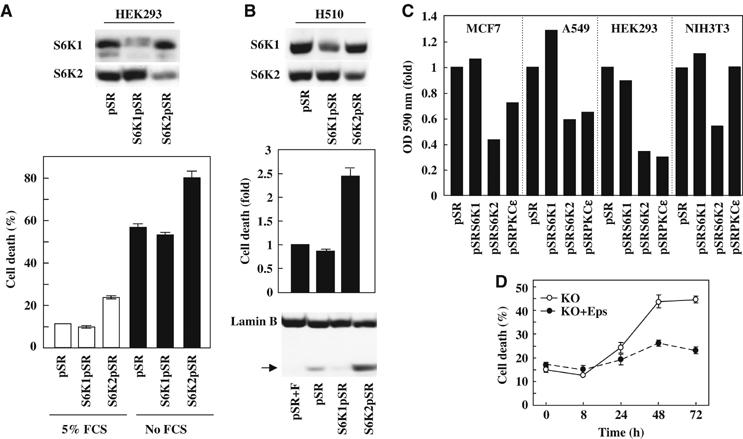
S6K2 and PKCɛ downregulation decreases cell viability and clonogenic cell growth in mammalian cells. (A) HEK293 cells were transfected with empty-vector (pSR), or pSR encoding for S6K1 (S6K1pSR) or S6K2 (S6K2pSR) RNAi sequences. Cells were grown in 5% FCS or serum-free medium for 18 h and cell viability was determined by trypan blue exclusion. (B) Lysates from H510 cells expresssing pSR, S6K1pSR or S6K2pSR were Western blotted as indicated (upper panel). The baseline cell death in 10% FCS was determined by Annexin V staining (middle panel). Lamin B cleavage was used as readout for caspase 3 activity (lower panel). pSR cells treated with FGF-2 (pSR+F) were used as negative control. (C) MCF-7, A549, HEK293 and NIH3T3 cells were transfected with the indicated pSR shRNAi constructs and grown in 5% FCS for 10 days. The OD of crystal violet-stained colonies was determined at 590 nm. For each cell line, results were normalised for absorbance found in pSR empty-vector cells. (D) KO or KO+ɛ MEFs were plated in the absence of FCS for the times indicated and the proportion of Trypan blue-positive cells determined. For (A, lower panel) and (B, middle panel) results represent the average of triplicates±s.e.m. For (A–C), the results shown are representative of at least three independent experiments.
In H510 cells, S6K1pSR and S6K2pSR also induced specific downregulation of the corresponding protein (Figure 5B, upper panel). Moreover, while expression of S6K1pSR had no effect on cell survival as compared to empty vector control (pSR), S6K2 downregulation increased basal cell death by greater than two-fold over control (Figure 5B lower panel). This correlated with an increase in the cleavage of lamin B, a substrate of caspase 3 and 7 (Figure 5B lower panel). In addition, S6K2pSR-H510, unlike the pSR- or S6K1pSR-H510 cells, could not be propagated in culture owing to cell death (data not shown). These results support our earlier findings that S6K2 but not S6K1 plays a crucial role in promoting cell survival.
As an additional approach to examine the specific effects of S6K2, we employed clonogenic assays, which at least in part reflect cell survival. Figure 5C demonstrates that only RNAi-mediated knockdown of S6K2 and not S6K1 expression inhibited the clonogenic growth of HEK293 cells. Similar findings were seen with PKCɛ knockdown by shRNAi (Figure 5C). Importantly, these results were not specific to HEK293 cells but could be reproduced in A549 human non-SCLC (NSCLC) and in MCF7 human breast carcinoma cell lines (Figure 5C). The effect of S6K2 downregulation was also not species specific, as downregulation of S6K2 in NIH3T3 murine fibroblasts using the same vector (which targets a conserved sequence) led to a decrease of clonogenic growth (Figure 5C). In contrast, the PKCɛ targeting sequences used here, which are not conserved between human and mouse, did not reduce NIH3T3 cell PKCɛ levels or clonogenic growth (Figure 5C and data not shown). However, the importance of PKCɛ in mediating prosurvival effects in murine cells was seen in the KO+ɛ cells which survived serum withdrawal much better than the KO cells (Figure 5D). Taken together, these data show that S6K2, but not S6K1, promotes cell survival of HEK293 and H510 SCLC cells and might be widely involved in regulating mammalian cell clonogenic growth.
S6K2 but not S6K1 downregulation inhibits the antiapoptotic effects of FGF-2
The preceding results suggest that S6K2 mediates the prosurvival effects of FGF-2. To substantiate this, we targeted S6K1 and -2 in H510 cells using the retroviral RNAi vectors described above and subjected the resulting cell lines to etoposide treatment with or without FGF-2. Empty vector (pSR) and S6K1-downregulated (S6K1pSR) H510 cells underwent an equivalent amount of cell death in response to etoposide and were both rescued by preincubation with FGF-2 (Figure 6A). As previously described (Pardo et al, 2002, 2003), this rescue was mirrored by an increase in Bcl-XL and XIAP protein levels (Figure 6B). In contrast, H510 cells knocked down for S6K2 (S6K2pSR) demonstrated a higher background death rate and were almost entirely depleted by etoposide (Figure 6A). Moreover, pretreatment with FGF-2 failed to upregulate XIAP or Bcl-XL and could not rescue these cells from etoposide killing (Figure 6A and B). These results support the hypothesis that S6K2 mediates FGF-2-induced chemoresistance.
Figure 6.
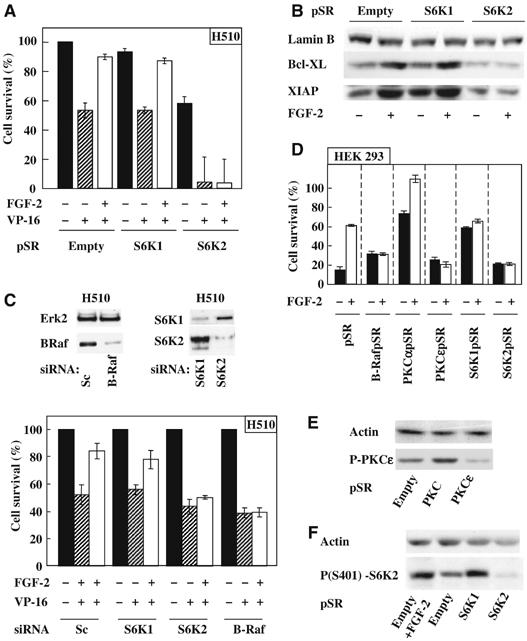
S6K2, but not S6K1, downregulation prevents FGF-2-mediated survival of H510 and HEK293 cells. (A–C) H510 cells were subjected to downregulation of the indicated proteins either by pSR RNAi retroviral vectors (A, B) or oligonucleotide RNAi (C). (A) Cells were preincubated with or without FGF-2 before etoposide treatment (VP-16). (B) Lysate from H510 cells infected with the indicated vectors and treated as shown were Western blotted for XIAP and Bcl-XL. (C) H510 cells treated with oligonucleotide RNAis were (upper panel) lysed and Western blotted as indicated or (lower panel) treated as described in (A). (D–F) HEK293 cells were subjected to downregulation of the indicated proteins by transfection of RNAi-encoding pSR vectors. (D) Cells were preincubated with or without FGF-2 before serum depletion. (A and D) Survival was determined by trypan blue exclusion. (E) HEK293 cells transfected as indicated were Western blotted for phosphoS729-PKCɛ (P-PKCɛ). (F) Transfected HEK293 cells incubated with or without FGF-2 were Western blotted for phosphoS412-S6K2. (A, C, D) Results are averages±s.e.m. of quadruplicates and (A, C) normalised to pSR (A) or Sc (C). (B, E, F) LaminB or actin immunodetection were used as loading control. (A–F) Results are representative of at least three independent experiments.
However, the overwhelming cell death induced by etoposide, as well as the elevated background death rate in the H510 S6K2pSR cells could interfere with the ability to observe the prosurvival effects of FGF-2. Therefore, we repeated these experiments using pooled siRNA's, targeting sequences within B-Raf, S6K2 or S6K1 distinct from those recognised by the pSR shRNAi's. Figure 6C (upper panel) shows that B-Raf, S6K2 and S6K1 were selectively downregulated in H510 cells with the respective pooled siRNA's similar to results seen in HEK293 cells (Supplementary Figure 3A). Moreover, transient downregulation of S6K2 completely blocked FGF-2-triggered rescue from etoposide killing (Figure 6C, lower panel). Similar results were obtained with the B-Raf siRNA (Figure 6C, lower panel). In contrast, transient knockdown of S6K1 failed to block FGF-2-induced chemoresistance. Similar results were seen when these experiments were repeated using individual siRNA to distinct target sequences (Supplementary Figure 5). Thus, B-Raf, S6K2 but not S6K1 are required for FGF-2 to provide prosurvival signals that prevent etoposide killing in H510 SCLC cells.
To demonstrate the importance of S6K2 and B-Raf in FGF-2-induced prosurvival signalling in a distinct cell system, we transiently transfected the same pSR constructs in HEK293 cells followed by serum deprivation in the presence or absence of FGF-2. In addition, RNAi vectors for PKCɛ, PKCα and S6K1 were tested in parallel. While similar amounts of cell death were induced by serum deprivation of pSR and S6K2pSR cells, FGF-2 increased the survival of pSR cells alone (Figure 6D). Similarly, FGF-2 failed to rescue cells downregulated for B-Raf or PKCɛ, the two proteins shown to interact with S6K2. In contrast, FGF-2 completely rescued HEK293 cells knocked down for PKCα (Figure 6D). Intriguingly, these cells demonstrated a high basal survival rate in the absence of serum and FGF-2. This could potentially be explained by an increase in PKCɛ phosphorylation at S729, a site linked to its kinase activity (Figure 6E). Alternatively, PKCα might be required for the induction of cell death in HEK293 cells. Surprisingly, S6K1 downregulation resulted in cell death levels comparable to those observed in pSR cells treated with FGF-2 (Figure 6D). This might reflect involvement of S6K1 in the induction of cell death. However, downregulation of S6K1 enhanced phosphorylation of S6K2 on S401, a site known to correlate with S6K2 activity, similar to that seen in response to FGF-2 stimulation (Figure 6F). In contrast, the basal phosphorylation levels of S401 were greatly reduced in the presence of the RNAi targeting S6K2 when compared to control pSR cells (Figure 6F). Thus, downregulation of S6K1 probably increases S6K2 basal activity mimicking the prosurvival effects induced by FGF-2. Moreover, this likely explains why the addition of FGF-2 fails to further improve the survival of cells knocked down for S6K1, as these cells already have activated S6K2. Taken together, these data demonstrate that S6K2 is the mediator of FGF-2 prosurvival effects. They also reveal the nonoverlapping roles of S6K1 and S6K2 in cell survival.
Discussion
Most common cancers such as SCLC are incurable principally because of chemoresistance. Therefore elucidation of the mechanism(s) involved is urgently required. We have previously shown that FGF-2 prevents apoptosis induced by the chemotherapeutic agent etoposide (Pardo et al, 2002). This prosurvival effect is mediated by MEK/ERK signalling which enhances the translation of several antiapoptotic proteins including Bcl-XL and XIAP (Pardo et al, 2002; Pardo et al, 2003). However, the kinases coupling FGFRs to MEK/ERK and the translational upregulation of these antiapoptotic proteins was unclear. Fortuitously, some SCLC cell lines (e.g. H510) have FGFRs that couple to MEK/ERK and prosurvival signaling, while other lines (e.g. H69) fail to elicit these responses despite having functional receptors (Pardo et al, 2001; Pardo et al, 2002; Pardo et al, 2003). This provided us with a useful opportunity to further dissect the signalling molecules involved in FGF-2-triggered prosurvival.
The PKC family including PKCɛ were potential candidates in this process as they are known to couple to MEK/ERK signalling (Schonwasser et al, 1998; Soh et al, 1999). Moreover, PKCɛ had already been shown to induce prosurvival effects in several cancers (Tillman et al, 2003; Wu et al, 2004) including lung cancer cells where it prevents caspase 3-mediated apoptosis induced by various chemotherapeutic agents (Ding et al, 2002). However, the underlying mechanisms involved had not been further defined. Here, we show that PKCɛ is both necessary and sufficient to couple FGFRs to MEK/ERK, Bcl-XL and XIAP upregulation and prosurvival effects in both SCLC and HEK293 cells (Figures 1, 5 and 6 and Supplementary data). Thus, (1) comparison of PKC family member expression levels in a panel of SCLC cell lines revealed that only PKCɛ correlated with the ability of FGF-2 to induce MEK/ERK signalling and upregulation of Bcl-XL and XIAP, (2) overexpression of PKCɛ was sufficient to induce MEK/ERK signalling, upregulation of Bcl-XL and XIAP and prosurvival effects, (3) selective suppression of PKCɛ function or expression prevented these FGF-2-induced effects.
The mechanism by which PKCɛ might couple to MEK/ERK signalling has been previously investigated. In endothelial cells, PKCɛ and PKCα can be co-immunprecipitated with Raf-1 in stress-mediated MEK/ERK activation (Cheng et al, 2001). In NIH-3T3 cells, PKCɛ can reside in a latent and inactive complex with Raf-1, which can be stimulated by phorbol ester to trigger MEK/ERK signalling (Hamilton et al, 2001). In contrast to these reports, we were unable to demonstrate a complex between PKCɛ or PKCα with Raf-1 in either SCLC or HEK293 cells. However, our results showed that FGF-2 inducibly triggered the association of PKCɛ with B-Raf (Figures 2 and 3 and Supplementary Figure 3). Moreover, downregulation of B-Raf with various selective RNAi species blocked FGF-2-induced MEK/ERK signalling and prosurvival effects (Figure 6, Supplementary Figure 3 and 5).
So how might PKCɛ, BRaf and MEK/ERK signalling modulate the translational machinery to upregulate the expression of Bcl-XL and XIAP, thereby promoting chemoresistance? Our previous work provided potential clues, in that FGF-2-induced MEK/ERK signalling, which was necessary for prosurvival effects (Pardo et al, 2002, 2003), was also required for the activation of S6K2 but not S6K1 (Pardo et al, 2001). Several other findings suggested that S6K2 might have discrete functions as discussed in the introduction. Thus, we postulated that S6K2 might associate with the PKCɛ/BRaf complex and mediate chemoresistance.
Our exciting new findings presented here show that S6K2, but not S6K1, does indeed associate with the FGF-2-induced PKCɛ/B-Raf complex, a finding common to both SCLC, HEK293 and MEF cells. Intriguingly, it was not possible to reproducibly show the presence of either MEK or ERK in this complex perhaps because the association was weak, transient or blocked antibody recognition. Nevertheless, RNAi knockdown studies in intact cells and coassociation studies of purified kinases in vitro indicate that the association of PKCɛ with S6K2 is direct and results in phosphorylation of S6K2. In contrast, B-Raf only associates with S6K2 in the presence of PKCɛ in intact cells and cannot directly phosphorylate S6K2 in the absence of PKCɛ. However, incubation of all three enzymes together further enhances the phosphorylation of S6K2 raising the possibility that B-Raf might phosphorylate S6K2 when PKCɛ is present. Alternatively, B-Raf might alter the conformation of PKCɛ and/or S6K2 providing further PKC sites on S6K2. A recent report examining phorbol ester-stimulated HEK293 cells suggest that S486 within the C-terminal domain of S6K2 is likely to be one of the PKC-regulated sites (Valovka et al, 2003). Clearly, the nature of the S6K2 phosphorylation sites regulated by PKCɛ and/or B-Raf following physiological stimulation with FGF-2 now warrants further investigation. Regardless of the nature of these sites, although, our results for selective overexpression or inhibition of PKCɛ function or expression indicate that this kinase mediates FGF-2-induced S6K2 activation (Figure 2).
So can S6K2 mediate the upregulation of Bcl-XL/XIAP and prosurvival signalling? In this paper, using tetracycline inducible kinase active mutants of S6K2 and S6K1, we demonstrate that only S6K2 triggers the upregulation of XIAP and Bcl-XL and induced prosurvival effects in HEK293 cells (Figure 4). Moreover, RNAi knockdown studies in both HEK293 and SCLC cells shows that downregulation of S6K2 but not S6K1 prevents survival (Figure 5, Supplementary Figure 4). In addition, S6K2 is also important for supporting clonogenic growth in several different cell lines (Figure 5). Crucially, the selective downregulation of S6K2 but not S6K1 blocks FGF-2-induced upregulation of Bcl-XL, XIAP in both SCLC and HEK293 cells and also inhibits death in response to etoposide and serum withdrawal, respectively (Figure 6, Supplementary Figure 5). Thus, S6K2 is both necessary and sufficient to mediate FGF-2-induced prosurvival signalling.
So is there any evidence that S6K2 might be implicated in prosurvival/chemoresistance in patients with SCLC? Intriguingly, we have recently found that increased protein expression levels of S6K2 in both SCLC and NSCLC biopsies appear to correlate with the development of chemoresistance (Supplementary Figure 6).
In summary, this paper identifies a novel FGF-2-induced signalling complex comprising PKCɛ/BRaf and S6K2 but excluding S6K1. The formation of this complex may explain how S6K1 and S6K2 can be guided to different cellular compartments to target distinct substrates despite their high homology within the kinase domains. Indeed, this complex, via S6K2 (but not S6K1), upregulates Bcl-XL and XIAP protein expression thereby promoting survival/chemoresistance. Thus, the discrete function of S6K2 as opposed to S6K1 has been revealed. Further investigation of the molecular mechanisms by which S6K2 might selectively interact with the translational machinery of the cell to differentially control a subset of antiapoptotic proteins is now required. Importantly, the targeting of individual members of the PKCɛ/BRaf/S6K2 signalling complex or their associations could enable the development of novel therapeutic strategies to reverse chemoresistance. Moreover, expression levels of S6K2 and possibly other members of the complex may also provide novel prognostic biomarkers.
Materials and methods
Cell culture
SCLC cell lines were maintained as previously described (Pardo et al, 2001). For experimental purposes, cells were grown in SFM (RPMI 1640 supplemented with 5 μg/ml insulin, 10 μg/ml transferrin, 30 nM sodium selenite, 0.25% bovine serum albumin) and used after 3–7 days. A549, HEK293, HEK293Tet, NIH-3T3, MCF-7 and Cos 7 cells were grown in DMEM medium containing 10% FCS at 37°C, 10% CO2. For experimental purpose, HEK293 cells were placed in serum-free DMEM for 6 h before growth factor stimulation.
Establishment of transgene-expressing cell lines
H69 cells were transfected with pEGFP constructs encoding wild-type PKCɛ using Lipofectin as per the manufacturer's instructions and cells were selected in 1 mg/ml G418. RNAi-expressing H510 cells were established by infecting H510 cells with an amphotropic virus coding for the murine ecotropic receptor (EcoR). Following selection with G418, cells were infected using murine retroviruses encoding for PKCα, PKCɛ, S6K1, S6K2, B-Raf or Raf-1 shRNAi. Stable gene downregulation was achieved by culturing the cells in the presence of 2μg/ml puromycin. Transient expression of B-Raf, Raf-1, PKCɛ, S6K1 or S6K2 RNAi was achieved by transfecting A549, HEK293, NIH-3T3, MCF-7 and Cos 7 cells with the relevant pSR construct using Lipofectamin Plus. Transgene expression or downregulation of target proteins were assessed by Western blotting.
Establishment of tetracycline-inducible S6K1 and S6K2 cell lines
HEK293Tet-on cells (Invitrogen) at 70% confluency were transfected with 25 μg of pCDNA4-S6K2-T412D, pCDNA4-S6K1-T401D or pCDNA4 (control) using calcium phosphate precipitation. Cells were selected in 50 mg/ml zeocin. In all, 15 colonies from each transfection were isolated using cylinders, and clonal cell lines were established and tested for expression of S6K1 or S6K2 upon incubation with 1 mg/ml of tetracycline by Western blot analysis.
Cell death assay
SCLC cells (5 × 104 cells/ml SFM) were pretreated with or without 0.1 ng/ml FGF-2 for 4 h before treatment with 0.1 μM etoposide and incubated at 37°C for 96 h. HEK293-Tet cells were plated in 48-well plates (104 cells/well), pretreated with or without 0.1 ng/ml FGF-2 for 4 h and cell death was induced by serum removal for 18 h. The proportion of cell death was either determined by trypan blue exclusion or by Annexin V staining and flow cytometry as previously described (Pardo et al, 2002).
Cell-permeable PKC_ɛ_ and PKC_α_ translocation inhibitor peptide
The PKCɛ translocation inhibitor (EAVSLKPT) and PKCα translocation inhibitor (SLNPEWNET) (Yedovitzky et al, 1997; Souroujon and Mochly-Rosen, 1998) were made cell permeable by linkage to the HIV-derived TITAT sequence (GRKKRRQRRRPPQ). H510 cells in RPMI were incubated for 4 h with 40 μM of either translocation inhibitor peptides or TITAT before further treatments. The activity of these inhibitors on ERK phosphorylation was assessed by Western blotting.
Co-immunoprecipitation experiments
SCLC cells grown in SFM were washed in RPMI 1640, and 2 × 106 cell aliquots were incubated in this medium for 30 min at 37°C. HEK293 cells were washed and incubated in DMEM for 6 h. Cells were then stimulated using FGF-2 for the time shown in the figure legends. Cells were lysed at 4°C in 1 ml of lysis buffer, lysates clarified by centrifugation at 15 000 g for 10 min and immunoprecipitation was performed for 1.5 h using the relevant antibody together with either Protein A or G.
S6K1/2 immune complex and in vitro kinase assays
See Supplementary data.
Clonogenic growth assays
A549, HEK293, NIH-3T3, MCF-7 and Cos 7 cells transfected with the relevant construct were plated in six-well plates (2 × 103 cells/plate) and left to grow for 10 days 37°C/10% CO2 in DMEM/5% FCS. Cells were then stained with crystal violet, colonies solubilised using a 10% acetic acid solution, and absorbance was measured at 595 nm.
RNAi sequences
RNAi-mediated downregulation of PKCα, PKCɛ, S6K1, S6K2, B-Raf and Raf-1 was achieved using short-hairpin sequences cloned into pSUPER Retro constructs or oligonucleotide siRNA. See Supplementary information for sequences.
Oligonucleotide nucleofection
1.5 × 106 SCLC cells grown in serum medium were transfected following the manufacturer's instructions with 6 μl of 20 μM siRNA in 100 μl of Nucleofector Solution V using the program T-16 on the Amaxa Nucleofector. Following transfection, cells were transferred into RPMI/10% FCS overnight before they were used for analysis.
Reagents
See Supplementary data.
Supplementary Material
Supplementary Data
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Acknowledgments
This work has been supported by grants from CR-UK and NTRAC.
References
- Calipel A, Lefevre G, Pouponnot C, Mouriaux F, Eychene A, Mascarelli F (2003) Mutation of B-Raf in human choroidal melanoma cells mediates cell proliferation and transformation through the MEK/ERK pathway. J Biol Chem 278: 42409–42418 [DOI] [PubMed] [Google Scholar]
- Cenni V, Doppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A (2002) Regulation of novel protein kinase C epsilon by phosphorylation. Biochem J 363: 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JJ, Wung BS, Chao YJ, Wang DL (2001) Sequential activation of protein kinase C (PKC)-alpha and PKC-epsilon contributes to sustained Raf/ERK1/2 activation in endothelial cells under mechanical strain. J Biol Chem 276: 31368–31375 [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon TJ, Karpitski V, Wetzel SA, Parker DC, Shaw AS, Stork PJ (2003) Ectopic B-Raf expression enhances extracellular signal-regulated kinase (ERK) signaling in T cells and prevents antigen-presenting cell-induced anergy. J Biol Chem 278: 35940–35949 [DOI] [PubMed] [Google Scholar]
- Ding L, Wang H, Lang W, Xiao L (2002) Protein kinase C-epsilon promotes survival of lung cancer cells by suppressing apoptosis through dysregulation of the mitochondrial caspase pathway. J Biol Chem 277: 35305–35313 [DOI] [PubMed] [Google Scholar]
- Dufner A, Thomas G (1999) Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 253: 100–109 [DOI] [PubMed] [Google Scholar]
- Erhardt P, Schremser EJ, Cooper GM (1999) B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol 19: 5308–5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout I, Minami T, Hara K, Tsujishita Y, Filonenko V, Waterfield MD, Yonezawa K (1998) Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase β containing a proline-rich sequence. J Biol Chem 273: 30061–30064 [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F (1994) Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun 199: 93–98 [DOI] [PubMed] [Google Scholar]
- Hamilton M, Liao J, Cathcart MK, Wolfman A (2001) Constitutive association of c-N-Ras with c-Raf-1 and protein kinase C epsilon in latent signaling modules. J Biol Chem 276: 29079–29090 [DOI] [PubMed] [Google Scholar]
- Ivaska J, Whelan RD, Watson R, Parker PJ (2002) PKC epsilon controls the traffic of beta1 integrins in motile cells. EMBO J 21: 3608–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi K, Lazarus AH, Sanghera JS, Man GL, Pelech SL, Delovitch TL (1996) Regulation of BCR- and PKC/Ca(2+)-mediated activation of the Raf1/MEK/MAPK pathway by protein-tyrosine kinase and -tyrosine phosphatase activities. Mol Immunol 33: 287–296 [DOI] [PubMed] [Google Scholar]
- Kermorgant S, Zicha D, Parker PJ (2004) PKC controls HGF-dependent c-Met traffic signalling and cell migration. EMBO J 23: 3721–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Frumen KK, Kuo CJ, Lippincott J, Terada N, Blenis J (1999) Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 18: 5108–5114 [DOI] [PubMed] [Google Scholar]
- Martin KA, Schlam SS, Romanelli A, Keon KL, Blenis J (2001) Ribosomal S6 kinase 2 inhibition by a potent C-terminal repressor domain is relieved by mitogen-activated protein-extracellular signal-related kinase kinase regulated phosphorylation. J Biol Chem 276: 7892–7898 [DOI] [PubMed] [Google Scholar]
- Pardo OE, Arcaro A, Salerno G, Raguz S, Downward J, Seckl MJ (2002) Fibroblast growth factor-2 induces translational regulation of Bcl-XL and Bcl-2 via a MEK-dependent pathway: correlation with resistance to etoposide-induced apoptosis. J Biol Chem 277: 12040–12046 [DOI] [PubMed] [Google Scholar]
- Pardo OE, Arcaro A, Salerno G, Tetley TD, Valovka T, Gout I, Seckl MJ (2001) Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene 20: 7658–7667 [DOI] [PubMed] [Google Scholar]
- Pardo OE, Lesay A, Arcaro A, Lopes R, Ng BL, Warne PH, McNeish IA, Tetley TD, Lemoine NR, Mehmet H, Seckl MJ, Downward J (2003) Fibroblast growth factor 2-mediated translational control of IAPs blocks mitochondrial release of Smac/DIABLO and apoptosis in small cell lung cancer cells. Mol Cell Biol 23: 7600–7610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh D, Ziegler W, Yonezawa K, Hara K, Parker PJ (1999) Mammalian TOR controls one of two kinase pathways acting upon nPKCdelta and nPKCepsilon. J Biol Chem 274: 34758–34764 [DOI] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G (2004) S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol 24: 3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraldi P, Frodin M, Barnier JV, Calleja V, Scimeca JC, Filloux C, Calothy G, Van Obberghen E (1995) Regulation of the MAP kinase cascade in PC12 cells: B-Raf activates MEK-1 (MAP kinase or ERK kinase) and is inhibited by cAMP. FEBS Lett 357: 290–296 [DOI] [PubMed] [Google Scholar]
- Richardson CJ, Broenstrup M, Fingar DC, Julich K, Ballif BA, Gygi S, Blenis J (2004) SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol 14: 1540–1549 [DOI] [PubMed] [Google Scholar]
- Ruotsalainen T, Joensuu H, Mattson K, Salven P (2002) High pretreatment serum concentration of basic fibroblast growth factor is a predictor of poor prognosis in small cell lung cancer. Cancer Epidemiol Biomarkers Prev 11: 1492–1495 [PubMed] [Google Scholar]
- Schonwasser DC, Marais RM, Marshall CJ, Parker PJ (1998) Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol 18: 790–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufferlein T, Rozengurt E (1996) Galanin, neurotensin and phorbol esters rapidly stimulate activation of mitogen activated protein kinase in small cell lung cancer cells. Cancer Res 56: 5758–5764 [PubMed] [Google Scholar]
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC (1998) Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 17: 6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh JW, Lee EH, Prywes R, Weinstein IB (1999) Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol Cell Biol 19: 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souroujon MC, Mochly-Rosen D (1998) Peptide modulators of protein–protein interactions in intracellular signaling. Nat Biotechnol 16: 919–924 [DOI] [PubMed] [Google Scholar]
- Tillman DM, Izeradjene K, Szucs KS, Douglas L, Houghton JA (2003) Rottlerin sensitizes colon carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis via uncoupling of the mitochondria independent of protein kinase C. Cancer Res 63: 5118–5125 [PubMed] [Google Scholar]
- Valovka T, Verdier F, Cramer R, Zhyvoloup A, Fenton T, Rebholz H, Wang ML, Gzhegotsky M, Lutsyk A, Matsuka G, Filonenko V, Wang L, Proud CG, Parker PJ, Gout IT (2003) Protein kinase C phosphorylates ribosomal protein S6 kinase betaII and regulates its subcellular localization. Mol Cell Biol 23: 852–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives E, Brodin P, Lebleu B (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 272: 16010–16017 [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116: 855–867 [DOI] [PubMed] [Google Scholar]
- Wang L, Gout I, Proud CG (2001) Cross-talk between the ERK and p70 S6 kinase (S6K) signaling pathways. MEK-dependent activation of S6K2 in cardiomyocytes. J Biol Chem 276: 32670–32677 [DOI] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL (2000) Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21: 181–187 [DOI] [PubMed] [Google Scholar]
- Wu D, Thakore CU, Wescott GG, McCubrey JA, Terrian DM (2004) Integrin signaling links protein kinase Cepsilon to the protein kinase B/Akt survival pathway in recurrent prostate cancer cells. Oncogene 23: 8659–8672 [DOI] [PubMed] [Google Scholar]
- Yedovitzky M, Mochly-Rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, Cerasi E, Nesher R (1997) Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic beta-cells. J Biol Chem 272: 1417–1420 [DOI] [PubMed] [Google Scholar]
- Zou Y, Komuro I, Yamazaki T, Aikawa R, Kudoh S, Shiojima I, Hiroi Y, Mizuno T, Yazaki Y (1996) Protein kinase C, but not tyrosine kinases or Ras, plays a critical role in angiotensin II-induced activation of Raf-1 kinase and extracellular signal-regulated protein kinases in cardiac myocytes. J Biol Chem 271: 33592–33597 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6