Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence (original) (raw)
Short abstract
Rapid computational heuristics have been developed based on BLAST analysis to detect segmental duplications, as well as regions containing potential sequence misassignments in the human genome assemblies. Most of the sequences in the human genome that have undergone recent segmental duplications have now been identified.
Abstract
Background
Previous studies have suggested that recent segmental duplications, which are often involved in chromosome rearrangements underlying genomic disease, account for some 5% of the human genome. We have developed rapid computational heuristics based on BLAST analysis to detect segmental duplications, as well as regions containing potential sequence misassignments in the human genome assemblies.
Results
Our analysis of the June 2002 public human genome assembly revealed that 107.4 of 3,043.1 megabases (Mb) (3.53%) of sequence contained segmental duplications, each with size equal or more than 5 kb and 90% identity. We have also detected that 38.9 Mb (1.28%) of sequence within this assembly is likely to be involved in sequence misassignment errors. Furthermore, we have identified a significant subset (199,965 of 2,327,473 or 8.6%) of single-nucleotide polymorphisms (SNPs) in the public databases that are not true SNPs but are potential paralogous sequence variants.
Conclusion
Using two distinct computational approaches, we have identified most of the sequences in the human genome that have undergone recent segmental duplications. Near-identical segmental duplications present a major challenge to the completion of the human genome sequence. Potential sequence misassignments detected in this study would require additional efforts to resolve.
Background
Segments of DNA with near-identical sequence (segmental duplications or duplicons) in the human genome can be hot spots or predisposition sites for the occurrence of non-allelic homologous recombination or unequal crossing-over leading to genomic mutations such as deletion [1], duplication [1], inversion [2] or translocation [3,4]. These structural alterations, in turn, can cause dosage imbalance of genetic material or lead to the generation of new gene products resulting in diseases defined as genomic disorders [5].
Previous studies to identify segmental duplications in the human genome have analyzed older versions of the genome assembly, which contained higher amounts of unfinished sequence and incorrectly mapped regions, and have used different computational approaches all performed by the same group [6-8]. With the human genome sequence now nearing completion, we have examined its content for segmental duplications using two distinct computational methods. In the first, we utilized the rapid BLAST2 [9] algorithms that allow direct chromosomal-wide sequence comparisons to be made. All BLAST results reported in table formats can be subsequently grouped, parsed and analyzed for the detection of duplicated sequences. In addition, we have shown previously that there is a strong correlation between ambiguously mapped SNPs (ambSNPs), as well as the density of SNPs, and segmental duplications [10]. AmbSNPs are SNPs that were annotated to map to two locations on a particular chromosome in the NCBI dbSNP. A subset of these ambSNPs are not true SNPs but are likely to be computer-generated nucleotide mismatches from paralogous copies of duplicated sequences and should be more appropriately labeled as paralogous sequence variants (PSVs) [10]. Another subset is likely to be false ambSNPs of genomic sequences that have been misassigned in genome assemblies. Here, we report our analysis of all potential PSVs in the human genome and their correlation with segmental duplications as detected by our BLAST analysis. Furthermore, we provide a critical assessment on the three latest human genome assemblies from our analysis of sequence misassignments as identified from this study.
Results and discussion
Human genome segmental duplication content
On the basis of the June 2002 (NCBI Build 30) human genome assembly, a total of 107.4 Mb (3.53%) of the human genome content (3,043.1 Mb) were found to be involved in recent segmental duplications by our BLAST analysis criteria (Table 1). This content is composed of more than 1,530 distinct intrachromosomal segmental duplications (80.3 Mb or 2.64% of the total genome, Figure 1) and 1,637 distinct interchromosomal duplications (43.8 Mb or 1.44% of the total genome). In addition, 29% of all duplications are located in unfinished regions of the current genome assembly. Our results are shown using the Generic Genome Browser [11,12]. We have also found that 38% of the duplications (52.3 Mb) can be considered as tandem duplications - defined here as two related duplicons separated by less than 200 kb.
Table 1.
Segmental duplication content of the human genome
| Chromosome | Size (bp) | Intrachromosomal duplication (bp) | % chromosome (previous)* | Interchromosomal duplication (bp) | % chromosome (previous)* | Total duplications (bp) | % chromosome (previous)** | Errors† (bp) | % chromosomes |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 246,874,334 | 5,278,549 | 2.1 (4.4) | 2,854,898 | 1.2 (2.3) | 7,056,274 | 2.9 (5.7) | 4,369,406 | 1.8 |
| 2 | 240,681,600 | 4,917,160 | 2.0 (2.4) | 3,298,723 | 1.4 (1.6) | 6,892,585 | 2.9 (3.2) | 2,311,522 | 1.0 |
| 3 | 194,908,136 | 2,128,493 | 1.1 (2.3) | 1,654,201 | 0.8 (2.0) | 3,146,570 | 1.6 (3.2) | 3,979,610 | 2.0 |
| 4 | 192,019,378 | 2,599,650 | 1.4 (2.3) | 2,164,382 | 1.1 (2.2) | 4,061,432 | 2.1 (3.4) | 2,482,740 | 1.3 |
| 5 | 180,966,400 | 3,519,480 | 1.9 (2.0) | 1,464,945 | 0.8 (1.3) | 4,530,406 | 2.5 (2.8) | 2,297,998 | 1.3 |
| 6 | 170,309,517 | 2,358,252 | 1.4 (2.3) | 743,875 | 0.4 (1.3) | 2,877,392 | 1.7 (3.4) | 569,918 | 0.3 |
| 7 | 157,432,793 | 8,636,434 | 5.5 (6.3) | 2,614,326 | 1.7 (2.9) | 10,139,669 | 6.4 (7.8) | 205,130 | 0.1 |
| 8 | 143,874,322 | 2,318,984 | 1.6 (2.2) | 1,125,241 | 0.8 (2.0) | 2,612,280 | 1.8 (3.0) | 3,956,756 | 2.8 |
| 9 | 132,438,756 | 7,248,232 | 5.5 (7.1) | 4,801,871 | 3.6 (4.7) | 8,341,767 | 6.3 (8.2) | 1,589,734 | 1.2 |
| 10 | 134,416,750 | 5,279,301 | 3.9 (4.3) | 1,375,341 | 1.0 (1.9) | 6,334,458 | 4.7 (5.7) | 1,250,157 | 0.9 |
| 11 | 137,442,545 | 3,622,080 | 2.6 (3.3) | 1,670,412 | 1.2 (1.8) | 4,363,619 | 3.2 (4.4) | 2,028,875 | 1.5 |
| 12 | 131,300,572 | 1,894,547 | 1.4 (2.3) | 971,490 | 0.7 (1.2) | 2,816,187 | 2.1 (3.3) | 3,383,730 | 2.6 |
| 13 | 113,446,104 | 918,255 | 0.8 (1.9) | 1,202,102 | 1.1 (2.3) | 1,855,806 | 1.6 (3.4) | 146,198 | 0.1 |
| 14 | 104,324,908 | 531,219 | 0.5 (0.7) | 820,880 | 0.8 (1.6) | 1,335,177 | 1.3 (2.1) | 13,814 | 0.0 |
| 15 | 99,217,355 | 4,593,233 | 4.6 (6.2) | 2,344,618 | 2.4 (3.9) | 5,634,201 | 5.7 (8.2) | 1,739,894 | 1.8 |
| 16 | 81,671,585 | 4,917,218 | 6.0 (8.3) | 2,228,116 | 2.7 (3.9) | 6,012,178 | 7.4 (9.8) | 2,113,843 | 2.6 |
| 17 | 80,052,782 | 4,775,137 | 6.0 (7.1) | 646,968 | 0.8 (2.5) | 5,274,195 | 6.6 (8.5) | 2,145,614 | 2.7 |
| 18 | 77,516,809 | 525,636 | 0.7 (1.2) | 700,654 | 0.9 (1.9) | 1,226,290 | 1.6 (3.1) | 1,443,775 | 1.9 |
| 19 | 60,013,307 | 2,700,984 | 4.5 (6.8) | 704,757 | 1.2 (2.6) | 3,156,687 | 5.3 (8.1) | 335,190 | 0.6 |
| 20 | 62,842,997 | 592,441 | 0.9 (1.1) | 873,152 | 1.4 (1.8) | 1,052,248 | 1.7 (2.1) | 147,940 | 0.2 |
| 21 | 44,626,493 | 481,879 | 1.1 (1.4) | 1,303,776 | 2.9 (5.1) | 1,504,333 | 3.4 (5.2) | 0 | 0.0 |
| 22 | 47,748,585 | 1,741,766 | 3.6 (6.7) | 1,374,363 | 2.9 (7.4) | 2,770,386 | 5.8 (10.9) | 0 | 0.0 |
| X | 14,924,9818 | 2,625,206 | 1.8 (3.6) | 2,927,714 | 2.0 (2.3) | 5,518,712 | 3.7 (5.5) | 2,185,046 | 1.5 |
| Y | 58,368,225 | 5,959,836 | 10.2 (28.4) | 3,524,276 | 6.0 (25.0) | 8,461,355 | 14.5 (40.7) | 56,204 | 0.1 |
| Un‡ | 1,391,854 | 179,709 | 12.9 (20.4) | 378,110 | 27.2 (32.6) | 407,013 | 29.2 (36.5) | 116,923 | 8.4 |
| Total | 3,043,135,925 | 80,343,681 | 2.6 (3.8) | 43,769,191 | 1.4 (2.6) | 107,381,220 | 3.5 (5.2) | 38,870,017 | 1.3 |
Figure 1.

Intrachromosomal segmental duplications identified in the human genome. Three panels of results are displayed for each chromosome. Left, graphical views of the paralogous relationships between recent segmental duplications (graphics produced using GenomePixelizer [29,30]; each line represents a duplicated module; coloring scheme, red = 99% to 100% sequence identity, purple = 96% to 98%, green = 93% to 95%, and blue = 90% to 92%). Middle panel: segmental duplications as detected by BLAST analysis (size of duplication in kb plotted against the length of chromosome in Mb). Right panel: ambSNPs density plot (number of ambSNPs plotted against the length of chromosome in Mb). All analyses were done using the June 2002 human genome sequence assembly.
In this study, we only analyzed large (size ≥ 5 kb) and recent (sequence identity ≥ 90%) duplications because we can achieve higher confidence and to prioritize those regions for their potential involvement in diseases. Previously, Bailey and colleagues [8] reported a total of 5.2% of the human genome involved in recent segmental duplications. The 1.6% discrepancy between our findings could be due to the difference in our detection criteria (size cutoff of 5 kb used in this study versus 1 kb used in Bailey et al. [8]). Moreover, we have identified 38.9 Mb of sequences (1.28% of the June 2002 genome assembly) likely to be artifactual duplications resulting from sequence misassignment errors present in the assembly. By comparing our results with those published previously [8], we found that 482/2579 clones that we identified to be involved in duplication were novel.
The molecular mechanism by which segmental duplications are created is still unclear at the moment. A recent report has suggested that Alu repeat clusters had a role as mediators of recurrent chromosomal rearrangements [13]. We have examined whether elevated amounts of repetitive elements could be found in duplicon junctions. We inspected all duplication borders from our results and calculated the occurrence of different repeat types within the 500 bp window outside each duplicon junction. The whole-genome average frequencies were determined by sampling random 500 bp windows across the genome (excluding gap regions). Overall, we found that there are significant enrichment (or relative fold increase) for the presence of small ribonucleoprotein RNA (srpRNA), satellite, long terminal (LTR) and SINE/Alu repeats (see Additional data file). In addition, our data also showed that for some chromosomes the amount of duplicated sequence is higher in the pericentromeric and subtelomeric regions of chromosomes (Figure 1), supporting the hypothesis that these repeat-dense regions have made an important contribution to the evolution of the human genome [14].
Regions containing recently occurring segmental duplications can harbor rapidly evolving hominoid-specific genes, as well as novel gene families that are unique to primates [15,16]. Using the National Center for Biotechnology Information RefSeq annotation, we identified 1,152 human genes that were mapped to duplicated regions. Of these, 475 genes were fully contained within duplicated regions and were best candidates for recent whole-gene duplication. We have carried out functional analysis of these 475 genes using the Gene Ontology Consortium database [17] and found that there is a significant increase in gene duplications for genes involved in immune defense (antibodies, blood-group antigens) and reproduction (pregnancy, sex differentiation) (see Additional data file).
Sequence misassignment errors in the human genome sequence assembly
We were aware that in silico detection methods, such as the ones used in this study, would not allow us to distinguish completely true duplications from artifactual duplications arising from misassigned sequences, especially in cases where sequence identity between two detected duplications exceeded 99.5% over a substantial length (> 5 kb) in regions composed of draft sequences. Although a small proportion of such results (duplications with > 99.5% identity) might represent unfinished regions of the genome that contain true duplications that have arisen very recently in the evolution of the human genome (such as the large and nearly perfect palindromic repeats located in the AZFc region on chromosome Yq11.223 involved in male infertility [18]), we suspect that most of the duplications (> 99.5% identity and contain draft sequences) are in fact sequence misassignment errors in the genome assembly. An explanation for such errors would be when two identical sequences belonging to the same genomic location were misassigned to distinct regions in the genome assembly.
We have used the NCBI e-PCR [19] to evaluate our results (potential sequence misassignment errors) from the June 2002 human genome assembly. Using some of the largest interchromosomal misassignment errors detected in our study, we found that none of the STS markers located within these misassigned sequences maps to their incorrectly assigned chromosomes. For example, AC121339 is incorrectly mapped to 3q13.13 in the June 2002 genome assembly, as supported by a consensus number of chromosome X sequence-tagged site (STS) markers (Figure 2, Table 2).
Figure 2.
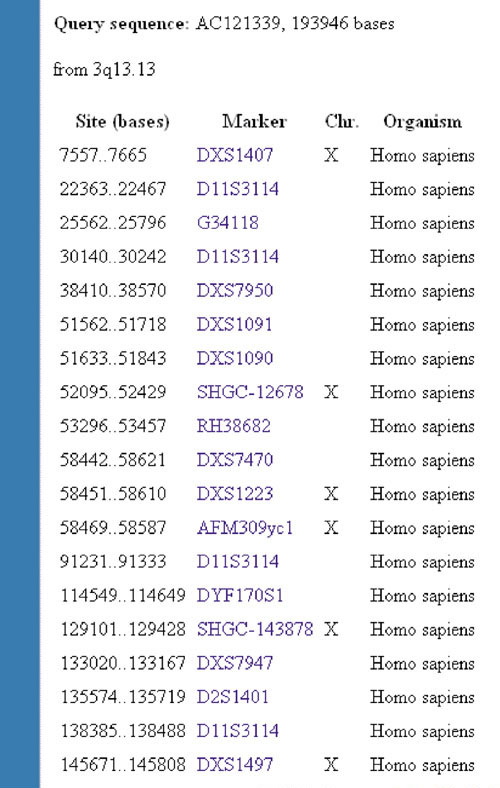
An example of sequence misassignment error as indicated by e-PCR analysis. AC121339 is incorrectly mapped to 3q13.13 in the June 2002 human genome assembly as shown by a consensus number of chromosome X STS markers.
Table 2.
Examples of sequence misassignment errors
| Clone* | Location | Size of region involved (bp) | e-PCR results |
|---|---|---|---|
| AC121339† | 3q13.13 | 193,190 | chrX |
| AC016003 | 17q21.31 | 181,582 | chr9 |
| AC119723 | 3q22.1 | 159,924 | chr6 |
| AC093007 | 3q12.1 | 169,882 | chr6 |
| AC110578 | 8p23.2 | 160,554 | chr15 |
| AC108862 | 11p15.3 | 156,150 | chr18 |
| AC113009 | 8q23.1 | 155,171 | chr11 |
| AC104765 | 8q12.1 | 150,029 | chr18 |
| AC105412 | 2p13.1 | 144,924 | chr5 |
| AC092744 | 12p12.3 | 144,009 | chr4 |
| AC099061 | 1p21.3 | 140,516 | chr15 |
| AC108735 | 3p24.3 | 136,005 | chr16 |
| AC122689 | 3q23 | 120,057 | chr12 |
| AC017027 | 1q32.1 | 116,265 | chr5 |
| AC013530 | 3q26.1 | 99,768 | chr8 |
| AC115093 | 11p15.4 | 98,715 | chr1 |
| AC112921 | Xp22.22 | 96,272 | chr3 |
| AC108094 | 16q21 | 94,953 | chr17 |
| AC079186 | 8q12.1 | 78,771 | chr7 |
| AC024573 | Unmapped | 56,016 | chr2 |
| AC115093 | 11p15.4 | 53,858 | chr1 |
From this genome assembly, we identified that a total of 38.9 Mb of sequences, representing 1.28% of the total sequence content, are involved in such potential errors (a full list of potentially misassigned sequences can be obtained from [12]) that would require additional effort and further sequencing to achieve resolutions. We also analyzed an additional two previous versions of the human genome assemblies, December 2001 and April 2002, and our results showed that there has been a dramatic reduction in potential errors in the latest human genome assembly compared to the two previous genome assemblies (Table 3). Furthermore, we examined the distribution of the amount of duplications in five different categories on the basis of their level of sequence identity to each other (Table 3). We observed a large reduction in duplications that fall within the 98-100% category, supporting the fact that the genome assemblies continue to improve and correct errors made. In addition, our data showed that there have been major improvements for chromosomes 5, 6, 7, 13, 14 and 19 over the last three genome assemblies. And for chromosomes that had reached finished status, such as chromosomes 20, 21 and 22, the number of errors was negligible.
Table 3.
Comparison of duplications and potential sequence misassignment errors in genome assemblies
| December 2001 | April 2002 | June 2002 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Length | Duplications | Errors | Length | Duplications | Errors | Length | Duplications | Errors | |
| Chr1 | 2,564 | 99 | 115 | 2,459 | 68 | 60 | 2,469 | 71 | 44 |
| Chr2 | 2,413 | 70 | 45 | 2,468 | 79 | 57 | 2,407 | 69 | 23 |
| Chr3 | 2,048 | 49 | 90 | 2,047 | 29 | 73 | 1,949 | 31 | 40 |
| Chr4 | 1,914 | 39 | 44 | 1,970 | 51 | 49 | 1,920 | 41 | 25 |
| Chr5 | 1,848 | 55 | 90 | 1896 | 55 | 112 | 1,810 | 45 | 23 |
| Chr6 | 1,783 | 58 | 56 | 1,828 | 43 | 153 | 1,703 | 29 | 6 |
| Chr7 | 1,638 | 130 | 48 | 1,605 | 119 | 27 | 1,574 | 101 | 2 |
| Chr8 | 1,457 | 35 | 66 | 1,484 | 33 | 43 | 1,439 | 26 | 40 |
| Chr9 | 1,330 | 83 | 38 | 1,291 | 75 | 27 | 1,324 | 83 | 16 |
| Chr10 | 1,421 | 74 | 51 | 1,385 | 72 | 39 | 1,344 | 63 | 13 |
| Chr11 | 1,414 | 51 | 84 | 1,341 | 43 | 36 | 1,374 | 44 | 20 |
| Chr12 | 1,396 | 30 | 83 | 1,342 | 24 | 32 | 1,313 | 28 | 34 |
| Chr13 | 1,151 | 29 | 21 | 1,136 | 22 | 15 | 1,134 | 19 | 1 |
| Chr14 | 1,065 | 27 | 8 | 1,054 | 23 | 10 | 1,043 | 13 | 0 |
| Chr15 | 991 | 62 | 30 | 1,000 | 54 | 20 | 992 | 56 | 17 |
| Chr16 | 938 | 65 | 44 | 932 | 67 | 32 | 817 | 60 | 21 |
| Chr17 | 839 | 66 | 46 | 811 | 46 | 29 | 801 | 53 | 21 |
| Chr18 | 818 | 16 | 59 | 809 | 12 | 32 | 775 | 12 | 14 |
| Chr19 | 769 | 45 | 28 | 730 | 34 | 12 | 600 | 32 | 3 |
| Chr20 | 630 | 10 | 5 | 628 | 12 | 4 | 628 | 11 | 1 |
| Chr21 | 446 | 18 | 3 | 446 | 16 | 2 | 446 | 15 | 0 |
| Chr22 | 478 | 28 | 0 | 477 | 29 | 1 | 477 | 28 | 0 |
| ChrX | 1,517 | 54 | 40 | 1,518 | 61 | 23 | 1,492 | 55 | 22 |
| ChrY | 584 | 86 | 2 | 584 | 95 | 2 | 584 | 85 | 1 |
| ChrUn | 74 | 10 | 1 | 125 | 11 | 43 | 14 | 4 | 1 |
| Total | 31,526 | 1,290 | 1,097 | 31,366 | 1,175 | 932 | 30,431 | 1,074 | 389 |
| % range* | Duplication | Error | Duplication | Error | Duplication | Error | |||
| 90-92% | 135 | 0 | 137 | 0 | 117 | 0 | |||
| 92-94% | 334 | 0 | 334 | 0 | 311 | 0 | |||
| 94-96% | 391 | 0 | 382 | 0 | 367 | 0 | |||
| 96-98% | 451 | 0 | 444 | 0 | 418 | 0 | |||
| 98-100% | 884 | 1,097 | 724 | 932 | 665 | 389 |
Paralogous sequence variants in the human genome
We have previously shown a strong correlation between ambSNPs with segmental duplications [10]. AmbSNPs are SNPs that were annotated to map to two locations on a particular chromosome in the NCBI dbSNP. Here we show on a genome scale that ambSNPs most specifically correlate with intrachromosomal segmental duplications, suggesting they are paralogous sequence variants (PSVs) (Figure 1). These PSVs were perhaps mistakenly introduced into dbSNP by automated _in silico_-generated analysis, arising from nucleotide mismatches in paralogous copies of duplicated sequences. Overall, a surprisingly high proportion, 8.6% (199,965 of 2,327,473), of the refSNPs were annotated as ambSNPs from dbSNP (Build 108). A significant number of the ambSNPs (139,974 of 199,965 or 70.0%) are located within duplicated regions as identified by BLAST and should be regarded as PSVs (see Additional data file).
The non-identification by BLAST analysis of regions that contain ambSNPs could be due to one of three possibilities. First, the duplicated copy(s) could have been removed from the sequence assembly or the two have been conflated, that is, mistakenly thought to be the same sequence owing to their high sequence similarity. A second possibility is that the duplication is smaller than 5 kb and was excluded in our BLAST analysis. A third possibility is that a collection of ambSNPs could have been generated from misassigned sequences (identical sequences but misassigned to two difference locations in the genome) in older assembly builds due to sequencing errors or true SNPs (with high polymorphism rate) in the sequence. We also observed that the density of ambSNPs generally correlates with the size of the putative duplication, although this might be affected by the level of sequence identity between duplications. For example, two duplicated sequences sharing 98% sequence identity compared to 95% over the same length might contain fewer PSVs as the number of base-pair mismatches would be fewer in the former. In addition, we observed that regions identified by our BLAST method but that do not contain ambSNPs often correspond to artifactual duplications generated from assembly errors.
Duplicons related to genomic disorders
The size, orientation, and contents of segmental duplications are highly variable and most of them show great organizational complexity. This is perhaps due to successive transposition and rearrangement events leading to the creation of segmental duplications [14]. In many cases, a contiguous duplicon is organized into multiple modules with different orientations and sizes. For example, one of the largest segmental duplicons detected in this study was 359 kb in size at the Williams-Beuren locus on 7q11.23 [20,21]. In this case, the two duplicons are separated by 1.6 Mb of intervening sequence with the telomeric duplicon comprising several separate smaller modules as compared to the primary duplicon. The results presented in our study (provided in tables available at [12]) would also allow rapid identification of new duplicons that are potentially responsible for chromosome rearrangements and genomic disorders. For example, the location of the duplicons on chromosomes 9q34/22 q11 that have been suggested to mediate recombination leading to the Philadelphia chromosome [4] was identified in our analysis, as were other medically relevant chromosomal regions (Table 4) [22,23].
Table 4.
Segmental duplications involved in known genomic disorders and chromosome rearrangements identified by BLAST and ambSNP analyses
| First copy | Second copy(s) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Disorders | Band | Start* | Size* | ambSNPs† | Start* | Size* | ambSNPs† | Identity | _Celera C4_‡ |
| Gaucher disease | 1q22 | 148108965 | 10,649 | 7 | 152776301 | -10,479 | 10 | 95.19 | S |
| Spinal muscular atrophy | 5p14/5q13 | 21621854 | 79,183 | 1,032 | 69175603 | -79,149 | 1,190 | 98.22 | M |
| Williams-Beuren syndrome | 7q11.23 | 70970126 | 359,416 | 380 | 72927299 | 111,773 | 56 | 99.60 | P |
| 73383317 | -227,260 | 355 | 99.20 | P | |||||
| t(4;8)(p16;p23) Wolf-Hirschhorn syndrome | 4p16/8p23 | 8769778 | 99,609 | 3† | 7156209 | -51,677 | 18 | 95.65 | P |
| 7470072 | -82,189 | 387 | 95.81 | P | |||||
| inv dup(8p) der(8)(8p23.1::p23.2-pter) del(8)(p23.1p23.2) | 8p23.1 | 7084847 | 138,560 | 123 | 7756853 | -126,769 | 229 | 99.16 | M |
| 7651975 | 54,807 | 463 | 96.93 | M | |||||
| Prader-Willi syndrome and Angelman syndrome | 15q11/15q13 | 19709020 | 75,325 | 102 | 19961243 | 34,902 | 55 | 98.70 | P |
| 20029574 | 41,965 | 83 | 98.79 | P | |||||
| 19802418 | 251,245 | 548 | 20064937 | 74,780 | 65 | 99.01 | P | ||
| Polycystic kidney disease 1 | 16p13 | 2164789 | 38,034 | 136 | 16249164 | 24,076 | 243 | 98.32 | P |
| Charcot-Marie-Tooth1A/Hereditary neuropathy with pressure palsies | 17p12/17p12 | 14440158 | 23,599 | 272 | 15837032 | 23,585 | 286 | 98.42 | P |
| Smith-Magenis syndrome/ dup(17)(p11.2-p11.2) | 17p12 | 18524425 | 152,700 | 547 | 20492073 | -147,255 | 539 | 99.06 | M |
| 25811482 | 28,239 | 24 | 99.20 | M | |||||
| Neurofibromatosis type 1 | 17q11.2 | 28686414 | 63,356 | 163 | 28952984 | -32,619 | 129 | 98.65 | P |
| DiGeorge syndrome and velocardiofacial syndrome | 22q11.21 | 15662253 | 155,811 | 471 | 18221385 | 155,996 | 322 | 99.42 | P |
| 17742343 | 9,740 | 62 | 97.84 | P | |||||
| 18164371 | -39,696 | 21 | 99.37 | P | |||||
| Chronic myeloid leukemia t(9;22)(pq34;q11) | 9q34/ 22q11 | 123263651 | 36,956 | NA | 20552124 | 26,424 | NA | 91.81 | S |
| Emery-Dreifuss muscular dystrophy | Xq28 | 147627873 | 11,030 | 2 | 147676529 | 11,034 | 2 | 99.61 | S |
| Shwachman-Diamond syndrome | 7q11.21 | 65091051 | 325,140 | 665 | 70647188 | 302,881 | 652 | 97.43 | P |
| Red green color blindness | Xq28 | 148439480 | 21,144 | 61 | 148476598 | 21,834 | 58 | 99.82 | S |
| BRCA1 duplication | 17q21 | 40983970 | 43,221 | 66 | 62252214 | 431,52 | 66 | 99.85 | P |
| Male infertility AZFc microdeletion region 2 | Yq11.22 | 23322362 | 190,336 | 391§ | 23680552 | -185,149 | 393§ | 99.88 | P |
| Yq11.22 | 23908727 | 94,194 | 282§ | 24794944 | -93,690 | 284§ | 99.92 | P | |
| Yq11.22 | 24794944 | 93,690 | 247§ | 27460935 | -94,218 | 248§ | 99.93 | P |
The characterization of most large segmental duplications is complicated by the fact that many of them (29% of all duplications) are only represented as draft sequences from the current genome assembly. Despite the fact that both BLAST and PSVs analyses allowed us to identify most segmental duplications involved in known genomic disorder mutations (Table 4), estimations of the size of rearranged regions were different from those previously reported [23]. In fact, with the exception of several small duplications and the segmental duplications on chromosome 22 [24], other regions containing duplications involved in genomic disorders were often erroneously assembled and misplaced. Furthermore, we have searched the Celera human genome C3 (publicly released version [25]) and C4 (subscription-based version) sequence assemblies for large duplications found on chromosome 7. We observed that most of them were not represented in large scaffolds, but instead were located in their sequence gaps, or only partially found at ends of scaffolds leading into gaps (see Table 4) [26]. This suggests that the whole-genome assembly approach [25] alone might not be able to finish such duplicated regions in mammalian genomes.
Conclusions
We have used two different computational approaches to identify the locations of all recent segmental duplications in the current human genome draft sequence. The fidelity of the results reflects the quality of the assembly examined and the parameters used. In addition, our approach has detected numerous potential sequence misassignment errors in the current genome annotation, allowing rapid error detection in future sequence assemblies. The segmental duplication map of the human genome should serve as a guide for investigation of the role of duplications in genomic disorders, as well as their contributions to normal human genomic variability [2,3,27]. It is clear that genomic regions containing segmental duplications present a major challenge to the completion of the human genome sequence by April 2003. Focused efforts including targeted sequencing of allele-specific clones, high-resolution fluorescence in situ hybridization, and expert curation would be required to validate the actual (or proposed) organization of these complex regions as well as to complete the human genome reference sequence.
Materials and methods
Genome sequence and chromosome-wide BLAST
We obtained the December 2001, April 2002, and June 2002 (NCBI Build 28, 29 and 30 respectively) human genome assemblies through the University of California, Santa Cruz Human Genome Browser [28]. All chromosome sequences were lower-case masked for highly repetitive elements by RepeatMasker (A.F.A. Smit and P. Green, unpublished). For each assembly build, each of the 25 masked chromosome sequences (including one unmapped chromosome sequence 'ChrUn') was compared against itself by chromosome-wide BLAST2 [9] to detect intrachromosomal segmental duplications (25 comparisons made), as well as pairwise comparisons to each of the other 24 chromosomes to detect interchromosomal segmental duplications (600 comparisons made). All BLAST results were subsequently parsed to eliminate low-quality and fragmented alignments under the following criteria: BLAST results having ≥ 90% sequence identity, ≥ 80 bp in length, and with expected value ≤ 10-30.
BLAST results parsing and duplication detection
Each BLAST report was sorted by chromosomal coordinates. All identical hits (same coordinate alignments), including suboptimal BLAST alignments recognized by multiple, overlapping alignments, as well as mirror hits (reverse coordinate alignments) from the BLAST results of the intrachromosomal set were removed. Contiguous alignments separated by a distance of less than 3 kb, then 5 kb, and subsequently 9 kb were joined (stepwise) into modules in order to traverse masked repetitive sequences and to overcome breaks in the BLAST alignments caused by insertions/deletions and sequence gaps. Such contiguous sequence alignment modules represent sequence similarity between the subject and query chromosome sequence in question (at their respective positional coordinates). This pairwise sequence comparison procedure serves as a rapid and robust way to detect duplication relationships. However, because of the use of masked sequences, our method would only yield a poor (on average 0.1-0.5 kb) resolution for the determination of the precise duplication alignment boundaries. Results were classified as either duplications or 'questionable' results based on sequencing status of the region and the percent sequence similarity between the detected alignments. Questionable duplications are results that fall within regions containing draft sequences with > 99.5 % detected sequence identity with another region. We consider these questionable duplications to be involved in potential sequence misassignment errors in the human genome assembly and would require further effort to achieve resolution.
Fine mapping of segmental duplications
Detailed information regarding segmental duplications as well as potential sequence misassignment errors identified by our analysis were presented using the Generic Genome Browser [11,12]. We have also summarized our results in table formats [12] that include information on size of duplications, chromosomal band locations, level of identity between duplicated copies, sequenced clones (accession numbers) and their sequencing status, as well as genes mapped to these regions. In addition, we have plotted the size of each intrachromosomal duplication (_y_-axis) against its chromosome position (_x_-axis) along each chromosome to indicate the intrachromosomal segmental duplication content of each chromosome (Figure 1) using the publicly available visualization tool GenomePixelizer [29,30]. Results generated from the detection of segmental duplications were subsequently converted into coordinate files as input for display using GenomePixelizer.
Paralogous sequence variants (PSV) density map
SNP mapping data from dbSNP were obtained through the NCBI ftp site [31]. Each chromosome SNP table, containing annotation regarding ambSNPs that have appeared twice in a particular chromosome, were extracted and sorted along with their corresponding chromosomal positions. The number of ambSNPs was tabulated along a 10-kb window to produce density plots of ambSNPs along the length of each chromosome (Figure 1).
Additional data file
The following additional data are availiable as a single file with this article: a table describing the relative frequency increase of repeat types at duplicon junctions (additional data file 1); a table giving the number of ambiguousgly mapped SNPs within segmental duplications (number of PSVs) (additional data file 1); a figure (additional data file 1) showing functional profiling of genes involved in recent whole-gene duplication vs human genome average.
Supplementary Material
Additional data file 1
Additional data file 1
Acknowledgments
Acknowledgements
We thank L. Zaifman, J. Zhang and G. Duggan for expert programming assistance. This work was supported by the Canadian Institutes of Health Research (CIHR) and Genome Canada to S.W.S. and L.-C.T. X.E. is a Senior Scientist at the Centre de Regulació Genòmica (CRG) and a Visiting Scientist of the Hospital for Sick Children Research Institute. L.-C.T is a Distinguished Scientist of CIHR and Sellers Chair of Cystic Fibrosis Research. S.W.S. is a Scholar of CIHR and International Scholar of the Howard Hughes Medical Institute.
Contributor Information
Xavier Estivill, Email: xavier.estivill@crg.es.
Stephen W Scherer, Email: steve@genet.sickkids.on.ca.
References
- Reiter LT, Murakami T, Koeuth T, Pentao L, Muzny DM, Gibbs RA, Lupski JR. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat Genet. 1996;12:288–297. doi: 10.1038/ng0396-288. [DOI] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, et al. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet. 2001;68:874–883. doi: 10.1086/319506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M, et al. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8) (p16; p23) translocation. Am J Hum Genet. 2002;71:276–285. doi: 10.1086/341610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglio G, Storlazzi CT, Giugliano E, Surace C, Anelli L, Rege-Cambrin G, Zagaria A, Jimenez Velasco A, Heiniger A, Scaravaglio P, et al. A 76-kb duplicon maps close to the BCR gene on chromosome 22 and the ABL gene on chromosome 9: possible involvement in the genesis of the Philadelphia chromosome translocation. Proc Natl Acad Sci USA. 2002;99:9882–9887. doi: 10.1073/pnas.152171299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Estivill X, Cheung J, Pujana MA, Nakabayashi K, Scherer SW, Tsui LC. Chromosomal regions containing high-density and ambiguous-mapped single nucleotide polymorphisms (SNPs) correlate with segmental duplications in the human genome. Hum Mol Genet. 2002;11:1987–1995. doi: 10.1093/hmg/11.17.1987. [DOI] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCAG - Human Segmental Duplication Homepage http://chr7.ocgc.ca/humandup
- Kolomietz E, Meyn MS, Pandita A, Squire JA. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer. 2002;35:97–112. doi: 10.1002/gcc.10111. [DOI] [PubMed] [Google Scholar]
- Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2002;3:65–72. doi: 10.1038/nrg705. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, Rocchi M, Eichler EE. Positive selection of a gene family during the emergence of humans and African apes. Nature. 2001;413:514–519. doi: 10.1038/35097067. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium http://www.geneontology.org
- Kuroda-Kawaguchi T, Skaletsky H, Brown LG, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Silber S, Oates R, Rozen S, et al. The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat Genet. 2001;29:279–286. doi: 10.1038/ng757. [DOI] [PubMed] [Google Scholar]
- Electronic PCR http://www.ncbi.nlm.nih.gov/genome/sts/epcr.cgi
- Valero MC, de Luis O, Cruces J, Perez Jurado LA. Fine-scale comparative mapping of the human 7q11.23 region and the orthologous region on mouse chromosome 5G: the low-copy repeats that flank the Williams-Beuren syndrome deletion arose at breakpoint sites of an evolutionary inversion(s). Genomics. 2000;69:1–13. doi: 10.1006/geno.2000.6312. [DOI] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, et al. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–325. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel BS, Shaikh TH. Segmental duplications: an 'expanding' role in genomic instability and disease. Nat Rev Genet. 2001;2:791–800. doi: 10.1038/35093500. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Cheung J. Discovery of the human genome sequence in the public and private databases. Curr Biol. 2001;11:R808–R811. doi: 10.1016/s0960-9822(01)00490-0. [DOI] [PubMed] [Google Scholar]
- Gratacos M, Nadal M, Martin-Santos R, Pujana MA, Gago J, Peral B, Armengol L, Ponsa I, Miro R, Bulbena A, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106:367–379. doi: 10.1016/s0092-8674(01)00447-0. [DOI] [PubMed] [Google Scholar]
- University of California, Santa Cruz, Genome Informatics http://genome.ucsc.edu
- Kozik A, Kochetkova E, Michelmore R. GenomePixelizer - a visualization program for comparative genomics within and between species. Bioinformatics. 2002;18:335–336. doi: 10.1093/bioinformatics/18.2.335. [DOI] [PubMed] [Google Scholar]
- GenomePixelizer: genome visualization tool http://www.atgc.org/GenomePixelizer/GenomePixelizer_Welcome.html
- NCBI ftp site ftp://ftp.ncbi.nih.gov/snp/human/chr_rpts
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional data file 1
Additional data file 1