Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division (original) (raw)
Abstract
Cells expressing indoleamine 2,3-dioxygenase (IDO), an enzyme which catabolizes tryptophan, prevent T-cell proliferation in vitro, suppress maternal antifetal immunity during pregnancy and inhibit T-cell-mediated responses to tumour-associated antigens. To examine the mechanistic basis of these phenomena we activated naïve murine T cells in chemically defined tryptophan-free media. Under these conditions T cells expressed CD25 and CD69 and progressed through the first 12 hr of G0/G1 phase but did not express CD71, cyclin D3, cdk4, begin DNA synthesis, or differentiate into cytotoxic effector cells. In addition, activated T cells with their growth arrested by tryptophan deprivation exhibited enhanced tendencies to die via apoptosis when exposed to anti-Fas antibodies. Apoptosis was inhibited by caspase inhibitor and was not observed when T cells originated from Fas-deficient mice. These findings suggest that T cells activated in the absence of free tryptophan entered the cell cycle but cell cycle progression ceased in mid-G1 phase and T cells became susceptible to death via apoptosis, in part though Fas-mediated signalling. Thus, mature antigen-presenting cells expressing IDO and Fas-ligand may induce antigen-specific T-cell tolerance by blocking T-cell cycle progression and by rapid induction of T-cell activation induced cell death in local tissue microenvironments.
Introduction
T-cell responses are regulated to optimize immunity against infectious pathogens and to minimize the risk of autoimmunity by tolerizing self-reactive T cells. T-cell repertoires are tailored to individual sets of tissue antigens displayed by thymic antigen-presenting cells (APCs), which eliminate self-reactive thymocytes via negative selection. Nevertheless, self-reactive T cells emerge into the peripheral T-cell repertoire and immunoregulatory processes prevent autoimmunity by eliminating self-reactive T cells or suppressing T-cell responses.1,2 Immunosuppressive processes that ensure T-cell tolerance in vivo depend on the integrity of tissue microenvironments. Disruption of tissue microenvironments releases signals and activates processes that rapidly alter the functional and phenotypic characteristics of cells, complicating attempts to define the tolerogenic processes that operate at the cellular and molecular level in vivo. Nevertheless, considerable evidence suggests that cells of myeloid or lymphoid origin are potent regulators of T-cell responsiveness in vivo both by promoting proliferation and differentiation of effector T cells and by inducing tolerance through activation-induced cell death (AICD).3–6 Myeloid dendritic cells are antigen-processing and -presenting cells and are potent activators of T cells. However, under certain conditions human myeloid precursor cells can be differentiated in vitro into cells that suppress T-cell responses7,8 and in mice lymphoid dendritic cells suppress T-cell responses.3,6
Several mechanisms contribute to the induction and maintenance of peripheral T-cell tolerance including Fas/Fas-ligand-mediated cell death and specific patterns of cytokine production, which influence the functional outcome of encounters between T cells and APCs.3,5,6,9 Multiple studies show that immunological outcomes following T-cell/APC encounters differ according to the dominant functional characteristics acquired by activated T cells, in particular the nature of effector functions elaborated during the activation process, such as the T helper type 1 (Th1) and type 2 (Th2) T-cell functions.10 The identity and functional status of APCs is likely to be a critical factor influencing the outcome of T-cell/APC encounters.
Depletion of essential nutrients from tissue microenvironments where T cells activate may also contribute to tolerance induction by limiting the proliferative potential of T cells at critical periods during activation. Indeed, cultured human macrophages expressing the tryptophan-degrading enzyme indoleamine 2,3 dioxygenase (IDO) completely prevent T-cell proliferation in vitro.7 Similar results were reported for cultured human dendritic cells, which can be induced to express IDO when stimulated to mature using lipopolysaccharide (LPS) and other stimuli.8 These findings suggest that cells expressing IDO in vivo may act as tolerizing APCs that limit access to tryptophan during T-cell activation. We tested this hypothesis experimentally by showing that tryptophan catabolism is essential during allogeneic murine pregnancies to protect the developing fetuses from the lethal maternal T-cell immunity provoked by fetal alloantigens.11,12 In the current study we examine the phenotypic and functional consequences of activating murine T cells in chemically defined media containing no tryptophan. In the absence of tryptophan, activated T cells enter G1 phase but fail to enter S phase and become highly susceptible to Fas-ligand-mediated cell death. Furthermore, we show that expression of IDO could be induced and Fas-ligand was expressed in a CD11c+ subset isolated from healthy mouse spleen tissues.
Materials and methods
Mice
CBA mice and four lines of transgenic mice prepared on the CBA genetic background, BM3, DES, A1 and CBK, were bred and maintained in our colony at the Medical College of Georgia. All procedures involving mice were carried out in compliance with institutional, state and federal regulations. BM313 and DES14 mice were transgenic for the productively rearranged recombinant T-cell receptor (TCR) α and β genes derived from alloreactive H-2Kb-specific cytotoxic T-lymphocyte clones and CBK mice carry a complete H-2Kb transgene expressed in most cells.13 A1 mice were transgenic for TCR α and β genes, which recognize the minor histocompatibility antigen H-Y in the context of H2-Ek.15 B6.MRL-Fas_lpr_ (B6-lpr) mice were obtained from the Jackson Laboratory (Bar Harbor, ME).
Media
Chemically defined, serum-free medium selectively deficient in tryptophan was prepared using tryptophan-free RPMI-1640 (Select-amine kit; Gibco BRL, Gland Island, NY) as previously described.11 This medium was supplemented with insulin (10 μg/ml), iron-saturated transferrin (5 μg/ml), bovine albumin crystalline (400 mg/l, Gibco BRL; measured concentration of free tryptophan <5 nm), HEPES buffer (10 mm), β-mercaptoethanol (50 μm), and sodium pyruvate (1 mm).
T-cell activation
BM3 or DES T cells were stimulated by co-culturing with APCs from CBK mice and A1 T cells with spleen cells from male CBA mice. In brief, responder spleen cells (4 × 106 cells/ml) were co-cultured with irradiated (3000 Rads) CBK spleen cells (4 × 106 cells/ml) in flat-bottomed, microtitre plates. In some experiments, T cells were stimulated with anti-CD3 monoclonal antibody (mAb) immobilized on plastic tissue culture wells [0·5 μg/cm2 in phosphate-buffered saline (PBS), pH 7·4] and soluble anti-CD28 mAb (1 μg/ml). T-cell proliferation was assessed using a standard thymidine incorporation assay in which cells were pulsed with 1 μCi of [3H]thymidine per well for 4 hr.7 All assays were conducted in triplicate and some in quadruplicate.
Flow cytometry
Three- or four-colour fluorescence-activated cell sorter (FACS) analysis was performed using directly conjugated mAbs as previously described.7,16 BM3 T cells were identified by gating on cells expressing both CD8 and TCR clonotype (Ti98) cells. Expression of CD25, CD69, CD71 and 1B11 was measured in the second or third colour. CD8, Strepavidin–CyChrome, CD25, CD69, CD71, CD43 (1B11), anti-Fas, anti-FasL antibodies and fluorescein isothiocyanate (FITC)-conjugated Annexin-V were purchased from Pharmingen (San Diego, CA). Anti-clonotypic antibody (Ti98) to TCR of BM3 T cells was produced, purified, and biotinylated in our laboratory.13
Cytotoxicity assays
Cytotoxicity was measured in standard 51Cr-release assays after co-culture of BM3 spleen cells and irradiated CBK spleen cells as described previously.16 Assays were performed in triplicate in round-bottomed microtitre plates at responder : target ratios of 90 : 1, 30 : 1, 10 : 1, 3 : 1, and 1 : 1. Target cells were incubated for 90 min at 37° with 100 μCi [51Cr]sodium chromate before being mixed with responder cells. After 5 hr at 37° 25 μl of supernatant was collected from each well and counted (Pharmacia Betaplate 1205, Piscataway, NJ). EL-4 (H-2Kb+) cells were used as targets.
Western blots and immunoprecipitation
Cell lysates were prepared from activated T cells and subjected to immunoblot analyses according to standard procedures17 using monoclonal anti-cyclin D3 (Lab-vision, Fremont, CA) and polyclonal anti-cdk4 antibodies (C22, Santa Cruz, CA). In some cases, cell lysates were immunoprecipitated with anti-cdk4 antibody and immunoblotted with anti-cyclin D3 antibody.
Fas-mediated apoptosis
Twenty-four-well tissue culture plates were coated with antibodies by incubating 360 μl/well of PBS containing 2·5 μg/ml of anti-CD3 mAb with or without anti-Fas mAb (anti-CD95, Jo2, 10 μg/ml) for 1 hr at 37°, as described.18,19 Wells were then washed, and BM3 or B6-lpr spleen cells (4 × 106 cells/well) were plated immediately into coated wells in defined media and then cultured at 37°. For optimal activation of T cells, soluble anti-CD28 antibody was also added. Apoptotic cells were assessed by staining with Annexin V-FITC and propidium iodide (PI) as described.19 To inhibit activation of the caspase cascade after Fas ligation, T cells were activated in the presence of a caspase inhibitor (z-VAD-FMK, 10 μm; Calbiochem, La Jolla, CA).20
Splenocyte fractionation
Dendritic cells and macrophages were enriched by collagenase treatment, plastic adherence, and magnetic cell separation, according to previously described methods with modifications.21–23 Spleens were teased apart in the presence of collagenase D (100 U/ml) and then incubated with additional collagenase D (400 U/ml) for 60–90 min at 37°. Cell suspensions were diluted immediately in Ca2+-free Hanks' solution (1 : 10), centrifuged and suspended in RPMI-1640 medium containing 5% fetal calf serum at ∼107 cells/ml. Then, 10 ml of the cell suspension was plated in T-75 tissue culture flasks and incubated for 90 min at 37°, to allow cells to adhere. Further enrichments of CD11c+ and CD11b+ cells were prepared from these adherent cells by re-suspending them in PBS (pH 7·2) supplemented with 0·5% bovine serum albumin and 2 mm ethylenediaminetetraacetic acid, incubating with CD11c or CD11b microbeads (10 μl of beads per 107 cells; Miltenyl Biotec, Auburn, CA) at 4°. CD11c+ and CD11b+ cells were positively selected by AutoMACS (Miltenyl Biotec). To evaluate the purity positively selected cells were also incubated with phycoerythrin (PE)-conjugated CD11c or CD11b mAbs (Pharmingen) for 15 min at 4° before separation. The purity of CD11c+ cells was ∼90% and that of CD11b+ cells was ∼95%.
Reverse transcription–polymerase chain reaction (RT-PCR)
To examine IDO transcription, adherent or CD11c+- or CD11b+-enriched subpopulations of splenocytes were cultured for up to 24 hr at 37°, and total RNA samples were extracted using RNA STAT-60 solution (Tel-Test, Inc., Friendswood, TX) according to the manufacturer's recommendations. A 0·2-μg sample of total RNA was reverse transcribed and PCR amplified for 30–35 cycles by Access RT-PCR system (Promega, Madison, WI). Primer sequences were as follows: GAPDH, 5′-AACGGATTTGGCCGTATT-3′, 5′-TCTGGGATGGAAATTGTGAG-3′; β-actin, 5′-AGCAAGAGAGGTATCCTG-3′, 5′-CTTTACGGATGTCAACGTC-3′; IDO, 5′-GTACATCACCATGGCGTATG-3′, 5′-GCTTTCGTCAAGTCTTCATTG-3′; and Fas ligand, 5′-CTACCACCGCCATCACAACC-3′, 5′-CCTCTTCTCCTCCATTAGCAC-3′. PCR products were size-fractionated by agarose electrophoresis and normalized according to the amount of GAPDH or β-actin detected in the same RNA sample.
Results
Tryptophan starvation blocks T-cell entry into S phase
To evaluate the effect of tryptophan deprivation on activated mouse T cells we stimulated freshly isolated splenocytes and monitored their responses over time by measuring thymidine incorporation and assessing their activation status by flow cytometry. Responder splenocytes were isolated from BM3,13 DES,14 or A115 TCR transgenic mice, which harbour cohorts of H-2Kb-specific CD8+ T cells (BM3 and DES mice) or male (H-Y) antigen-specific, H-2Ek-restricted CD4+ T cells (A1 mice). Splenocytes from these mice were activated with CD3 and CD28 antibodies or co-cultured with irradiated splenocytes from CBK transgenic mice, which express H-2Kb on all cell types, or from male CBA mice, respectively. All transgenic mouse lines were generated on the inbred CBA strain background and have been characterized extensively.
When activated in tryptophan-free medium T cells from BM3, DES and A1 TCR transgenic mice failed to incorporate thymidine when assayed 72 hr after cultures were established (Fig. 1a). In contrast, T cells activated in tryptophan-sufficient (RPMI-1640) medium incorporated thymidine. BM3 T cells began to incorporate thymidine after 24 hr and incorporation increased progressively at later time-points (Fig. 1b). T cells cultured in tryptophan-free medium did not form cell aggregates or transform into blasts (data not shown). Similar outcomes were obtained when T cells from DES (H-2Kb-specific, CD8+) and A1 (H-Y-antigen-specific, CD4+) TCR transgenic mice were cultured with appropriate APCs (data not shown). These data show that T cells failed to enter S phase when activated in tryptophan-free medium.
Figure 1.

T-cell proliferation in tryptophan-deficient medium. (a) Splenocytes from BM3, A1 and DES mice were co-cultured with irradiated splenocytes from CBK (BM3, DES) or CBA male (A1) mice in RPMI (Trp+) and tryptophan-deficient (Trp−) medium. Proliferation was assessed by thymidine incorporation after 72 hr as described in the Materials and methods. (b) Thymidine incorporation by activated BM3 T cells over time. (c) Effect of tryptophan (•) concentration or combined isoleucine/leucine (▪) concentrations on thymidine incorporation by activated A1 T cells 48 hr after stimulation. Assays shown are representative of at least two separate experiments. Outcomes were assayed in triplicate and standard deviations were <5% from the mean values shown.
To evaluate the effect of tryptophan availability on T-cell proliferation, splenocytes from A1 TCR transgenic mice were cultured in media containing various concentrations of tryptophan. Thymidine incorporation was measured after 48 hr when T cells activated in tryptophan-sufficient media incorporated thymidine at maximal rates (Fig. 1c and data not shown). Half maximal DNA synthesis rates were achieved at approximately 1 μm tryptophan. Comparable outcomes were obtained when splenocytes from BM3 TCR transgenic mice were activated in various concentrations of tryptophan (data not shown). In contrast, half maximal T-cell DNA synthesis rates were obtained at much lower combined concentrations of isoleucine and leucine (∼50 nm), which are also essential amino acids, when BM3 and A1 T cells were activated in defined media (Fig. 1). Similar outcomes were obtained irrespective of whether T cells were activated by adding mitogenic antibodies (anti-CD3 and anti-CD28) or co-culturing with irradiated antigen-specific APCs from mice. These outcomes show that activated T cells are considerably more sensitive to low tryptophan than isoleucine/leucine concentrations by a factor of ∼20-fold.
Tryptophan deprivation blocked expression of late but not early activation markers
Following co-culture with APCs from CBK transgenic mice, BM3 T cells were analysed by flow cytometry to evaluate expression of activation markers (Fig. 2a). BM3 T cells activated in tryptophan-deficient medium expressed CD25 and CD69 surface activation markers. Most BM3 T cells activated in tryptophan-free medium expressed CD25 and CD69 from 24 hr after initial stimulation but mean expression levels were three to five times lower than the levels detected on control T cells activated in tryptophan-sufficient medium. This differential was maintained at 48 hr after activation. In contrast, BM3 T cells activated in tryptophan-free medium did not express CD71 when assayed up to 72 hr after initial activation (Fig. 2a and data not shown). Activated BM3 T cells cultured in tryptophan-sufficient medium all expressed CD71 from 24 hr. Unstimulated BM3 cells expressed no detectable activation markers during the periods assayed (data not shown). H-Y-antigen-specific CD4+ T cells from A1 TCR transgenic mice also expressed CD25 and CD69 but not CD71 when activated in tryptophan-free medium (data not shown).
Figure 2.

Characterization of activated T cells. BM3 T cells were cultured with CBK splenocytes (a) or activated with anti-CD3 monoclonal antibody (b) in tryptophan-free (heavy lines) and tryptophan-sufficient, RPMI-1640 (fine lines) media for 24 and 48 hr. (a) Cells were stained with anti-CD8, anti-(TCR) clonotypic antibody (Ti-98), and antibodies against CD25, CD69 and CD71 and analysed by flow cytometer. Histograms show the expression of the activation markers on gated CD8+ Ti-98+ BM3 T cells. Markers indicate staining intensity of non-activated control BM3 cells. (b) BM3 cells were cultured for 24 (lanes 1, 2) or 48 (lanes 3, 4) hours in tryptophan-sufficient (lanes 1, 3) or tryptophan-free (lanes 2, 4) medium. Lanes 5 and 6 are positive controls for cdk4 (LS.174 cells) and cyclin D3 (Jurkatt cells), respectively. Cell lysates were fractionated by gel electrophoresis and then immunoblotted with anti-cyclin D3 (upper panel) and anti-cdk4 antibodies (centre panel) as described in the Materials and methods. Anti-cdk4 immunoprecipates were also immunoblotted with anticyclin D3 antibody (lower panel). Results shown are representative of two independent experiments.
To evaluate further cell cycle progression lysates prepared from activated T cells 24 and 48 hr after stimulation were immunoblotted with antibodies against two proteins induced during cell cycle progression, cyclin D3 and cdk4 (Fig. 2b). Neither of these proteins was detected at either time-point in lysates made from T cells activated in tryptophan-free media while both proteins were present in lysates made from T cells activated in tryptophan-sufficient media. No cyclin D3 was associated with cdk4 in these T cells when cdk4 was immunoprecipated. Taken together, these data show that activated T cells entered the cell cycle but cell cycle progression ceased before S phase when they were activated in tryptophan-deficient medium.
Tryptophan deprivation caused cell cycle arrest of activated T cells in mid-G1 phase
To evaluate how far from G0 into G1 phase BM3 T cells progressed when deprived of tryptophan, we stimulated BM3 T cells with anti-CD3 and soluble anti-CD28 antibodies in tryptophan-free medium. After various times (6–18 hr) tryptophan was added and the time of entry into S phase was assessed by pulsing cultures with [3H]thymidine (Fig. 3). Addition of tryptophan after 6 or 12 hr had no significant effect on entry into S phase at 24 hr or on the rate of DNA synthesis after S phase began relative to T cells cultured entirely in tryptophan-sufficient medium. Thus, tryptophan deprivation during the first 12 hr of G1 phase had no measurable effect on cell cycle progression. When tryptophan was added to cultures after 18 hr entry into S phase was delayed by approximately 6 hr. However, once DNA synthesis began rates of thymidine incorporation were comparable with those observed for BM3 T cells activated in tryptophan-sufficient medium. These data suggest that, as for activated human polyclonal T cells,7 tryptophan deprivation caused murine T-cell cycle progression to halt approximately 12 hr after they first received activation signals through the TCR and CD28 complexes. Moreover, tryptophan addition several hours after T cells had entered the growth-arrested state allowed T cells to continue their transition from G0 through G1 to S phase, suggesting that the arrest point was relatively stable. When tryptophan was added 18 hr after initial stimulation, DNA synthesis was first detected approximately 14–16 hr later. This suggests that activated T cells deprived of tryptophan entered the cell cycle but ceased to progress through the cell cycle ∼8–10 hr before S phase because naïve T cells (in G0) took longer (∼24 hr) to begin synthesizing DNA following stimulation.
Figure 3.

Analysis of T-cell cycle arrest point in tryptophan-free media. BM3 T cells were activated by anti-CD3 and anti-CD28 antibodies in tryptophan-free medium and tryptophan was added to cultures at the times indicated by the arrowheads. Thymidine incorporation was assessed in triplicate at the time-points shown on the graph (plotted as the start of the 4-hr pulse period with [3H]thymidine). Top panel indicates G0/G1 phase (white bar), S phase (black bar) and G1/S transition period (grey bar) for BM3 T cells cultured in RPMI-1640. Results are representative of three separate experiments. Standard deviations from the mean values shown were <10%.
Tryptophan deprivation blocked T-cell differentiation into cytolytic effector cells
We evaluated if BM3 T cells activated in tryptophan-free medium acquired cytolytic activity in chromium release assays using EL4 target cells that express H-2Kb (Fig. 4a). BM3 T cells cultured with CBK APCs for 2–4 days in tryptophan-free medium did not kill EL-4 target cells. In contrast, BM3 T cells activated in tryptophan-sufficient medium developed potent cytolytic activity after only 48 hr in culture. In addition, activated BM3 T cells were stained with 1B11 monoclonal antibody,24 a surface marker expressed by cytotoxic CD8+ T cells (Fig. 4b). BM3 T cells stimulated in tryptophan-free medium expressed no detectable 1B11, even after 72 hr of culture with CBK APCs, whereas BM3 T cells activated in tryptophan-sufficient medium expressed high levels of 1B11 from 48 hr onwards. Thus, BM3 T cells did not acquire cytolytic effector functions when activated in the absence of tryptophan.
Figure 4.

Tryptophan is required for effector cell differentiation. (a) Chromium release assays conducted in triplicate for antigen-activated BM3 T cells cultured in tryptophan-deficient and RPMI-1640 media after 48 hr. T cells were incubated with 51Cr-labelled EL-4 target cells at different effector : target cell ratios for 6 hr when target cell lysis was determined by standard procedures. Standard deviations were <10% from mean values shown. (b)Flow cytometric analyses of activated BM3 T cells stained with 1B11 antibody after culture in tryptophan-free (heavy line) and RPMI-1640 (fine line) media. Dotted line indicates staining profile of non-activated BM3 T cells. Results are representative of four separate experiments.
Fas-mediated apoptosis of activated T cells was enhanced by tryptophan deprivation
Next, we evaluated whether tryptophan deprivation enhanced T-cell susceptibility to Fas-mediated apoptosis following activation. BM3 T cells were activated with anti-CD3/CD28 antibodies in the presence of anti-Fas (CD95) antibody and stained with Annexin V to assess apoptosis after 18 hr (Fig. 5). Significantly increased numbers (23%) of BM3 T cells activated in tryptophan-free medium stained with Annexin V if anti-Fas antibody was present (Fig. 5c). Similar outcomes were obtained when T cells were stained 65 hr after initial activation except that more than 50% of BM3 T cells activated in tryptophan-free medium stained with Annexin V following Fas engagement at this time (Fig. 5c). Less than 5% of activated BM3 T cells stained with Annexin V when T cells were activated in tryptophan-sufficient medium or if anti-Fas antibody was not included in cultures. Comparable basal levels of Annexin V staining (∼5%) were detected in parallel cultures of BM3 T cells that were not activated. Surface expression of Fas did not increase when T cells were activated in tryptophan-free media (Fig. 5b). In contrast, Fas expression increased when T cells were activated in tryptophan-sufficient media, suggesting that increased Fas expression is a late event compromised by lack of tryptophan. However, RNA transcripts of Fas were detected in RNA samples prepared from T cells activated in tryptophan-free media, suggesting that basal expression of Fas still occurred in the absence of tryptophan (data not shown). These data show that tryptophan deprivation increased T-cell susceptibility to death via apoptosis following activation but only when anti-Fas antibody was present.
Figure 5.
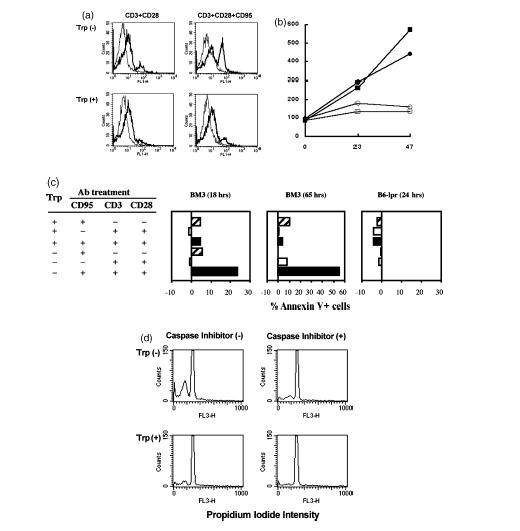
Tryptophan deprivation induces sensitivity to Fas (CD95)-mediated cell death by apoptosis. (a) BM3 T cells activated by anti-CD3 and anti-CD28 antibodies in the presence or absence of immobilized anti-CD95 (anti-Fas) antibodies were cultured in tryptophan-free (Trp−) and RPMI-1640 (Trp+) media, stained with Annexin V and analysed by flow cytometry (heavy lines). Fine lines indicate unstained control cells. (b) Mean fluorescent intensities over time for T cells from BM3 (▪, □) or CBA (•, ○) mice activated in tryptophan-sufficient (▪, •) or tryptophan-free (□, ○) and stained with anti-Fas (CD95) antibody. Data are representative of two experiments (c) Annexin V staining expressed as mean percentage of Annexin V+ cells in each treatment group after subtracting background staining detected in control (non-activated) cultures (5–10%). Data acquired after longer culture periods (65 hr) or after culturing splenocytes from B6-lpr (Fas-deficient) mice for 18 hr were analysed in the same way. Data were compiled from six separate experiments, including the results shown in (a). Statistically significant outcomes (Wilcoxon rank-sum test) were obtained when T cells were activated in tryptophan-free versus tryptophan-sufficient media with all three antibodies present and when T cells were activated in tryptophan-free media plus anti-CD3/anti-CD28 antibodies, with versus without addition of anti-CD95 antibody (P < 0·05). (d) Flow cytometric analyses of activated (anti-CD3 and anti-CD28) BM3 cells treated with anti-CD95 antibody in the presence and absence of caspase inhibitor following culture in tryptophan-deficient (Trp−) and RPMI-1640 (Trp+) media. Cells were stained with propidium iodide as described in the Materials and methods.
To evaluate further if enhanced susceptibility to apoptosis was Fas dependent we repeated these experiments using T cells from B6-lpr mice, which are defective in Fas expression (Fig. 5c). T cells from B6-lpr mice did not undergo apoptosis when activated in tryptophan-free medium and treated with anti-Fas antibody. In addition, we included a wide-spectrum caspase inhibitor in cultures of BM3 T cells activated in tryptophan-free medium and treated with anti-Fas antibody (Fig. 5d). Inclusion of caspase inhibitor reduced the proportion of apoptotic T cells to levels comparable with controls cultured in tryptophan-sufficient medium, showing that T-cell apoptosis depended on activation of the caspase cascade.
CD11c+ cells express IDO in mouse spleen
To identify cells expressing IDO in vivo, we isolated cell subpopulations from mouse spleens and evaluated IDO gene transcription by semiquantitative RT-PCR analyses (Fig. 6). RNA samples prepared from freshly isolated, plastic-adherent splenocytes (enriched in macrophages and dendritic cells) did not contain detectable IDO transcripts but IDO transcripts were detected after these cells were cultured in vitro (Fig. 6a). IDO transcription was detected after 6 hr and levels peaked after 18 hr in culture (Fig. 6B). IDO transcripts were not detected in RNA samples isolated from non-adherent splenocytes, which contain mostly T and B cells and few myeloid cells, even after 24-hr culture with LPS or interferon-γ (Fig. 6a and data not shown). CD11c+ and CD11b+ cells were further enriched from the adherent cell pool by antibody-coated magnetic bead sorting. IDO transcript levels were significantly higher in RNA samples from CD11c+- than CD11b+-enriched cell populations (Fig. 6c). IDO transcripts were barely detectable in RNA samples from CD11b+ and CD11c− populations of splenocytes (Fig. 6c and data not shown). Thus, IDO transcripts were preferentially expressed in CD11c+ splenocytes, which consist of mature and immature dendritic cells. We also evaluated Fas-ligand transcription in RNA samples isolated from enriched CD11c+ and CD11b+ splenocyte populations (Fig. 6c). Fas-ligand RNA transcripts were detected in RNA prepared from CD11c+- but not CD11b+-enriched cell populations, suggesting that cells expressing IDO and Fas-ligand co-purify with CD11c+, CD11b− subpopulations.
Figure 6.

RT-PCR analyses of IDO gene transcription in splenocytes. (a) RNA extracted from spleens of CBA mice as described in the Materials and methods after 24 hr in culture; total splenocytes (lane 1); non-adherent splenocytes (lane 2), three separate preparations of adherent cells (lanes 3–5). Lane 6, no RNA; lane 7, epididymis RNA. (b) RNA samples extracted from adherent splenocytes after culture for the number of hours indicated. Asterisk indicates RNA samples from non-adherent splenocytes after 18 hr culture. (c) RNA samples from CD11c+- and CD11b+-enriched cell populations selected by magnetic cell sorting after overnight (18 hr) culture. Samples were screened using primers for IDO and Fas ligand sequences and PCR products were of the size expected for mRNA transcripts.
Discussion
In this study we show that cell cycle progression in murine T cells ceased midway between G0 phase and the start of DNA synthesis, approximately 12 hr after initial activation, and T cells became sensitized to Fas-mediated apoptosis following stimulation in the absence of tryptophan. We have previously shown that polyclonal human T cells exhibit a similar tryptophan-sensitive arrest point midway through G1.7 Our current findings reveal that murine CD8+ and CD4+ T cells of defined antigen specificity behaved in a similar manner when deprived of tryptophan during activation. Thus, while T cells activated in the absence of tryptophan did not acquire effector functions, they were nonetheless predisposed to die via AICD as if they had undergone full proliferation and functional maturation. We propose that these experimental outcomes mimic events in tissue microenvironments where cells expressing IDO accumulate, which are therefore regions where T-cell tolerance is imposed.
Our findings that murine T cells activated in the absence of tryptophan uniformly expressed the activation markers CD69 and CD25 and down-regulated TCR suggest that T cells received and responded to TCR-mediated signals normally by exiting G0 and entering G1 phase. The absence of CD71, cyclin D3 and cdk4 or of increased Fas expression is consistent with the conclusion that T cells ceased cell cycle progression in mid-G1 phase based on outcomes from studies in which tryptophan was added at specific times after initial activation. The simplest explanation for the inhibitory effects of tryptophan deprivation on T-cell proliferation is cessation of protein synthesis. However, murine T cells were less sensitive to low concentrations of isoleucine/leucine (half maximal DNA synthesis rates achieved at ∼50 nm combined) than they were to low tryptophan concentrations (∼1 µm). These outcomes suggest that global inhibition of protein synthesis is unlikely to explain the more potent effects of tryptophan deprivation on T-cell cycle progression since isoleucine and leucine combined account for ∼10% of amino acids in cell proteins while tryptophan is the rarest amino acid (∼1%). In addition, studies with human T cells showed that complete absence of isoleucine and leucine during T-cell activation did not reproduce the effects of tryptophan deprivation on blocking T-cell entry into S phase (Munn et al. unpublished results). Thus, it is unlikely that the effects of tryptophan deprivation on T-cell cycle progression are explained simply by an inability to synthesize proteins. Rather, we propose that human and murine T cells are specifically sensitive to the concentration of free tryptophan available to them at a critical checkpoint during cell cycle progression.
A role for cells expressing IDO in inhibiting in vivo T-cell responses is implied by studies on murine pregnancy.11,12 However, IDO expression is not limited to the maternal–fetal interface. We propose that cells expressing IDO have a more general role in regulating in vivo T-cell responses, specifically acting as a peripheral tolerance mechanism. Mechanistic links between tryptophan catabolism and regulation of immune responses are implied by observations that localized tryptophan catabolizing activity mediated by cells expressing IDO is enhanced by tissue inflammation.25,26 Inflammation induces IDO expression in many tissues, although within these tissues only a few cells are actually induced to express IDO.27 Until recently, the prevailing view was that the IDO mechanism was a component of innate host defence that allowed infected cells, such as macrophages, to limit the proliferation of intracellular micro-organisms by degrading tryptophan. Based on observations that human macrophages expressing IDO prevent in vitro T-cell proliferation, we proposed the alternative hypothesis that cells expressing IDO suppressed T-cell activation by depriving them of access to tryptophan as they progressed through the cell cycle.7 We validated this hypothesis in vivo by showing that tryptophan catabolism during gestation protected allogeneic murine fetuses from rejection by T-cell-dependent maternal immune processes.11,12 More recently, others have suggested that regulatory dendritic cells expressing IDO suppress in vivo T-cell responses (delayed type hypersensitivity) to peptides6 and that allogeneic pancreatic islet cells genetically modified to express IDO are more resistant to host immunity following transplantation.28 In addition, we have shown that cells transfected to express IDO inhibit T-cell proliferation in vitro and eliminate priming of allogeneic T-cell responses to allogeneic tumour cells in immunized mice.29 Collectively, these experimental outcomes support the notion that cells expressing IDO mediate regulatory functions resulting in suppressed T-cell responses.
Several tolerogenic mechanisms, including clonal deletion, anergy, ignorance, deviation and suppression, have been proposed to explain why self-reactive T cells do not drive autoimmunity in most individuals.1,2 AICD is considered to be an important pathway that limits the ability of activated T cells to sustain prolonged effector functions, thereby preventing autoimmunity or collateral damage to healthy cells and tissues during episodes of inflammation. AICD is usually mediated by Fas/Fas-ligand interactions that activate cell death pathways by inducing the caspase cascade. In most, if not all, experimental systems described to date, Fas-dependent AICD was observed only after several rounds of T-cell division.3,5,6 In contrast, our findings show that T cells were rendered highly susceptible to apoptotic cell death before their first division when they were deprived of access to tryptophan. In the absence of Fas engagement, human T cells in this arrested state were stable for over 4 days without tryptophan (ref. 7 and Munn et al. unpublished data). In the current study, tryptophan deprivation prevented entry of murine cells into S phase but the proportion of apoptotic T cells did not increase significantly over background levels for up to 56 hr in tryptophan-free medium. However, activated, growth-arrested murine T cells were rapidly sensitized to induction of the caspase cascade leading to apoptosis when Fas antibody was added, indicating that activation under tryptophan-limiting conditions and Fas engagement were necessary to sensitize T cells to AICD. Failure to up-regulate Fas expression under tryptophan-limiting conditions suggests that Fas-mediated signalling through death domains to induce the caspase cascade occurred even though surface levels of Fas did not increase. Furthermore, increased Fas expression by T cells activated under tryptophan-sufficient conditions was not an accurate predictor of susceptibility to Fas-mediated induction of apoptosis via the caspase cascade, suggesting that Fas protein may not be associated with proteins containing death domains under these conditions. It is unclear if these observations apply to T cells that activate in tissues while associated with APCs expressing IDO and further experiments to address this question are in progress. Nevertheless, our observations on isolated T cells suggest that T cells associated intimately with APCs expressing IDO may exert profound influences on the subsequent course of T-cell responses to TCR triggering.
We interpret these outcomes to propose that T cells activated in tissue microenvironments where access to tryptophan is limited by neighbouring cells expressing IDO will exhibit enhanced susceptibility to die via AICD. This model further predicts that cells expressing both IDO and Fas-ligand would be potent suppressors of T-cell activation. If such cells also processed and presented antigens to T cells, they would function as antigen-specific suppressors of T-cell responses. Our findings suggest that IDO gene expression is not constitutive in normal mouse spleen but is induced several hours after CD11c+ cells are placed in culture. It has recently been shown that murine lymphoid CD8α+, CD11c+ dendritic cells isolated from spleen are immunosuppressive APCs that employ tryptophan catabolism to suppress in vivo T-cell responses to tumour-associated peptides.6 Recently, these immunoregulatory dendritic cells have been shown to express IDO activity when purified from spleen.30 Our findings concur with these analyses since inducible IDO transcription was associated with the CD11c+ subset but not the CD11b+ subset in mouse spleen. Moreover, Fas-ligand transcription has also been associated with the CD11c+ subset since CD8+ dendritic cells have been reported to express Fas ligand and kill CD4+ T cells by Fas-mediated apoptosis.3 Taken together, these findings support our hypothesis that cells expressing IDO and Fas ligand may act as potent immunoregulatory APCs that suppress immunity and impose tolerance.
Acknowledgments
We thank our colleagues in the IMMAG Molecular Immunology Program for stimulating discussions relating to these studies. We thank Doris McCool and Anita Wylds, for assistance with mouse breeding and typing, Jun Fang Tsai for technical assistance, Brendan Marshall for help with RT-PCR, and Angie Compton and Phyllis McKie for help with manuscript preparation. Studies described in this manuscript were funded by NIH grants awarded to A.L.M. (AI44219) and D.H.M. (HL60137) and by generous support from the Carlos and Marguerite Mason Trust and the MCG Departments of Medicine, Pediatrics and IMMAG.
References
- 1.Fairchild PJ, Waldmann H. Dendritic cells and prospects for transplantation tolerance. Curr Opin Immunol. 2000;12:528–35. doi: 10.1016/s0952-7915(00)00134-5. [DOI] [PubMed] [Google Scholar]
- 2.Stockinger B. T lymphocyte tolerance: from thymic deletion to peripheral control mechanisms. Adv Immunol. 1999;71:229–65. doi: 10.1016/s0065-2776(08)60404-6. [DOI] [PubMed] [Google Scholar]
- 3.Suss G, Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J Exp Med. 1996;183:1789–96. doi: 10.1084/jem.183.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 5.Li XC, Wells AD, Strom TB, Turka LA. The role of T cell apoptosis in transplantation tolerance. Curr Opin Immunol. 2000;12:522–7. doi: 10.1016/s0952-7915(00)00133-3. [DOI] [PubMed] [Google Scholar]
- 6.Grohmann U, Fallarino F, Silla S, et al. CD40 ligation ablates the tolerogenic potential of lymphoid dendritic cells. J Immunol. 2001;166:277–83. doi: 10.4049/jimmunol.166.1.277. [DOI] [PubMed] [Google Scholar]
- 7.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–72. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol. 2000;164:3596–9. doi: 10.4049/jimmunol.164.7.3596. [DOI] [PubMed] [Google Scholar]
- 9.Bronte V, Wang M, Overwijk WW, Surman DR, Pericle F, Rosenberg SA, Restifo NP. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J Immunol. 1998;161:5313–20. [PMC free article] [PubMed] [Google Scholar]
- 10.Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal Malefyt R, Liu YJ. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283:1183–6. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 11.Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 12.Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, Munn DH. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2:64–8. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 13.Sponaas A-M, Tomlinson PD, Antoniou J, et al. Induction of tolerance to self MHC class I molecules expressed under the control of milk protein or β globin gene promoters. Intl Immunol. 1994;6:277–87. doi: 10.1093/intimm/6.2.277. [DOI] [PubMed] [Google Scholar]
- 14.Schulz R, Mellor AL. Self major histocompatiblity complex class I antigens expressed solely in lymphoid cells do not induce tolerance in the CD4+ T cell compartment. J Exp Med. 1996;184:1573–8. doi: 10.1084/jem.184.4.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zelenika D, Adams E, Mellor A, et al. Rejection of H-Y disparate skin grafts by monospecific CD4+ Th1 and Th2 cells: no requirement for CD8+ T cells or B cells. J Immunol. 1998;161:1868–74. [PubMed] [Google Scholar]
- 16.Tarazona R, Sponaas A-M, Mavria G, et al. Effects of different antigenic microenvironments on the course of CD8+ T cell responses in vivo. Intl Immunol. 1996;8:351–8. doi: 10.1093/intimm/8.3.351. [DOI] [PubMed] [Google Scholar]
- 17.Munn DH, Beall AC, Song D, Wrenn RW, Throckmorton DC. Activation-induced apoptosis in human macrophages: developmental regulation of a novel cell death pathway. J Exp Med. 1995;181:127–36. doi: 10.1084/jem.181.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desbarats J, Freed JH, Campbell PA, Newell MK. Fas (CD95) expression and death-mediating function are induced by CD4 cross-linking on CD4+ T cells. Proc Natl Acad Sci USA. 1996;93:11014–18. doi: 10.1073/pnas.93.20.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desbarats J, Wade T, Wade WF, Newell MK. Dichotomy between naive and memory CD4 (+) T cell responses to Fas engagement. Proc Natl Acad Sci USA. 1999;96:8104–9. doi: 10.1073/pnas.96.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ko SC, Johnson VL, Chow SC. Functional characterization of Jurkat T cells rescued from CD95/Fas-induced apoptosis through the inhibition of caspases. Biochem Biophys Res Commun. 2000;270:1009–15. doi: 10.1006/bbrc.2000.2565. [DOI] [PubMed] [Google Scholar]
- 21.Inaba K, Pack M, Inaba M, Sakuta H, Isdell F, Steinman RM. High levels of a major histocompatibility complex II-self peptide complex on dendritic cells from the T cell areas of lymph nodes. J Exp Med. 1997;186:665–72. doi: 10.1084/jem.186.5.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grohmann U, Belladonna ML, Bianchi R, Orabona C, Ayroldi E, Fioretti MC, Puccetti P. IL-12 acts directly on DC to promote nuclear localization of NF-kappaB and primes DC for IL-12 production. Immunity. 1998;9:315–23. doi: 10.1016/s1074-7613(00)80614-7. [DOI] [PubMed] [Google Scholar]
- 23.Vremec D, Zorbas M, Scollay R, Saunders DJ, Ardavin CF, Wu L, Shortman K. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med. 1992;176:47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrington LE, Galvan M, Baum LG, Altman JD, Ahmed R. Differentiating between memory and effector CD8 T cells by altered expression of cell surface O-glycans. J Exp Med. 2000;191:1241–6. doi: 10.1084/jem.191.7.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–22. [PubMed] [Google Scholar]
- 26.Varga J, Yufit T, Brown RR. Inhibition of collagenase and stromelysin gene expression by interferon-gamma in human dermal fibroblasts is mediated in part via induction of tryptophan degradation. J Clin Invest. 1995;96:475–81. doi: 10.1172/JCI118058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20:469–73. doi: 10.1016/s0167-5699(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 28.Alexander AM, Crawford M, Bertera S, Rudert WA, Takikawa O, Robbins PD, Trucco M. Indoleamine 2,3-dioxygenase expression in transplanted NOD islets prolongs graft survival after adoptive transfer of diabetogenic splenocytes. Diabetes. 2002;51:356–65. doi: 10.2337/diabetes.51.2.356. [DOI] [PubMed] [Google Scholar]
- 29.Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing indoleamine 2,3 dioxygenase inhibit T cell responses. J Immunol. 2002;168:3771–6. doi: 10.4049/jimmunol.168.8.3771. [DOI] [PubMed] [Google Scholar]
- 30.Fallarino F, Vacca C, Orabona C, et al. Functional expression of indoleamine 2,3-dioxygenase by murine CD8alpha (+) dendritic cells. Intl Immunol. 2002;14:65–8. doi: 10.1093/intimm/14.1.65. [DOI] [PubMed] [Google Scholar]