Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange (original) (raw)
Abstract
Molecular studies on double-strand break (DSB) repair in mitosis are usually performed with enzymatically induced DSBs, but spontaneous DSBs might arise because of replication failures, for example when replication encounters nicks. To study repair of replication-born DSBs, we defined a system in Saccharomyces cerevisiae for the induction of a site-specific single-strand break. We show that a 21-base pair (bp) HO site is cleaved at only one strand by the HO endonuclease, with the resulting nick being converted into a DSB by replication during the S phase. Repair of such replication-born DSBs occurs by sister-chromatid exchange (SCE). We provide molecular evidence that cohesins are required for repair of replication-born DSBs by SCE, as determined in smc3, scc1 and scc2 mutants, but not for other recombinational repair events. This work opens new perspectives to understand the importance of single-strand breaks as a source of recombination and the relevance of cohesion in the repair of replication-born DSBs.
Keywords: cohesins, DSB repair, replication-born DSBs, sister-chromatid exchange
Introduction
Double-strand breaks (DSBs) can be repaired by either homologous recombination (HR) or non-homologous end joining. HR with the sister chromatid (sister-chromatid exchange, SCE), which results in perfect accurate repair, has been shown to be the preferred pathway for the repair of induced DSBs (Kadyk & Hartwell, 1992; Johnson & Jasin, 2000; Gonzalez-Barrera et al, 2003). Although in vivo studies on DSB repair in mitosis have been performed with enzymatically induced DSBs, experimental evidence linking HR to replication suggests that DSBs might occur naturally during replication, presumably as a consequence of impairment of replication-fork progression (Michel et al, 2001; Branzei & Foiani, 2005). DSBs can arise when replication forks encounter single-strand breaks (Kuzminov, 1999), which are frequently occurring DNA lesions (Lindahl, 1993). It would therefore be important to have a system for the in vivo induction of replication-dependent DSBs in order to understand the role of HR, and in particular SCE, in DSB repair. Although it is expected that proteins directly involved in HR are also required for SCE, one intriguing question is whether there is a specific machinery to favour SCE over other forms of postreplicative DSB repair.
Cohesins consist of two SMC (structural maintenance of chromosomes) components (Smc1 and Smc3) and two non-SMC components (Scc1 and Scc3 in Saccharomyces cerevisiae) that form a ring-like structure. They are deposited in G1 in a process that is dependent on the Scc2 factor, and holds sister chromatids together after replication until the onset of anaphase, ensuring proper chromosome segregation (Nasmyth, 2002). Several observations point to a role of cohesins in DSB repair: the sensitivity of cohesin mutants of different organisms to DNA-damaging agents (Sonoda et al, 2001; Lehmann, 2005); the requirement of chromatid cohesion for recovery of chromosome integrity after X-ray irradiation (Sjogren & Nasmyth, 2001); and the loading of cohesins at sites of DNA damage (Kim et al, 2002; Strom et al, 2004; Unal et al, 2004). However, whether the role of cohesins in DNA repair is specific for SCE is not known.
The purpose of this study was to define an in vivo system that mimicked a natural scenario for the origin of DSBs in dividing cells, that is, single-strand DNA breaks that are converted into DSBs during replication, and that could permit physical analysis of their repair. We show that, at a 21-base pair (bp) HO site, the HO endonuclease primarily causes nicks in the DNA that become DSBs during S phase in a replication-dependent manner. Such DSBs are repaired preferentially by SCE; cohesins are necessary for repair of these replication-born DSBs by SCE, but not by other forms of recombination.
Results
A system to induce a site-specific nick in vivo
Using plasmid substrates containing a suboptimal 21-bp long HO-cleavage site—the cleavage efficiency of which was below 10%—we recently demonstrated in the yeast S. cerevisiae that DSBs are primarily repaired by SCE in dividing cells (Gonzalez-Barrera et al, 2003). The question raised here was whether the high levels of SCE could indicate that DSBs at the 21-bp HO site appeared specifically during the S phase of the cell cycle, owing to nicks converted into DSBs by replication. To assay whether the 21-bp HO site was nicked at one DNA strand rather than cleaved at both strands, the kinetics of formation of DSBs and DNA nicks was analysed in centromeric monocopy plasmid pRS316-TINV by using native and alkaline gel electrophoresis. Only DSBs are detected in native gels, whereas both DSBs and nicks are resolved in alkaline gels; therefore, nicks are calculated from the difference between alkaline and native gels. Cleavage products were detected by the specific 1.4- and 2.4-kb fragments obtained in _Xho_I/_Spe_I-digested plasmids (Fig 1A). The analysis was made in asynchronous or G1-arrested yeast cultures expressing HO under the control of the GAL10 promoter. As can be seen in Fig 1B, the number of plasmids containing a single-strand break in asynchronous cultures accumulated at a faster rate and to a level about double that of DSBs in 2% galactose (more than 9% of the plasmids contained a nick at 2 h of HO induction, whereas only 4% accumulated a DSB). In cells arrested in G1 with α-factor, nicks accumulated with the same kinetics and reached the same levels as in asynchronous cultures (8% of total DNA molecules after 2 h), whereas DSBs accumulated at levels below 1%, suggesting that the 21-bp HO site was preferentially cleaved at one strand. Southern blot analysis with specific single-stranded DNA probes showed that both strands were cleaved indistinctly, although the Watson DNA strand showed a slight cleavage preference (Fig 2). This could be explained by the structural asymmetry of the HO site, and further supports the conclusion that HO creates nicks rather than DSBs at the 21-bp site.
Figure 1.
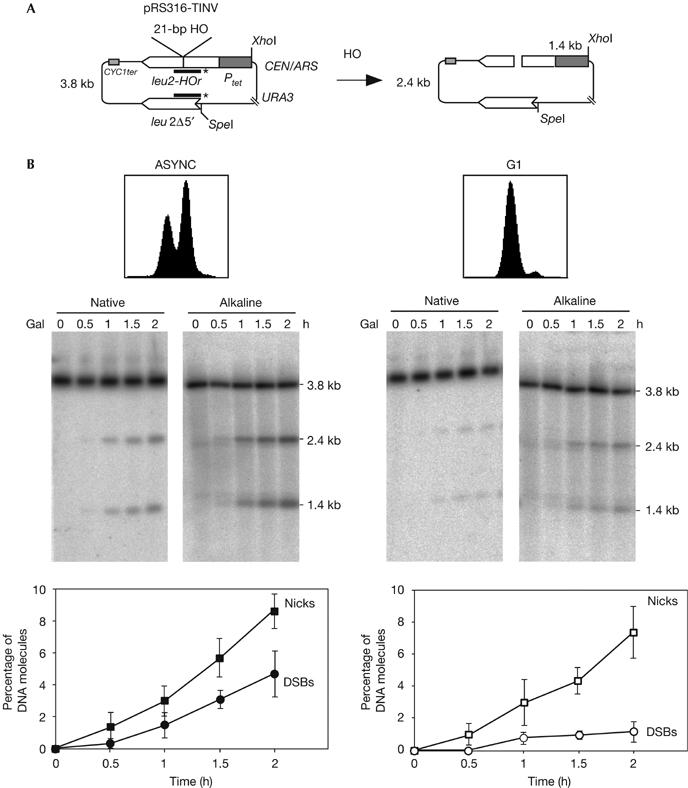
Breaks at the 21-bp HO site are primarily single-strand nicks. (A) Scheme of plasmid pRS316-TINV before and after the HO-induced DSB. Lines with asterisks indicate the LEU2 probe used for hybridization experiments. (B) Kinetics of HO cleavage as assayed by native and alkaline gel electrophoresis of DNA from asynchronous (ASYNC) and G1-arrested cultures of WS-bar1 strains. DNA samples were taken at the indicated time after HO induction and cut with _Xho_I and _Spe_I before electrophoresis. Fluorescence-activated cell sorting pattern of cultures and quantification data of nicks and DSBs are shown. The percentage of plasmids containing nicks was calculated as twice the difference between the number of broken molecules observed in the alkaline and native gels and normalized with respect to the total DNA fragments. Plasmids containing DSBs were determined directly from the native gels. DSBs, double-strand breaks; Gal, galactose.
Figure 2.

Nicks are made in both strands. Breaks in each DNA strand were determined in alkaline gels by hybridization of DNA from G1-arrested cells with LEU2 single-stranded specific probes. For other details, see Fig 1. Gal, galactose.
Replication converts nicks into DSBs
To test the possibility that nicks are converted into DSBs after replication, we determined the kinetics of DSB formation in G1-arrested cells after different periods of release from arrest, both in the absence and presence of hydroxyurea (HU), which delays DNA replication by depleting the intracellular dNTP pools. By using fluorescence-activated cell sorting analysis we confirmed that 50 mM HU was sufficient to inhibit replication significantly, as cells entered S phase synchronously and much later than in the absence of HU (Fig 3A). Southern blot analysis after α-factor release showed that, in the absence of HU, DSBs accumulated with the same kinetics and to the same levels as asynchronous cultures (7% of total molecules 2 h after release). However, the kinetics of DSBs was clearly delayed in the presence of HU (Fig 3B): in the absence of HU, an increase in DSBs was evident 1 h after α-factor release, whereas in the presence of HU, induction was observed at 2 h, the levels of cleaved molecules being less than 5% even 4 h after release. As expected, no further accumulation of DSBs was observed in G1-arrested cells in which α-factor was present all the time. By contrast, the levels of nicks remained stable (around 10%) after release (data not shown). These results indicate that DSBs are formed at the 21-bp HO site after the replication fork encounters the nick, providing a unique system to study the role of SCE in the repair of replication-dependent DSBs that are initiated by a nick.
Figure 3.

HO-induced double-strand breaks at the 21-bp HO site are formed after DNA replication. (A) Fluorescence-activated cell sorting analysis of the WS-bar1 cells arrested in G1 with α-factor and at different periods after release. (B) Kinetics of DSB formation (native gel) after release from G1 arrest, as determined by Southern blot analysis. After 2 h of HO induction in galactose (Gal), WS-bar1 G1-arrested cells were released from α-factor in the presence or absence of 50 mM HU. Quantification data are plotted at the bottom. For other details, see Fig 1. DSB, double-strand break; HU, hydroxyurea.
Cohesins are required specifically for SCE
This HO site is located in a 1.2-kb leu2 inverted repeat; therefore, the system allows the study of DSB repair efficiency by SCE (Gonzalez-Barrera et al, 2003). As can be seen in Fig 4A, 2.9- and 4.7-kb bands in _Spe_I/_Xho_I-digested plasmids appear as the result of unequal SCE events, which can be used as an estimate of total SCE. In addition, an event between the intrachromatid repeats (intrachromatid recombination, ICR), which we have shown to occur by break-induced replication, can also result in a 2.9-kb band (F.C.-L. & A.A., unpublished data). Therefore, whereas the 4.7-kb fragment is specific for SCE events, ICR can be estimated from the difference between the 2.9- and 4.7-kb bands. Interestingly, SCE was shown to be the main repair pathway detected in these assays (Gonzalez-Barrera et al, 2003), which is not surprising in the light of the finding that DSBs appear specifically during replication.
Figure 4.
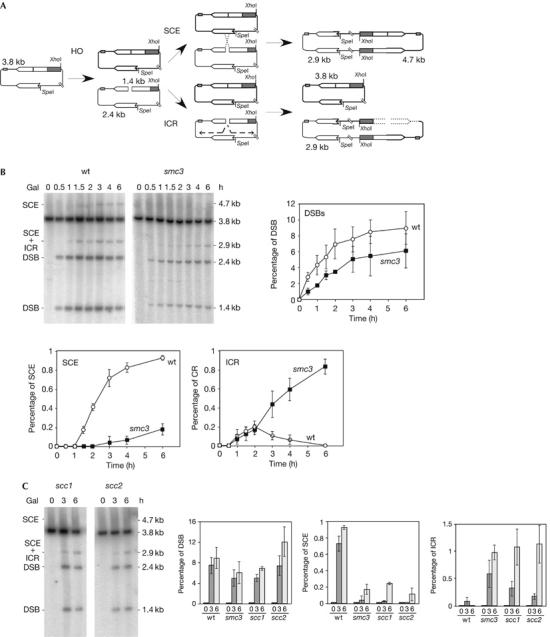
Cohesion is required for sister-chromatid exchange but not for intrachromatid recombination. (A) Schemes of plasmid pRS316-TINV and the intermediates produced by unequal SCE and ICR. Each band detected after _Xho_I–_Spe_I cleavage, using the LEU2 probe shown in Fig 1, is indicated with its corresponding size. Note that ICR events observed in these assays occur by break-induced replication, and gene conversions are not detected. (B) Kinetics of DSB formation, SCE and ICR intermediates in isogenic wild-type (wt) and smc3-42 strains. (C) Kinetics of DSB formation, SCE and ICR intermediates in isogenic scc1-73 and scc2-4 strains. Wt and smc3-73 data, taken from (B), are plotted for comparison. Cultures were incubated at restrictive temperature (37°C) for 30 min before HO induction. SCE levels were calculated as the ratio between the 4.7-kb band and the total plasmid DNA. ICR levels were calculated by subtracting the density value of the 4.7-kb band from that of the 2.9-kb band. For other details, see Fig 1. DSB, double-strand break; ICR, intrachromatid recombination; SCE, sister-chromatid exchange.
Knowing the role of cohesins in holding sister chromatids together after replication (Nasmyth, 2002), we wondered whether cohesins were specifically required for SCE compared with other forms of HR such as ICR. For this purpose, we determined the effect of a cohesin-subunit thermosensitive allele, smc3-42, on the repair by SCE and ICR of replication-born DSBs induced at the 21-bp HO site in plasmid pRS316-TINV. As can be seen in Fig 4B, the kinetics of DSB accumulation in smc3-42 cells at the restrictive temperature was similar and only slightly lower than in wild type, with 6% and 8% of molecules containing DSBs, respectively, after 6 h of HO induction. Remarkably, the kinetics of accumulation and the overall levels of SCE intermediates, as indicated by the 4.7-kb band, were severely reduced with respect to the wild type. Thus, whereas almost 1% of total DNA molecules underwent an SCE event in wild-type cells, this value was about 0.2% in smc3 cells. By contrast, smc3 did not reduce the relative intensity of the 2.9-kb band. ICR intermediates accumulated with wild-type kinetics in the first 2 h after HO induction, and significantly above wild-type levels after this time (0.8% at 6 h), in smc3 cells; however, in wild-type cells, ICR diminished until becoming undetectable after 2 h of HO induction. Therefore, SCE, but not ICR, was impaired in smc3-42 mutants. This suggests a specific role of cohesins in SCE, which is consistent with the reported intrachromosomal recombination proficiency of cohesin mutants (Unal et al, 2004).
To determine whether the effect of cohesins on SCE was not specific to mutants of the Smc3 structural subunit, we tested mutants of two other proteins: the Scc1 cohesin subunit, scc1-73, and of the Scc2 protein required for cohesin loading, scc2-4. As can be seen in Fig 4C, scc1-73 and scc2-4 decreased SCE efficiency similarly as smc3-42. Therefore, we can conclude that cohesins are required for general SCE.
Cohesins are required for equal SCE
Our results indicate that cohesins contribute to SCE, as detected by unequal events. Therefore, we wanted to demonstrate that this was also the case for equal SCE. For this purpose, we used centromeric monocopy plasmid pCM189-leu2HOr, which contains the 21-bp HO site at a unique leu2 copy. In this system, equal SCE generates a circular dimer (Fig 5A) that can be scored by Southern blot analysis of non-digested DNA (Gonzalez-Barrera et al, 2003). As can be seen in Fig 5B, equal SCE intermediates were reduced more than threefold in smc3 mutants, indicating that Smc3 is required for equal SCE. Therefore, we conclude that cohesins might ensure that recombinational repair takes place using the sister chromatid as the donor.
Figure 5.
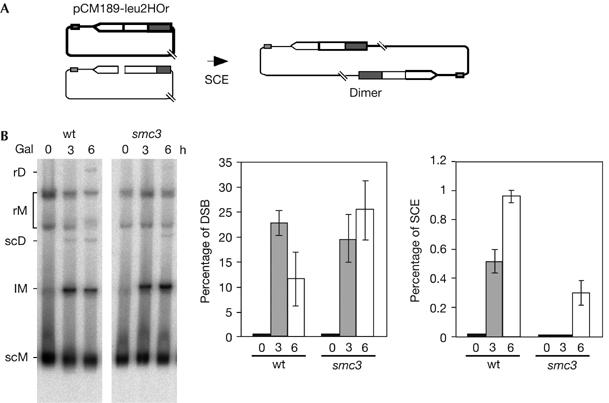
Cohesins are required for equal sister-chromatid exchange. (A) Schematic representation of the monomeric plasmid pCM189-leu2HOr, containing no DNA repeats, and the dimer obtained by equal SCE. (B) Kinetics of DSB formation and equal SCE intermediates in isogenic wild-type (wt) and smc3-42 strains. Plasmid DNA was not digested before electrophoresis. lM, linear monomers; rD, relaxed dimers; rM, relaxed monomers; scD, supercoiled dimers; scM, supercoiled monomers. Quantification of dimers (rD and scD) related to total plasmid DNA is shown. For other details, see Fig 1. DSB, double-strand break; SCE, sister-chromatid exchange.
Discussion
We provide an in vivo functional assay for the specific production of single-stranded breaks that are converted into DSBs after replication and for the analysis of their repair by SCE. Our 21-bp HO assay provides a useful experimental system for mimicking a natural scenario for the origin of DSBs in dividing cells, in which case DSBs appear after DNA replication failure (Michel et al, 2001; Branzei & Foiani, 2005). These replication-born DSBs are efficiently repaired by SCE. Interestingly, such repair by SCE strongly depends on cohesins, which do not have a role in other types of HR.
One relevant feature of the 21-bp HO site is that the two ends of the DSB are detected, whereas replication run-off at a nick could theoretically result in a one-ended DSB. This is similar to the results obtained with an Escherichia coli system in which the single-strand DNA cleavage site of the M13 phage DNA was inserted in the λ-phage chromosome (Kuzminov, 2001). Formation of double-ended DSBs after replication through a nick could be explained in two ways, depending on whether the nick is encountered on the lagging or on the leading strand (Fig 6). In the first case (Fig 6, left), the replication fork could traverse the nick leaving a DSB behind the DNA polymerase. In the second case (Fig 6, right), a replication fork coming from the other side would lead to a double-ended DSB.
Figure 6.

Model to explain the role of cohesins in the repair of double-strand breaks formed after replication forks encounter single-strand DNA breaks. Double-ended DSBs could be formed after replication through a nick in two ways depending on whether the nick is encountered at the lagging (left) or the leading strand (right). Cohesins (thin grey ovals), which are additionally loaded in the proximity of the DSB, favour HR with the sister. DSB, double-strand break; HR, homologous recombination.
It has been previously shown that induction of a nick in S. cerevisiae by the Gene II protein from bacteriophage f1 stimulates HR (Strathern et al, 1991). The fact that this increase was observed only in cycling cells (Galli & Schiestl, 1998), together with the pattern of HR products, was consistent with the events initiated by DSBs. Nevertheless, neither formation of DSBs nor any repair intermediate has been analysed at the molecular level in this system. Nicks can also be produced by the DNA topoisomerase I (Topo I) inhibitor camptothecin. In this case, Topo I remains bound to the DNA and could become a physical obstacle for the DNA polymerase. As a consequence, replication through such nicks leads to one-ended DSBs (Avemann et al, 1988; Strumberg et al, 2000; Saleh-Gohari et al, 2005). It is less likely that these one-ended DSBs are converted into two-ended DSBs by a fork coming from the opposite direction, because it is likely to encounter another Topo I cleavage complex in its way.
As cohesins hold chromatids after replication, their involvement in DNA repair, but not in ectopic HR, has led to the idea that their role in DSB repair would be to attach a broken chromatid to its sister to favour repair (Sjogren & Nasmyth, 2001; Sonoda et al, 2001; Kim et al, 2002; Strom et al, 2004; Unal et al, 2004). Molecular evidence that cohesins participate in DSB repair comes from the observation that they are deposited onto DNA in response to a DSB, also in an Scc2-dependent manner (Strom et al, 2004; Unal et al, 2004). Our system has allowed us to demonstrate that, in a cohesin-subunit mutant (smc3-42), repair of replication-generated DSBs by SCE is impaired. In agreement with previous reports (Unal et al, 2004), no defect in general HR is observed. The result was the same for the other cohesin-subunit mutant tested, scc1-73, and, interestingly, for the scc2-4 mutant affected in the Scc2 activity of cohesin loading. So far, physical detection of SCE in linear chromosomes is not possible, but the fact that cohesins also act in circular plasmids (Ivanov & Nasmyth, 2005) and that HR mechanisms do not depend on the linear or circular nature of the DNA molecule suggest that our results may be valid for chromosomes. The fact that scc2 reduces SCE as mutations of the structural subunits of cohesins is consistent with this defect being related to deficient cohesion. Our results indicate that cohesion is required to assist postreplicative repair of DNA breaks by holding sister chromatids together, ensuring the choice of the sister chromatid as HR partner, rather than having an enzymatic role in the HR reaction. When cohesion is defective, other homologous DNA substrates, including the allelic counterpart of the homologous chromosome or ectopic repeats, can become preferential repair templates, thus affecting genomic stability. Indeed, we show that cohesins are required for equal SCE, which is in agreement with the recent work of Kobayashi et al (2004) at the ribosomal DNA locus, who proposed that cohesins favour equal SCE events induced by the natural replication-fork barrier in the rDNA region.
In addition to cohesins, two other SMC complexes exist in the cell: condensins (Smc2–Smc4), with a main role in chromosome condensation, and the Smc5–Smc6 complex, the function of which is not well defined (Losada & Hirano, 2005). Both SMC complexes seem to affect DNA repair (Lehmann, 2005). In addition, mutants of Smc5–Smc6 are impaired in rDNA segregation (Torres-Rosell et al, 2005), and are affected in methyl methanesulphonate-induced HR (Onoda et al, 2004). It would be interesting to determine whether, as cohesins, these SMC complexes function specifically in SCE.
In summary, we provide a novel molecular assay for the analysis of repair of DSBs, which are specifically generated by replication through a site-specific single-strand break—the most natural scenario for the formation of DSBs—and we demonstrate that cohesion is required specifically for SCE and not for other forms of HR that use other partners. Altogether, this study opens new perspectives for the analysis and understanding of postreplicative DSB recombinational repair.
Methods
Strains, plasmids and oligonucleotides. The wild-type WS strain (MATa-inc ade2-1 his3-11,15 trp1-1 ura3-1 ade3_∷_GAL-HO leu2_∷_SFA1) was previously described (Gonzalez-Barrera et al, 2003), and the isogenic _bar1_∷ HphMX4 mutant was obtained by gene replacement after PCR transformation. Deletion was confirmed by PCR and Southern blot analysis. The smc3-42, kindly supplied by K. Nasmyth, and scc1-73 and scc2-4, kindly supplied by C. Sjögren, were introduced in the WS genetic background by genetic cross. Centromeric monocopy plasmids pRS316-TINV and pCM189-leu2HOr were described previously (Gonzalez-Barrera et al, 2003). The 25-mer oligonucleotides used to insert the 21-bp HO site into the _LEU2 Eco_RI site were AATTTCAGCTTTCCGCAACAGTATA (HO.1) and AATTTATACTGTTGCGGAAAGCTGA (HO.2).
Cell-cycle synchronization experiments. WS-bar1 (_bar1_Δ) cells, used as wild-type controls to enhance the effect of α-factor, were grown at 30°C in SC-ura with 2% raffinose as carbon source to an optical density of 0.4. The culture was divided into two halves, one of which was supplemented with 0.2 μM α-factor to arrest cells at G1. After 4 h, 2% galactose was added to induce HO expression. For α-factor release, cells were washed twice with pre-warmed fresh medium without α-factor, and 50 μg/ml Pronase (Sigma, St Louis, MO, USA) was added to remove any trace of α-factor. HO cleavage was recurrent in the experimental conditions used, as cells were maintained in 2% galactose after α-factor release. If 2% glucose was added, nicks were quickly repaired and DSBs did not form (data not shown).
Physical analysis of DNA breaks and HR intermediates. It was performed as described previously (González-Barrera et al, 2003) with modifications. For the analysis of nicks, DNA was electrophoresed in alkaline (50 mM NaOH, 1 mM EDTA) conditions. For analysis of strand specificity of nicks, strand-specific probes were obtained by primer extension of a LEU2 fragment using GTTCCACTTCCAGATGAGGC (Crick) and TAACGGAGGCTTCATCGGAG (Watson) oligonucleotides. In all cases, the average and standard deviation of 2–4 experiments are shown.
Acknowledgments
We thank K. Nasmyth and C. Sjögren for the gift of strains, F. Prado and R. Wellinger for reading the manuscript and D. Haun for style supervision. This work was supported by grants from the Ministry of Science of Education of Spain (SAF2003-00204) and Junta de Andalucía (CVI102). F.C.-L. was the recipient of a predoctoral training grant from the Spanish Ministry of Health.
References
- Avemann K, Knippers R, Koller T, Sogo JM (1988) Camptothecin, a specific inhibitor of type I DNA topoisomerase, induces DNA breakage at replication forks. Mol Cell Biol 8: 3026–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M (2005) The DNA damage response during DNA replication. Curr Opin Cell Biol 17: 568–575 [DOI] [PubMed] [Google Scholar]
- Galli A, Schiestl RH (1998) Effects of DNA double-strand and single-strand breaks on intrachromosomal recombination events in cell-cycle-arrested yeast cells. Genetics 149: 1235–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Barrera S, Cortes-Ledesma F, Wellinger RE, Aguilera A (2003) Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol Cell 11: 1661–1671 [DOI] [PubMed] [Google Scholar]
- Ivanov D, Nasmyth K (2005) A topological interaction between cohesin rings and a circular minichromosome. Cell 122: 849–860 [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19: 3398–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyk LC, Hartwell LH (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132: 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Krasieva TB, LaMorte V, Taylor AM, Yokomori K (2002) Specific recruitment of human cohesin to laser-induced DNA damage. J Biol Chem 277: 45149–45153 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Horiuchi T, Tongaonkar P, Vu L, Nomura M (2004) SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117: 441–453 [DOI] [PubMed] [Google Scholar]
- Kuzminov A (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol Mol Biol Rev 63: 751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A (2001) Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci USA 98: 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR (2005) The role of SMC proteins in the responses to DNA damage. DNA Repair (Amst) 4: 309–314 [DOI] [PubMed] [Google Scholar]
- Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362: 709–715 [DOI] [PubMed] [Google Scholar]
- Losada A, Hirano T (2005) Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev 19: 1269–1287 [DOI] [PubMed] [Google Scholar]
- Michel B, Flores MJ, Viguera E, Grompone G, Seigneur M, Bidnenko V (2001) Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci USA 98: 8181–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K (2002) Segregating sister genomes: the molecular biology of chromosome separation. Science 297: 559–565 [DOI] [PubMed] [Google Scholar]
- Onoda F, Takeda M, Seki M, Maeda D, Tajima J, Ui A, Yagi H, Enomoto T (2004) SMC6 is required for MMS-induced interchromosomal and sister chromatid recombinations in Saccharomyces cerevisiae. DNA Repair (Amst) 3: 429–439 [DOI] [PubMed] [Google Scholar]
- Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T (2005) Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol 25: 7158–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren C, Nasmyth K (2001) Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol 11: 991–995 [DOI] [PubMed] [Google Scholar]
- Sonoda E et al. (2001) Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev Cell 1: 759–770 [DOI] [PubMed] [Google Scholar]
- Strathern JN, Weinstock KG, Higgins DR, McGill CB (1991) A novel recombinator in yeast based on gene II protein from bacteriophage f1. Genetics 127: 61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom L, Lindroos HB, Shirahige K, Sjogren C (2004) Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell 16: 1003–1015 [DOI] [PubMed] [Google Scholar]
- Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y (2000) Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol 20: 3977–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rosell J, Machin F, Farmer S, Jarmuz A, Eydmann T, Dalgaard JZ, Aragon L (2005) SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat Cell Biol 7: 412–419 [DOI] [PubMed] [Google Scholar]
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D (2004) DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell 16: 991–1002 [DOI] [PubMed] [Google Scholar]