When loss is gain: reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. Talking Point on the role of presenilin mutations in Alzheimer disease (original) (raw)
Abstract
More than 100 missense mutations in presenilin 1 and 2 are associated with early-onset dominant Alzheimer disease. These proteins span the membrane several times and are ostensibly the catalytic component of the γ-secretase complex, which is responsible for producing the amyloid β-peptide (Aβ) that deposits in the Alzheimer brain. A common outcome of Alzheimer-associated presenilin mutations is an increase in the ratio of the more aggregation-prone 42-residue form of Aβ to the 40-residue variant, which is often referred to as a presenilin ‘gain of function'. An apparent paradox is that most of these mutant presenilins have reduced proteolytic efficiency, which forms part of the counter argument that presenilin ‘loss of function' can cause the neuronal dysfunction and death that lead to the disease. In this review, a unifying hypothesis is presented that puts forward a biochemical mechanism by which slower less-efficient forms of the protease can result in a greater proportion of 42-residue Aβ.
Keywords: amyloid β-peptide, enzyme mechanism, Notch receptor, pathogenesis, protease
Introduction
A century ago, Alois Alzheimer first described the neurological disease that bears his name. He detailed two characteristic pathological features that are observed post mortem: amyloid plaques, and neurofibrillary tangles in the cerebral cortex and limbic system. Many decades passed before the biochemical components of these two features were discovered. The plaques are composed primarily of the 39–43-residue amyloid β-peptide (Aβ), and the tangles include filaments of the otherwise microtubule-associated protein tau (Wolfe, 2006). Aβ is released from the amyloid precursor protein (APP) by the sequential action of β- and γ-secretases, with the latter cutting relatively heterogeneously (Esler & Wolfe, 2001). Most of the Aβ produced by γ-secretase is the 40-residue form (Aβ40); however, the major Aβ species deposited in the plaques is the 42-residue variant (Aβ42), although this peptide represents only 5–10% of all Aβ produced. In 1988, the APP gene was identified and found to reside on chromosome 21, which is triplicated in Down syndrome (trisomy 21). Indeed, most Down syndrome patients manifest Alzheimer disease (AD) by the age of 50, and post-mortem analyses of those who die young show diffuse intraneuronal deposits of Aβ in the absence of any tau pathology, suggesting that Aβ deposition is an early event in AD.
APP was the first gene to be associated with early-onset dominant AD, and the responsible mutations are located in and around the Aβ region of the encoded type I integral membrane glycoprotein. These mutations were soon determined to alter the amount of Aβ produced, the ratio of Aβ42 to Aβ40 or the amino-acid sequence of Aβ (Selkoe, 1994). Mutations near the β-secretase cleavage site make APP a better substrate for this enzyme and lead to increased production of all forms of Aβ. Mutations near the γ-secretase cleavage site lead to increases in the more aggregation-prone Aβ42 relative to Aβ40, and mutations in the Aβ region itself change the biophysical properties of the peptide to render it more likely to aggregate. These findings provided the first compelling evidence that Aβ might be involved in the pathogenesis of AD rather than simply being a marker. The recent discovery of an extra copy of the APP gene in familial AD (Rovelet-Lecrux et al, 2006) provides further support that increased Aβ production can cause the disease.
In 1995, mutations in the presenilins—PS1 and PS2—were also linked with early-onset familial AD (Levy-Lahad et al, 1995; Rogaev et al, 1995; Sherrington et al, 1995). The multi-pass membrane proteins encoded by these genes bore little resemblance to any other proteins known at the time, and their biochemical function was a complete mystery. However, the AD-associated mutations were soon determined to increase the ratio of Aβ42 to Aβ40 (Aβ42/Aβ40) in mice and humans (Borchelt et al, 1996; Citron et al, 1997; Scheuner et al, 1996), indicating that presenilins might modify the way in which γ-secretase cuts APP. Subsequently, the knockout of PS1 in mice was found to markedly reduce γ-secretase cleavage of APP (De Strooper et al, 1998), and follow-up studies showed that the knockout of both PS1 and PS2 eliminated γ-secretase cleavage completely (Herreman et al, 2000; Zhang et al, 2000). Presenilins are therefore essential for this proteolytic function. Parallel studies revealed that peptide-substrate mimics with classical aspartyl protease-inhibitory motifs could inhibit γ-secretase, suggesting that the enzyme belongs to this category of proteases (Wolfe et al, 1999a). This led to the identification of two conserved transmembrane aspartates in presenilin that are essential for γ-secretase activity, and the idea that presenilin is the catalytic component of the enzyme (Wolfe et al, 1999b,c). This hypothesis was soon confirmed through affinity labelling with transition-state analogue inhibitors (Esler et al, 2000; Li et al, 2000). More recently, three presenilin-associated co-factors for γ-secretase—the membrane proteins nicastrin (NCT), anterior pharynx-defective phenotype 1 (Aph1) and PS enhancer 2 (Pen2)—were discovered (Edbauer et al, 2003; Francis et al, 2002; Goutte et al, 2002; Kimberly et al, 2003; Takasugi et al, 2003; Yu et al, 2000), and purification of what is now known as the γ-secretase complex proved that presenilin and its three co-factors are necessary and sufficient for γ-secretase activity (Fraering et al, 2004). Furthermore, the discovery that presenilin homologues, such as signal peptide peptidase, apparently have proteolytic activity alone (Weihofen et al, 2002), without the need for other protein factors, cemented the concept of presenilin as the catalytic component of the γ-secretase complex. The implications for how presenilin mutations cause familial AD were clear: the mutations occur in the catalytic subunit of the protease responsible for determining the length of Aβ peptides (see Box). Nevertheless, it should be pointed out that presenilin also has nonproteolytic functions (Baki et al, 2004; Huppert et al, 2005; Kang et al, 2002), the disruption of which might also contribute to familial AD pathogenesis.
Concomitantly, however, it became clear that presenilin and γ-secretase have other substrates besides APP. After interacting with its cognate ligands, the Notch receptor undergoes a series of proteolytic events, in a manner similar to APP, ultimately releasing the intracellular domain that mediates the expression of genes controlling many types of cellular differentiation. We now know that the same γ-secretase cuts the transmembrane domain of the APP and Notch families of proteins, as well as a long list of other type I integral membrane proteins, including neuregulin and its receptor ErbB4, E-cadherins and N-cadherins, CD44, LDL-receptor-related protein (LRP), nectin-1 and growth hormone receptor. Cleavage of some of these substrates apparently has a role in cell signalling, but other cleavage events might simply be a means of clearing out membrane-bound stubs after ectodomain shedding (Kopan & Ilagan, 2004). The discovery of a new substrate has often led to suggestions that alterations in its proteolysis by presenilin mutations might underlie or contribute to AD. However, these speculations do not explain why APP mutations that change the production and/or properties of Aβ also cause the disease. This would require separate mechanisms of pathogenesis for presenilin and APP mutations, which is unlikely given that the former produces Aβ; furthermore, it does so at the crucial site that determines the length of Aβ and its aggregation properties.
In parallel with the discovery of presenilin as a protease that cleaves APP and Notch, AD-causing mutations in presenilin were found to have reduced proteolytic function. Yankner and colleagues first showed this effect with a range of mutant presenilins by using a Notch-based luciferase-reporter assay (Song et al, 1999). Several other groups have since noted this phenomenon (for example, Lewis et al, 2000), with the most recent being De Strooper and colleagues who showed that mutations in presenilin reduced its proteolytic function towards several different substrates (Bentahir et al, 2006). These findings raise an apparent paradox, in which AD-causing disease mutations cause both a ‘gain of function'—an increase in Aβ42/Aβ40—and a ‘loss of function'—a decrease in proteolytic activity. These seemingly opposing effects have elicited considerable debate over how the presenilin mutations cause AD, with some researchers suggesting that reducing Aβ production with candidate therapeutics might even exacerbate or cause the disease (for further details, see the online discussion at http://www.alzforum.org). Below, I offer a synthesis of these two phenomena, which, at first glance, seem to be antithetical.
To appreciate the resolution of this purported paradox, it should initially be noted that the presenilin-containing γ-secretase complex cuts the transmembrane domain of APP, and other substrates, in at least two positions: the γ-site that produces the carboxyl terminus of Aβ and the ε-site further downstream that produces the amino terminus of the APP intracellular domain (AICD; Table 1; Weidemann et al, 2002). Cleavage at the γ-site is heterogeneous, producing Aβ peptides of 39–43 residues, whereas the cut at the ε-site produces almost exclusively a 50-residue AICD. The same phenomenon occurs with Notch, involving heterogeneous cleavage in the middle of the transmembrane domain (the S4 site) and homogeneous cleavage further downstream (at the S3 site; Okochi et al, 2002). Interestingly, proteolysis at these two sites is affected by AD-causing mutations in APP and the presenilins, which lead to an increase in the proportion of Aβ42 relative to Aβ40 along with an increase in a new 51-residue AICD relative to the 50-residue product (Sato et al, 2003). These two proteolytic events are therefore not completely independent: a change in the cleavage site in one correlates with a change in the cleavage site of the other. However, it should be pointed out that in one study, several artificial mutations of Lys 166 in PS1 increased Aβ42 production without affecting AICD levels (Moehlmann et al, 2002), and in another report, inhibition of endocytosis altered AICD formation without changing Aβ42/Aβ40 (Fukumori et al, 2006).
Table 1.
Summary of evidence supporting increased amyloid β-peptide (Aβ) or Aβ42/Aβ40 in the pathogenesis of presenilin mutations
| Alzheimer disease (AD)-causing amyloid precursor protein (APP) mutations increase Aβ or Aβ42/Aβ40. |
|---|
| Triplication of the APP gene, either alone or with all of chromosome 21, leads to AD. |
| Brain Aβ42 deposits are observed early and presymptomatically in trisomy 21 |
| Presenilin is the catalytic component of γ-secretase, produces Aβ and determines the carboxy-terminal length of Aβ peptides. |
| AD-causing presenilin mutations increase Aβ42/Aβ40; the fact that presenilin and APP mutations cause AD by a common mechanism is parsimonious and logical, as presenilin cleaves APP to produce Aβ. |
| No AD-associated mutations have been found in any other γ-secretase substrate besides APP. |
| Among more than 100 AD-causing mutations in presenilins, complete loss of function of a single allele has not been found. |
| The Ihara model of processive proteolysis of APP by γ-secretase points to a biochemical mechanism by which a less-efficient protease (reduction of function) can lead to an increase in Aβ42/Aβ40 (gain of function). |
Recent evidence from Ihara and colleagues indicates that the ε-cleavage event might occur before proteolysis at the γ-site. First, analysis of intracellular Aβ reveals a small but significant amount of longer forms of this peptide, up to Aβ49, which is the proteolytic counterpart to the 50-residue AICD (Qi-Takahara et al, 2005); by contrast, longer AICDs, for example AICD counterparts to Aβ40 or Aβ42, have not been detected. Second, expression of Aβ49 leads to the secretion of Aβ40 and Aβ42 in the same proportion that is produced by γ-secretase (Funamoto et al, 2004). Third, swapping tryptophan residues into the γ-site within the APP transmembrane domain prevents γ-cleavage but allows ε-cleavage; however, swapping tryptophans into the ε-site leads to proteolysis between the γ-site and ε-site, at a so-called ζ-site (Fig 1, and see below; Sato et al, 2005). Installing tryptophans into the ζ-site prevents any transmembrane cleavage of APP. Therefore, with these tryptophan swaps, ε-cleavage can occur without γ-cleavage, but γ-cleavage is not seen without ε-cleavage or ζ-cleavage. Interestingly, longer Aβ peptides resulting from cleavage at the ζ-site are seen intracellularly on treatment with one particular γ-secretase inhibitor: a dipeptide analogue called DAPT (Yagishita et al, 2006). Finally, a mutation in PS1—M233T—leads to alternative ε-cleavage, producing the 51-residue AICD and its counterpart Aβ48 in a cell-free assay with detergent-solubilized membranes (Kakuda et al, 2006).
Figure 1.
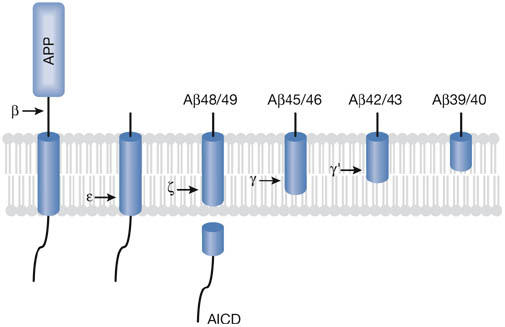
Ihara model of processive proteolysis of the amyloid precursor protein transmembrane domain by γ-secretase, beginning at the ε-cleavage site and cleaving every three residues. This model explains how reduction of proteolytic function owing to presenilin mutations might lower amyloid β-peptide (Aβ) production but increase the ratio of Aβ42 to Aβ40. Longer forms of Aβ, with more of the hydrophobic transmembrane domain, might be more likely to be retained in the active site of the protease, whereas the shorter forms are more likely to be released. Less catalytically efficient γ-secretase complexes would allow more time for the release of longer Aβ peptides. In addition, Alzheimer disease-causing presenilin mutations shift the initial ε-cleavage site to produce more Aβ48, which would lead to Aβ42. AICD, APP intracellular domain; APP, amyloid precursor protein.
One way to explain two major cleavage sites would be the presence of two pairs of catalytic aspartates within a presenilin dimer at the core of the γ-secretase complex. Several laboratories have reported evidence for presenilin–presenilin interaction (for example, Schroeter et al, 2003). However, recent evidence from our laboratory suggests that immunoprecipitation of one epitope-tagged presenilin does not bring down a co-expressed differentially tagged counterpart, but γ-secretase activity is nevertheless found in the immunoprecipitate (T. Sato & M.S.W., unpublished data). Therefore, two presenilin molecules per complex are not required for proteolytic activity: one presenilin suffices to generate the normal γ-cleaved Aβ and ε-cleaved AICD. Furthermore, cysteine mutagenesis and disulphide crosslinking experiments show that the key aspartate in transmembrane domain 6 is adjacent to the key aspartate in transmembrane domain 7, with no evidence for dimeric presenilin (Tolia et al, 2006). Together, these findings support a model of the γ-secretase complex in which one presenilin, and therefore one pair of aspartates, is sufficient to cut the transmembrane domain of APP, and other substrates, in at least two places.
Ihara and co-workers have suggested that these and other observations are consistent with successive cleavage events: initial proteolysis at the ε-site leads to the release of AICD, but the long Aβ products (Aβ49 or Aβ48) remain in the active site, and are successively cleaved every three residues upstream at the ζ-sites and then again at the γ-sites (Fig 1; Qi-Takahara et al, 2005). Specifically, they propose that Aβ49 is processed to Aβ46, Aβ43 and Aβ40, whereas Aβ48 is trimmed to Aβ45, Aβ42 and Aβ39. This model of successive proteolysis from the ε-site to the γ-sites elegantly explains how so many presenilin mutations can both reduce proteolytic activity—causing a loss of function—and increase the Aβ42/Aβ40 ratio—causing a gain of function. Mutant versions of the enzyme that are less proteolytically efficient also cut proportionately more at the alternative ε-site, producing Aβ48 (Sato et al, 2003). The slower mutant enzymes allow proportionately more release of Aβ42 before further trimming to Aβ39. The net result might be less total Aβ, including less Aβ40 (Bentahir et al, 2006), but the ratio of Aβ42 to Aβ40 is elevated. In cases in which the proteolytic efficiency of the enzyme is only slightly reduced, the resulting increase in substrate levels might ultimately lead to compensation and little change in total Aβ; nevertheless, the reduced efficiency would cause an increase in Aβ42/Aβ40. This same mechanism might also account for the changes in Aβ42/Aβ40 seen with Alzheimer-causing APP mutations that are located near the γ-secretase cleavage sites; in these cases, the mutant substrates might be processed less efficiently by the wild-type protease.
The scenario described above, which is supported by numerous reports, also provides an explanation for why the deletion of three out of four presenilin alleles in mice—with only one PS1 allele remaining—does not result in elevated Aβ42/Aβ40 (Lai et al, 2003; although see Refolo et al, 1999): the remaining γ-secretase complexes are wild type, have normal proteolytic activity and trim ε-cleaved Aβ efficiently. This is also consistent with the fact that although more than 100 missense mutations in PS1 and PS2 have so far been associated with AD, none are complete loss-of-function mutations in an allele, for example, complete deletion, loss of expression, mutation of one of the catalytic aspartates or severe truncations. Conditional knockout of presenilins in the brain can apparently result in memory impairment and age-dependent neurodegeneration (Saura et al, 2004); however, without either Aβ-containing plaques or tau-containing tangles, this is arguably not a phenocopy of AD. Apparently, presenilin is crucial for neuronal survival and/or replacement in the brain, and complete knockout of both alleles has serious consequences. However, many other genes that are unrelated to AD but are also needed for maintaining neuronal density would be expected to do the same on complete knockout in the adult brain.
As discussed earlier, the presenilin-containing γ-secretase complex processes many other type I integral membrane proteins besides APP. Could reduced or altered proteolysis of one of these other substrates be the true underlying molecular cause of AD, with reduced or altered APP proteolysis being an epiphenomenon? Could the AD-causing mutations in APP be only indirectly pathogenic by tying up more γ-secretase complexes and reducing the proteolysis of another key substrate? This possibility remains real but unlikely, unless AD-causing mutations are found in one of these alternative substrates with concomitant tau pathology but no change in either the level of Aβ or the ratio of Aβ42 to Aβ40. So, although reduced and/or altered processing of other substrates is often observed with mutant presenilins—and might exacerbate the AD phenotype in some cases—suggestions that such alterations might be the primary cause of these familial AD cases are speculative. These alterations—for example, reduced Notch signalling—might nevertheless be secondary contributors to neurodegeneration (Beglopoulos & Shen, 2006).
Finally, the emerging role of soluble Aβ assemblies in synaptic dysfunction and neuronal loss should be mentioned (Selkoe, 2002). A major objection to the amyloid hypothesis—with consequent searches for other reasons why presenilin mutations cause AD—is the lack of correlation between amyloid plaque counts with disease severity or onset. However, a clear correlation is observed under complete solubilization of Aβ from post-mortem brains using formic acid (Naslund et al, 2000). Moreover, soluble Aβ oligomers, but not monomers, secreted from cells in culture have substantial effects on long-term potentiation after their injection into the brains of living mice. Therefore, even subtle changes in Aβ levels or in the Aβ42/Aβ40 ratio might lead to soluble aggregates that are able to cause AD during the course of years or decades.
Some presenilin mutations might cause AD by simply decreasing Aβ40 as a net effect on Aβ production (Bentahir et al, 2006). Other evidence also indicates that Aβ40 might be protective to the degree that this shorter peptide helps to keep Aβ42 from aggregating (T.E. Golde, personal communication). Such evidence should not be taken to mean that γ-secretase inhibition will cause or exacerbate AD: inhibition should reduce Aβ42 as well as Aβ40, and therefore prevent Aβ aggregation and downstream toxic effects. Similarly, compounds that selectively decrease Aβ42, such as certain non-steroidal anti-inflammatory drugs (Weggen et al, 2001), should be beneficial. However, compounds that selectively lower Aβ40 would have the potential to promote Aβ42 aggregation. No such compounds have been reported so far, and any that are identified in the future should be avoided as therapeutic candidates. Inhibition of the presenilin-containing γ-secretase complex might indeed lead to mechanism-based toxic effects, for example, through reduced Notch signalling; however, the recent discovery of selective γ-secretase modulators that inhibit the processing of APP while allowing Notch processing to continue (Fraering et al, 2005; Netzer et al, 2003) promises to be an important therapeutic approach. Efforts to identify Aβ-decreasing agents that work at the level of γ-secretase should be vigorously pursued. Arguments to the contrary, on the basis of a simplistic idea of presenilin loss of function in AD pathogenesis, might impede the discovery and development of life-saving medicines that considerably slow or stop disease progression.


Michael S. Wolfe
Acknowledgments
M.S.W. is supported by the National Institutes of Health, the Alzheimer's Association and the Foundation for Neurologic Diseases. The author thanks T. Sato, H. Laudon and P. Osenkowski for critical reading of the manuscript.
References
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK (2004) PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J 23: 2586–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglopoulos V, Shen J (2006) Regulation of CRE-dependent transcription by presenilins: prospects for therapy of Alzheimer's disease. Trends Pharmacol Sci 27: 33–40 [DOI] [PubMed] [Google Scholar]
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, De Strooper B (2006) Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J Neurochem 96: 732–742 [DOI] [PubMed] [Google Scholar]
- Borchelt DR et al. (1996) Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron 17: 1005–1013 [DOI] [PubMed] [Google Scholar]
- Citron M et al. (1997) Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med 3: 67–72 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390 [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C (2003) Reconstitution of γ-secretase activity. Nat Cell Biol 5: 486–488 [DOI] [PubMed] [Google Scholar]
- Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai J-Y, Rahmati T, Xia W, Selkoe DJ, Wolfe MS (2000) Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat Cell Biol 2: 428–434 [DOI] [PubMed] [Google Scholar]
- Esler WP, Wolfe MS (2001) A portrait of Alzheimer secretases: new features and familiar faces. Science 293: 1449–1454 [DOI] [PubMed] [Google Scholar]
- Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, Van Dorsselaer A, Wang R, Selkoe DJ, Wolfe MS (2004) Purification and characterization of the human γ-secretase complex. Biochemistry 43: 9774–9789 [DOI] [PubMed] [Google Scholar]
- Fraering PC, Ye W, Lavoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS (2005) γ-secretase substrate selectivity can be modulated directly via interaction with a nucleotide binding site. J Biol Chem 280: 41987–41996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R et al. (2002) aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev Cell 3: 85–97 [DOI] [PubMed] [Google Scholar]
- Fukumori A et al. (2006) Presenilin-dependent γ-secretase on plasma membrane and endosomes is functionally distinct. Biochemistry 45: 4907–4914 [DOI] [PubMed] [Google Scholar]
- Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, Ihara Y (2004) Truncated carboxyl-terminal fragments of β-amyloid precursor protein are processed to amyloid β-proteins 40 and 42. Biochemistry 43: 13532–13540 [DOI] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR (2002) APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99: 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B (2000) Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol 2: 461–462 [DOI] [PubMed] [Google Scholar]
- Huppert SS, Ilagan MX, De Strooper B, Kopan R (2005) Analysis of Notch function in presomitic mesoderm suggests a γ-secretase-independent role for presenilins in somite differentiation. Dev Cell 8: 677–688 [DOI] [PubMed] [Google Scholar]
- Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, Ihara Y (2006) Equimolar production of amyloid β-protein and amyloid precursor protein intracellular domain from β-carboxyl-terminal fragment by γ-secretase. J Biol Chem 281: 14776–14786 [DOI] [PubMed] [Google Scholar]
- Kang D, Soriano S, Xia X, Eberhart C, De Strooper B, Zheng H, Koo E (2002) Presenilin couples the paired phosphorylation of β-catenin independent of axin. Implications for β-catenin activation in tumorigenesis. Cell 110: 751. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ (2003) γ-secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc Natl Acad Sci USA 100: 6382–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX (2004) γ-Secretase: proteasome of the membrane? Nat Rev Mol Cell Biol 5: 499–504 [DOI] [PubMed] [Google Scholar]
- Lai MT et al. (2003) Presenilin-1 and presenilin-2 exhibit distinct yet overlapping γ-secretase activities. J Biol Chem 278: 22475–22481 [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E et al. (1995) Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269: 973–977 [DOI] [PubMed] [Google Scholar]
- Lewis PA, Perez-Tur J, Golde TE, Hardy J (2000) The presenilin 1 C92S mutation increases Aβ 42 production. Biochem Biophys Res Commun 277: 261–263 [DOI] [PubMed] [Google Scholar]
- Li YM et al. (2000) Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: 689–694 [DOI] [PubMed] [Google Scholar]
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H (2002) Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Aβ 42 production. Proc Natl Acad Sci USA 99: 8025–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000) Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 283: 1571–1577 [DOI] [PubMed] [Google Scholar]
- Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P (2003) Gleevec inhibits β-amyloid production but not Notch cleavage. Proc Natl Acad Sci USA 100: 12444–12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi M, Steiner H, Fukumori A, Tanii H, Tomita T, Tanaka T, Iwatsubo T, Kudo T, Takeda M, Haass C (2002) Presenilins mediate a dual intramembranous γ-secretase cleavage of Notch-1. EMBO J 21: 5408–5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y (2005) Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J Neurosci 25: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refolo LM, Eckman C, Prada CM, Yager D, Sambamurti K, Mehta N, Hardy J, Younkin SG (1999) Antisense-induced reduction of presenilin 1 expression selectively increases the production of amyloid β42 in transfected cells. J Neurochem 73: 2383–2388 [DOI] [PubMed] [Google Scholar]
- Rogaev EI et al. (1995) Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376: 775–778 [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A et al. (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38: 24–26 [DOI] [PubMed] [Google Scholar]
- Sato T et al. (2003) Potential link between amyloid β-protein 42 and C-terminal fragment γ 49-99 of β-amyloid precursor protein. J Biol Chem 278: 24294–24301 [DOI] [PubMed] [Google Scholar]
- Sato T, Tanimura Y, Hirotani N, Saido TC, Morishima-Kawashima M, Ihara Y (2005) Blocking the cleavage at midportion between γ- and ε-sites remarkably suppresses the generation of amyloid β-protein. FEBS Lett 579: 2907–2912 [DOI] [PubMed] [Google Scholar]
- Saura CA et al. (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42: 23–36 [DOI] [PubMed] [Google Scholar]
- Scheuner D et al. (1996) Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2: 864–870 [DOI] [PubMed] [Google Scholar]
- Schroeter EH et al. (2003) A presenilin dimer at the core of the γ-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci USA 100: 13075–13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1994) Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Biol 10: 373–403 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789–791 [DOI] [PubMed] [Google Scholar]
- Sherrington R et al. (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375: 754–760 [DOI] [PubMed] [Google Scholar]
- Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA (1999) Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci USA 96: 6959–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T (2003) The role of presenilin cofactors in the γ-secretase complex. Nature 422: 438–441 [DOI] [PubMed] [Google Scholar]
- Tolia A, Chavez-Gutierrez L, De Strooper B (2006) Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the γ-secretase complex. J Biol Chem 14: 14. [DOI] [PubMed] [Google Scholar]
- Weggen S et al. (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414: 212–216 [DOI] [PubMed] [Google Scholar]
- Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G (2002) A novel var ε-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41: 2825–2835 [DOI] [PubMed] [Google Scholar]
- Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B (2002) Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296: 2215–2218 [DOI] [PubMed] [Google Scholar]
- Wolfe MS (2006) Shutting down Alzheimer's. Sci Am 294: 72–79 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Donkor IO, Selkoe DJ (1999a) Peptidomimetic probes and molecular modeling suggest Alzheimer's γ-secretases are intramembrane-cleaving aspartyl proteases. Biochemistry 38: 4720–4727 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, De Los Angeles J, Miller DD, Xia W, Selkoe DJ (1999a) Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer's disease. Biochemistry 38: 11223–11230 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999c) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398: 513–517 [DOI] [PubMed] [Google Scholar]
- Yagishita S, Morishima-Kawashima M, Tanimura Y, Ishiura S, Ihara Y (2006) DAPT-induced intracellular accumulations of longer amyloid β-proteins: further implications for the mechanism of intramembrane cleavage by γ-secretase. Biochemistry 45: 3952–3960 [DOI] [PubMed] [Google Scholar]
- Yu G et al. (2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 407: 48–54 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner BA (2000) Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat Cell Biol 2: 463–465 [DOI] [PubMed] [Google Scholar]