Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations (original) (raw)
Abstract
Activation-induced cytidine deaminase (AID) is required for the maturation of antibodies in higher vertebrates, where it promotes somatic hypermutation (SHM), class switch recombination and gene conversion. While it is known that SHM requires high levels of transcription of the target genes, it is unclear whether this is because AID targets transcribed genes. We show here that the human AID promotes C to T mutations in Escherichia coli which are stimulated by transcription. The mutations are strand-biased and occur preferentially in the non-transcribed strand of the target gene. Human AID purified from E.coli is active without prior treatment with a ribonuclease and deaminates cytosines in plasmid DNA in vitro. Further, the action of this enzyme is greatly stimulated by the transcription of the target gene in a strand-dependent fashion. These results confirm the prediction that AID may act directly on DNA and show that it can act on transcribing DNA in the absence of specialized DNA structures such as R-loops. It suggests that AID may be recruited to variable genes through transcription without the assistance of other proteins and that the strand bias in SHM may be caused by the preference of AID for the non-transcribed strand.
INTRODUCTION
Activation-induced cytidine deaminase (AID) was first described as a protein that is required for somatic hypermutations (SHM) and class switch recombination (CSR) in B lymphocytes (1). Subsequently, it was also found to be required for the third mutational process involved in antibody maturation, gene conversion (2,3). AID is a homolog of a known RNA-cytosine deaminase, Apobec-1 (1), and the human AID gene causes C to T mutations in Escherichia coli (4). AID protein purified from insect cells and pre-treated with a ribonuclease was recently shown to deaminate cytosines to uracil in DNA (5), providing an explanation for its mutator phenotype.
It is not clear how AID causes base substitutions other than C to T that are observed during SHM, or how it creates the double-strand breaks that are thought to be necessary for CSR (6). Additionally, it is known that high transcription is a requirement for SHM in B cells as well in non-B cells (7–10) and that there is a strand bias in mutations introduced during SHM (11). It is unclear whether these properties of antibody maturation can be explained by biochemical properties of AID. We show here that the properties of human AID expressed in E.coli, and of the protein purified from E.coli are consistent with the dependence of antibody maturation on transcription and that its mutagenic action is strand-biased.
MATERIALS AND METHODS
Escherichia coli strains and plasmids
Escherichia coli strains BH156 (relevant genotype ung-1) and BH158 (=BH156 mug::Tn10) have been described previously (12). BH214 is BH158 containing DE3 prophage, which contains the T7 RNA polymerase gene under the control of the lac promoter and was kindly provided by W. Franklin (Albert Einstein College of Medicine, New York, NY). BL21 (F′, _omp_T _hsd_S _ga_l) containing DE3 is from our collection.
Plasmid pUP21 was described previously (13), but was referred to as pUP21-op75. PΔUP21 was constructed from pUP21 by deleting an EcoRI–EcoO109I restriction fragment containing the UP-tac promoter from pUP21. To create pUP27, pUP21 was modified to introduce an XbaI site immediately upstream from the Pkan and an NheI site downstream from the kan-ble cassette. The XbaI–NheI fragment in the resulting plasmid, pUP25, was inverted to create pUP27. The plasmids pAB7 and pAB8 were constructed by cloning a restriction fragment containing Pkan and kanS-94D elements into pET11d (Novagen Corp.). The orientation of the kanS-94D allele is the opposite with respect to the PT7 promoter in the two plasmids.
Purification of human AID
Human AID cDNA (I.M.A.G.E. Consortium ID 4054915) was obtained from the American Type Culture Collection (Rockville, MD) and the gene was amplified using the primers 5′-CTCTGGACGAATTCCATGGACAGCCTCTTC-3′ and 5′-CCTGGAAGCTCGAGTCAAAGTCCCAAAGTA-3′. It was cloned into pGEX4T3 (Amersham Biosciences) as an EcoRI–XhoI fragment.
The plasmid clone containing GST-AID fusion, pGhAID1, was introduced into BL21 DE3 and the transcription of the fusion gene was induced by adding IPTG to the growth media to 0.2 mM. Cells were grown at 30°C for 4 h and harvested. The cell pellet was resuspended in 20 ml of 1× PBS and sonicated for 10 pulses of 20 s duration separated by 1 min on ice. The fusion protein was purified through a glutathione– Sepharose column according to the recommendations of the manufacturer (Amersham Biosciences) and the protein from the fractions was separated on 12% SDS–polyacrylamide gel to identifiy those containing GST-AID. The appropriate fractions were pooled, the protein was concentrated using a YM30 centricon filter (Amicon) and equilibrated with the storage buffer (25 mM Tris–HCl pH 7.5, 0.1 mM EDTA, 1.0 mM DTT and 10% glycerol).
Kanamycin-resistance reversion assays
The genetic assays for reversion to kanamycin-resistance (KanR) were performed as described previously (13), with the modification that two plasmids were maintained in the cells; one plasmid being pSU24 or pSU-AID and the other being pΔUP21, pUP21 or pUP27. Briefly, one culture was grown to early log phase and then diluted 1000-fold to create several independent cultures. IPTG was added to 0.5 mM in half of the cultures and all the cultures were grown to mid-log phase before plating. Plates containing carbenicillin were used to determine the total number of cells in each culture and plates with 50 µg/ml kanamycin were used to score the revertants.
To treat plasmid DNA in vitro, pAB7 or pAB8 DNA was included at 2 nM in transcription buffer [40 mM Tris–HCl, pH 7.9, 10 mM MgCl2, 10 mM NaCl, 2 mM spermidine, 100 mM potassium glutamate and 10 mM dithiothreitol (DTT)] containing 8 U of Topoisomerase II (Amersham Biosciences) and 87.5 nM T7 RNA polymerase in a 100 µl reaction. When nucleotide triphosphates were included in the reaction, they were at 3.75 mM each. GST-AID (1.2 µg) and 2 U of E.coli uracil-DNA glycosylase (UDG) (New England Biolabs) were included as indicated in the legend to Figure 3. The reactions were performed at 37°C for 4 h and terminated by the addition of EDTA to 10 mM and RNase A (Boehringer Mannheim). Following another 15 min incubation, the DNA was deproteinized and introduced into BH156 through electroporation, and colonies were scored for resistance to carbenicillin (CarbR) and kanamycin.
Figure 3.

The effect of transcription on cytosine deaminations caused by AID in vitro. Plasmid pAB7 (A) or pAB8 (B) DNA was transcribed with T7 RNA polymerase as described in the Materials and Methods. The revertant frequency is (total no. of KanR colonies/total no. of CarbR colonies).
Treatment of a bubble DNA duplex with AID
200 fmol of 32P end-labeled DNA was incubated with 2 µg GST-AID in a 10 µl reaction containing 50 or 20 mM Tris–HCl, pH 7.5 at 37°C for 45 min. Where indicated, EDTA or 1,10-phenanthroline were respectively added to 2 mM or 5 mM final concentration. The DNA was subsequently incubated with 1 U of E.coli UDG (New England Biolabs) for an additional 45 min and the reactions were terminated by the addition of NaOH to 0.1 M, followed by heating to 95°C for 7 min. The products of the reactions were separated on a 20% sequencing gel. The gel was scanned using a phosphorimager and the intensities of the bands were quantified.
RESULTS
Human AID is a DNA cytosine deaminase
Human AID was expressed in E.coli as a fusion with GST and purified over an affinity column. A 56 bp DNA duplex containing a 5 nt bubble (Fig. 1A) was sequentially treated with AID, UDG and NaOH. If AID deaminates cytosines in this DNA to uracil, UDG would excise these uracils, and cleavage at the resulting abasic sites by NaOH would create shorter DNA fragments. Treatment of this substrate with AID resulted in the conversion of 17% of labeled DNA into a single, shorter oligonucleotide (Fig. 1B, lane 3). This oligomer is 27 nt in length and its length is consistent with the conversion of one of the two cytosines in the bubble to uracil (Fig. 1B). Remarkably, under the reaction conditions used, the other cytosine in the bubble and the 12 cytosines in the rest of the DNA strand appeared untouched by AID.
Figure 1.

Deamination of cytosines in a DNA bubble by AID. (A) The sequence of the duplex used as substrate for the AID reaction is shown. The bottom strand is labeled at the 5′ end with 32P. The cytosine that is the target of AID is indicated by a solid arrow. (B and C) 32P-labeled substrate was incubated with the reagents indicated above each lane and then heated in the presence of NaOH. The buffer contained 50 mM Tris–HCl (B) or 20 mM Tris–HCl (C). Φ is 1,10-phenanthroline.
However, the other cytosine in the bubble and some of the other cytosines in the duplex become susceptible to AID under different reaction conditions (Fig. 1C) or if the bottom DNA strand is used as a substrate without its sequence complement (not shown). We also found that AID action is inhibited by 1,10-phenanthroline, a strong chelator, but not by the weaker chelator EDTA (Fig. 1B, lanes 4 and 5). These results show that AID has a strong preference for unpaired cytosines and requires a tightly bound metal ion for its action.
Cytosine deamination by AID in E.coli is transcription-dependent
We used an E.coli plasmid-based genetic reversion assay to study the transcription-dependence of AID mutagenesis. An allele of the kan gene, kanS-94D, reverts only through C to T changes in cells lacking UDG (genotype ung), conferring KanR phenotype upon its host (14). If AID deaminates cytosines in DNA to uracil, there would be an increase in KanR revertants and would be scored as such. In one of the plasmids used, pΔUP21, kan is transcribed from a weak constitutive promoter (Pkan), while in the other plasmids kan is either transcribed from an inducible UP-tac promoter or a combination of UP-tac and Pkan (15) (Fig. 2A). Previous studies have shown that transcription from an induced UP-tac promoter dominates over transcription from Pkan (15,16); thus different DNA strands of the gene are predominantly transcribed in pUP21 and pUP27 when IPTG is present in the growth media.
Figure 2.
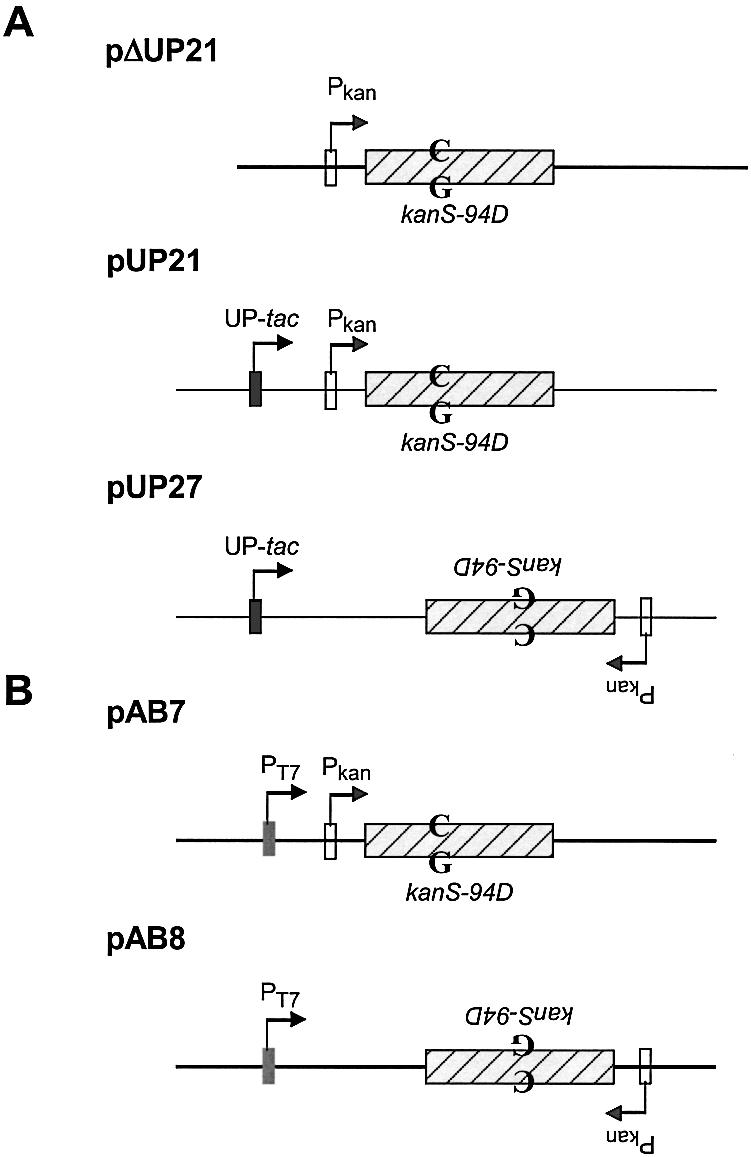
Structures of plasmids. (A) The relevant genetic elements in the three plasmids are shown. All the plasmids confer resistance to carbenicillin and contain the lacI Q gene (not shown). The product of this gene represses transcription from the promoter UP-tac, but not Pkan. The position of the C·G pair in kanS-94D that is the target for AID action is shown. To emphasize that kanS-94D is inverted in pUP27 compared to pUP21, the relevant text has been inverted. (B) pAB7 and pAB8 are respectively similar to pUP21 and pUP27 except the UP-tac promoter in the latter plasmids is replaced with a T7 RNA polymerase promoter.
The human AID gene was cloned into pSU24 (17) (pSU-AID) under the control of an inducible T7 promoter and introduced into E.coli. In cells containing pΔUP21, AID expression results in increased KanR revertants in the cultures. The Lea–Coulson method of the median (18) was used to determine the number of revertants per culture, m, and AID expression increased m by ∼12-fold compared to the uninduced cultures (310 compared to 25; Table 1). In control cultures lacking AID, there was little increase in m upon addition of IPTG to the growth media (Table 1).
Table 1. KanR revertants promoted by AID in vivo.
| | m (M)a | | | | | | | | ----------- | -------- | ---------- | --------- | ------------- | ------- | ---------- | | | pΔUP21 | pΔUP21 | pUP21 | pUP21 | pUP27 | pUP27 | | | | pSU24 | pSU-AID | pSU24 | pSU-AID | pSU24 | pSU-AID | | | No IPTG | 32 (123) | 25 (119) | 41 (167) | 91 (372) | 25 (96) | 21 (84) | | With IPTG | 55 (168) | 310 (1683) | 158 (535) | 4545 (30 455) | 30 (79) | 289 (1467) |
Similar experiments were also performed using cells containing pUP21. When transcription was induced from the strong promoter in front of kan in cells lacking AID, m increased ∼4-fold (Table 1). This increase in mutations is due to spontaneous cytosine deaminations in the non- transcribed strand during transcription from UP-tac, and has been described previously (16,19). When parallel experiments were performed using cells containing pUP21 and pSU-AID, induction of transcription increased m by ∼50-fold (Table 1). The number of reversions per culture obtained when transcription of both AID and kan genes was induced (4545) is in vast excess of the sum of revertants obtained as a result of overexpression of AID in E.coli (310) and transcription of kan (158). Thus, the extent of transcription of the target gene directly correlates with increased AID mutagenesis.
Further, this increase in mutations was dependent on which of the two DNA strands was being copied by the RNA polymerase. When the experiments were repeated using pUP27 instead of pUP21 (Fig. 2A), the increase in mutations caused by AID was modest (m = 289) and was comparable to the increase observed for kan in pΔUP21 (310; Table 1). The target cytosine is in opposite strands with respect to UP-tac in the two plasmids and m is ∼16-fold lower with pUP27 compared to pUP21 (Table 1). These data suggest that AID acts preferentially on cytosines in the non-transcribed strand of kan.
Sixty-two independent KanR revertants from different experiments were sequenced and all contained a C to T change in codon 94 of kan (not shown). Our observations are consistent with the spectra of rifampicin-resistance mutations promoted by AID in E.coli (4). AID increased C to T mutations in the non-transcribed strand of rpoB from 30 to 63%, while G to A transitions in this strand increased from 1.4 to only 16% (4).
In vitro cytosine deaminations by AID are also transcription-dependent
Plasmid pAB7 containing kanS-94D (Fig. 2B) was incubated with GST-AID under a variety of conditions and the DNA was introduced into ung E.coli to score KanR revertants. Incubation of the plasmid DNA with GST-AID increased the frequency of KanR revertants and the magnitude of this increase was dependent on the reaction conditions. While the increase was ∼5-fold in the absence of transcription, it was ∼1600-fold when the gene was actively transcribed by T7 RNA polymerase (Fig. 3A). The increase in KanR frequency was almost completely eliminated when E.coli UDG was included in the reaction suggesting that AID had converted the target cytosine to uracil. The residual increase in KanR frequency by AID in the presence of UDG is most likely due to some uracils escaping repair. If non-transcribing plasmid DNA is treated with GST-AID in the presence of UDG, no increase in revertants is oberved compared to the control reaction (not shown).
Treatment of pAB8 with GST-AID also increased KanR frequency, but this effect was not strongly dependent on transcription (Fig. 3B). In contrast to pAB7, transcription caused only a slight increase in KanR revertants; the maximum revertant frequency obtained with pAB8 was ∼50-fold lower than what was observed with pAB7 (Fig. 3). Total RNA was quantified from representative reactions and all the reactions were found to contain comparable amounts of RNA (not shown). Consequently, the differences in the mutagenicity of AID for pAB7 and pAB8 are not due to poor transcription of the latter plasmid and must be related to the transcriptional orientation of kan within these plasmids.
DISCUSSION
We have shown that the human AID protein is an active DNA cytosine deaminase both in vivo and in vitro, and AID mutageneis is stimulated by transcription. Further, the protein targets cytosines in the non-transcribed strand. After this work was completed, similar results were reported from other laboratories (5,20,21). Additionally, purified Apobec-1 was shown to deaminate cytosines in single-stranded DNA in vitro (22). While the results presented here are generally consistent with the results of those studies, they clarify the biochemical activity and substrate specificity of AID in important ways.
It was reported that GST-AID purified from an insect expression system was inactive unless treated with a ribonuclease (5). The sequence of the RNA bound to GST-AID is unknown, but it was speculated that it may have a regulatory role in AID action (5). In contrast to those results, we did not find it necessary to include a ribonuclease in AID reactions to detect its biochemical activity. To assess whether a significant fraction of GST-AID purified from E.coli was bound to RNA, the strand cleavage assay was repeated with the inclusion of ribonuclease A in the reaction. Ribonuclease treatment did not improve the activity of AID (Fig. 1C) and may have decreased it slightly. These data show that unlike the GST-AID purified from insect cells, the protein purified from E.coli does not contain RNA and is active. Further, if AID does bind RNA (5), it is likely to bind specific RNA sequences. It should also be noted that Chaudhari et al. (20) did not treat the partially purified AID from B-cells with ribonucleases to demonstrate its cytosine deaminase activity. Consequently, whether AID binds RNA and whether such binding has biological significance remains unclear.
It has been argued that the segment of DNA where CSR occurs (S region) may contain RNA–DNA hybrids (R-loops), and that such structures are essential for recombination (23–25). Further, Chaudhuri et al. (20) showed that partially purified AID targets the unpaired DNA strand when such R-loops are formed as a result of transcription in vitro. We have shown here that R-loop formation is not essential for AID action (Fig. 3). Normal transcription of DNA, which presumably creates transient transcription bubbles, may be all that is necessary for AID action.
Finally, Bransteitter et al. (5) have argued that the propensity by AID to cause mutations within WRCY sequences (W = A or T; R = purine and Y = pyrimidine) is the result of preferential targeting of AID at these sites. The data presented here show that such preferences may depend on specific reaction conditions (compare Fig. 1B with 1C). Only one of the cytosines in the DNA bubble is within a WRCY sequence (product 27 nt), but both cytosines are converted to uracil at about the same frequency under one reaction condition (Fig. 1C). Hence, conclusions regarding the specificity of AID must await a more thorough biochemical analysis than has been presented so far.
It has been argued that the dependence of SHM on the transcription of immunoglobulin genes (8,26) is the result of the action of a mutator protein that associates with the transcription elongation complex (9). The results presented here suggest that AID is likely to be this mutator. It is also unknown whether the targeting of the transcribing immunoglobulin genes by this mutator requires additional proteins (26,27). Our observation that AID targets a highly transcribed gene in vitro and in E.coli suggests that the targeting of AID may not require additional mammalian proteins. Once AID is brought to the region of the chromatin containing immunoglobulin genes, it may find the actively transcribed variable gene and help mutate it. The strand bias in SHM has variously been interpreted as evidence for transcription-dependent damage to DNA (28,29), or the involvement of transcription-coupled repair in eliminating DNA damage (9). While it is clear that downstream events may process uracils generated by AID and influence the mutation spectrum of SHM, it is tempting to suggest that the strand selectivity of AID with respect to transcription is the source of the mutational strand bias.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank T. Roy and J. McLellan for assistance with initial experiments and P. Gearhart (National Institute of Aging) for useful comments on the manuscript.
REFERENCES
- 1.Muramatsu M., Kinoshita,K., Fagarasan,S., Yamada,S., Shinkai,Y. and Honjo,T. (2000) Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell, 102, 553–563. [DOI] [PubMed] [Google Scholar]
- 2.Arakawa H., Hauschild,J. and Buerstedde,J.M. (2002) Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science, 295, 1301–1306. [DOI] [PubMed] [Google Scholar]
- 3.Harris R.S., Sale,J.E., Petersen-Mahrt,S.K. and Neuberger,M.S. (2002) AID is essential for immunoglobulin V gene conversion in a cultured B cell line. Curr. Biol., 12, 435–438. [DOI] [PubMed] [Google Scholar]
- 4.Petersen-Mahrt S.K., Harris,R.S. and Neuberger,M.S. (2002) AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature, 418, 99–103. [DOI] [PubMed] [Google Scholar]
- 5.Bransteitter R., Pham,P., Scharff,M.D. and Goodman,M.F. (2003) Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl Acad. Sci. USA, 21, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gearhart P.J. (2002) Immunology: the roots of antibody diversity. Nature, 419, 29–31. [DOI] [PubMed] [Google Scholar]
- 7.Bachl J., Carlson,C., Gray-Schopfer,V., Dessing,M. and Olsson,C. (2001) Increased transcription levels induce higher mutation rates in a hypermutating cell line. J. Immunol., 166, 5051–5057. [DOI] [PubMed] [Google Scholar]
- 8.Fukita Y., Jacobs,H. and Rajewsky,K. (1998) Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity, 9, 105–114. [DOI] [PubMed] [Google Scholar]
- 9.Storb U. (1996) The molecular basis of somatic hypermutation of immunoglobulin genes. Curr. Opin. Immunol., 8, 206–214. [DOI] [PubMed] [Google Scholar]
- 10.Yoshikawa K., Okazaki,I.M., Eto,T., Kinoshita,K., Muramatsu,M., Nagaoka,H. and Honjo,T. (2002) AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science, 296, 2033–2036. [DOI] [PubMed] [Google Scholar]
- 11.Wagner S.D. and Neuberger,M.S. (1996) Somatic hypermutation of immunoglobulin genes. Annu. Rev. Immunol., 14, 441–457. [DOI] [PubMed] [Google Scholar]
- 12.Lutsenko E. and Bhagwat,A.S. (1999) The role of the Escherichia coli Mug protein in the removal of uracil and 3,N(4)-ethenocytosine from DNA. J. Biol. Chem., 274, 31034–31038. [DOI] [PubMed] [Google Scholar]
- 13.Klapacz J. and Bhagwat,A.S. (2002) Transcription-dependent increase in multiple classes of base substitution mutations in Escherichia coli. J. Bacteriol., 184, 6866–6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wyszynski M., Gabbara,S. and Bhagwat,A.S. (1994) Cytosine deaminations catalyzed by DNA cytosine methyltransferases are unlikely to be the major cause of mutational hot-spots at sites of cytosine methylation in E. coli. Proc. Natl Acad. Sci. USA, 91, 1574–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beletskii A. and Bhagwat,A.S. (1998) Correlation between transcription and C to T mutations in the non-transcribed DNA strand. Biol. Chem., 379, 549–551. [PubMed] [Google Scholar]
- 16.Beletskii A. and Bhagwat,A.S. (2001) Transcription-induced cytosine-to-thymine mutations are not dependent on sequence context of the target cytosine. J. Bacteriol., 183, 6491–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartolome B., Jubete,Y., Martinez,E. and de la Cruz,F. (1991) Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene, 102, 75–78. [DOI] [PubMed] [Google Scholar]
- 18.Rosche W.A. and Foster,P.L. (2000) Determining mutation rates in bacterial populations. Methods, 20, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beletskii A. and Bhagwat,A.S. (1996) Transcription-induced mutations: increase in C to T mutations in the non-transcribed strand during transcription in Escherichia coli. Proc. Natl Acad. Sci. USA, 93, 13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhuri J., Tian,M., Khuong,C., Chua,K., Pinaud,E. and Alt,F.W. (2003) Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature, 422, 726–730. [DOI] [PubMed] [Google Scholar]
- 21.Ramiro A.R., Stavropoulos,P., Jankovic,M. and Nussenzweig,M.C. (2003) Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nature Immunol., 4, 452–456. [DOI] [PubMed] [Google Scholar]
- 22.Petersen-Mahrt S.K. and Neuberger,M.S. (2003) In vitro deamination of cytosine to uracil in single-stranded DNA by APOBEC1. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 23.Daniels G.A. and Lieber,M.R. (1995) RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucleic Acids Res., 23, 5006–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tracy R.B. and Lieber,M.R. (2000) Transcription-dependent R-loop formation at mammalian class switch sequences. EMBO J., 19, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Yu K., Chedin,F., Hsieh,C.L., Wilson,T.E. and Lieber,M.R. (2003) R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunol., 4, 442–451. [DOI] [PubMed] [Google Scholar]
- 26.Betz A.G., Milstein,C., Gonzalez-Fernandez,A., Pannell,R., Larson,T. and Neuberger,M.S. (1994) Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell, 77, 239–248. [DOI] [PubMed] [Google Scholar]
- 27.Peters A. and Storb,U. (1996) Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity, 4, 57–65. [DOI] [PubMed] [Google Scholar]
- 28.Neuberger M.S., Ehrenstein,M.R., Klix,N., Jolly,C.J., Yelamos,J., Rada,C. and Milstein,C. (1998) Monitoring and interpreting the intrinsic features of somatic hypermutation. Immunol. Rev., 162, 107–116. [DOI] [PubMed] [Google Scholar]
- 29.Storb U. (1998) Progress in understanding the mechanism and consequences of somatic hypermutation. Immunol. Rev., 162, 5–11. [DOI] [PubMed] [Google Scholar]