Significant Impact of Promoter Hypermethylation and the 540 C>T Polymorphism of CDKN2A in Cutaneous Melanoma of the Vertical Growth Phase (original) (raw)
Abstract
Promoter hypermethylation, mutations, and loss of heterozygosity in the CDKN2A gene as well as polymorphisms at the 3′-untranslated region were determined in vertical growth phase melanomas. Methylation-specific polymerase chain reaction in soluti and in situ showed that 19% of the cases were hypermethylated at the CDKN2A promoter region, and some of these cases were heterogeneous with both methylated and unmethylated tumor cells. Methylation was associated with increased tumor cell proliferation by Ki-67 expression (P = 0.01) and decreased patient survival (P = 0.025). Point mutations in CDKN2A were found in 4% of the cases, whereas 90% had loss of heterozygosity at one or more of 4 markers studied. Furthermore, presence of the 540 C>T polymorphism at the 3′-untranslated region of CDKN2A (23%) was associated with improved survival in multivariate analysis (hazard ratio, 2.6; P = 0.02). Our results suggest that promoter methylation of the CDKN2A gene is present in a subgroup of the tumors and associated with increased tumor cell proliferation and reduced survival. Further, the 540 C>T polymorphism might define a distinct subgroup of low-grade vertical growth phase melanomas. These findings support a significant role of the CDKN2A gene in melanoma progression.
The tumor suppressor gene CDKN2A (p16), which is located at chromosome 9p21 1 and encodes two cell cycle inhibitors, p16INK4a and p14ARF, from a partially shared genomic sequence, 2 is frequently altered in several tumor types. 3-5 Thus, p16 is inactivated by germline mutations in about 20% of melanoma-prone families studied, 6 and has been regarded a melanoma susceptibility gene. 7,8 In sporadic melanomas, various genetic alterations of p16 have been reported, such as point mutations (0 to 26%), promoter methylation (0 to 10%), and homozygous deletions (6 to 25%). 9 Point mutations are rare in most studies (∼7%), although they have been reported in up to 26% of the cases. 10
Hypermethylation of the p16 promoter is frequent in several cancer types, 11 being associated with gene silencing. 12 Only a few small studies report on its presence in melanomas, 13-16 and the frequency seems to be <10%. In contrast, loss of heterozygosity (LOH) at chromosome 9p21 has been reported in 47 to 71%. 10,15,17-19 The _CDKN2A_ gene also carries polymorphisms, and the two most common sites (500 C>G and 540 C>T) are located within the 3′-untranslated region (3′-UTR). 20 The functional importance of these polymorphisms is not known.
In a recent study, we found that loss of p16 expression in 45% of vertical growth phase melanomas was associated with significantly increased tumor cell proliferation and reduced survival, indicating a role of p16 protein as an important cell cycle regulator. 21 These findings are in line with studies on other tumor types, eg, pancreatic carcinoma, 22 squamous cell carcinoma of the esophagus, 23 lung carcinoma, 24 and leukemia. 25 The aim of our present study was to determine the frequency and relative importance of CDKN2A promoter hypermethylation, point mutations, LOH, and polymorphisms with respect to p16 protein expression and patient prognosis in this consecutive series of vertical growth phase cutaneous melanoma.
Materials and Methods
Patients and Follow-Up
The patient material of this series is described in detail elsewhere. 21 Briefly, 202 vertical growth phase melanomas occurring during 1981 to 1997 were initially included. The presence of a vertical growth phase, and the lack of a radial growth phase, ie, adjacent in situ or microinvasive component, were used as inclusion criteria for the present study. 26 In addition, 68 separate biopsies of local (skin, n = 17), regional (lymph nodes, n = 44), or distant (n = 7) metastases from 58 patients with recurrent disease were available for analyses. The hospital records were used to obtain follow-up information with respect to recurrences. Information about cause and date of death was obtained from the Cancer Registry of Norway and Statistics Norway. Complete information on patient survival, and time and cause of death was available in all 202 cases, and 69 patients died during follow-up. Last date of follow-up was December 18th, 1998, and median follow-up time for all survivors (total survival) was 76 months (range, 13 to 210 months).
Clinicopathological Variables
The following variables were recorded: date of histological diagnosis, sex, age at diagnosis, anatomical site of the primary tumor, and presence of local (skin), regional (lymph node), or distant metastases at diagnosis. The hematoxylin and eosin (H&E) slides were re-examined, and the following histological features were also included:tumor thickness according to Breslow, 27 level of invasion according to Clark, 28 microscopic ulceration, and vascular invasion (each case was closely examined using both H&E- and factor VIII-stained slides).
Immunohistochemistry
As previously described in detail, 21 staining was performed on formalin-fixed and paraffin-embedded archival tissue. Thin sections (5 μm) from 190 cases with sufficient tumor material left in the tissue blocks, were incubated with the rabbit polyclonal p16 antibody SC-468 (Santa-Cruz Biotechnology, Santa Cruz, CA) at 1:200 overnight in room temperature. Negative controls were incubated with the antibody and blocking peptide (SC-468P). The p53 staining protocol included microwave antigen retrieval (10 minutes at 750 W and 3 × 5 minutes at 500 W) and incubation for 1 hour (room temperature) with the DO-7 monoclonal antibody (code no. M-700; DAKO, Copenhagen, Denmark) diluted 1:100. After microwave antigen retrieval (10 minutes at 750 W and 4 × 5 minutes at 500 W), the sections were incubated for 1 hour (room temperature) with the polyclonal Ki-67 antibody (code no. A-047, DAKO) diluted 1:50.
Immunohistochemical staining of the p16 and p53 proteins was recorded as previously described, 21 considering both the staining intensity and proportion of positive tumor cells. (Intensity: 0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining. Area: 0, no positive tumor cells; 1, < 10% positive tumor cells; 2, 10 to 50% positive tumor cells; 3, > 50% positive tumor cells. Staining index: staining intensity times positive area.) The Ki-67 staining was assessed according to the approach of Weidner and colleagues. 29 Briefly, the tumors were scanned at low magnification (×40 and ×100) to identify the areas of most intense nuclear staining (hot spots). The percentage of immunoreactive tumor cell nuclei (proliferative rate) was then calculated by counting at least 500 cells at ×1000 within the selected areas.
Solution-Phase Methylation-Specific Polymerase Chain Reaction (MSP)
DNA was isolated as previously described, 20 and isolated DNA from 81 primary tumors and 12 metastases were modified by the CpGenome DNA Modification Kit and amplified by the CpGWIZ Amplification Kit (Intergen Company, New York, NY) according to the recommendations of the manufacturer. These 81 cases did not differ significantly from the rest of the patients in this series with respect to standard clinicopathological variables and survival.
The protocol used is based on the method described by Herman and colleagues. 30 Modified DNA was amplified with primers specific for unmethylated DNA (U-primers) and methylated DNA (M-primers), and the efficiency of the bisulfite modification was determined by primers specific for the wild-type p16 promoter region (W-primers). All cases analyzed were amplified by U-, M-, and W-primers. The products were electrophoresed on a 2% agarose gel containing ethidium bromide and visualized by UV light. Cases not showing amplification with any of the primers (n = 22) were regarded to have insufficient quality of DNA. These cases were analyzed separately with respect to clinicopathological variables and p16 expression to exclude any selection bias, and the DNA quality was also checked for by primers for other genes (p53) under optimal conditions. Positive and negative control DNA were included in the kit.
In Situ MSP and Hybridization
Cases showing a mixed pattern with bands after amplification with both U-primers and M-primers (n = 11) were analyzed by in situ MSP, as described by Nuovo and colleagues 31 with some modifications. After dewaxing, the sections were subjected to a 15-minute microwave treatment in citric buffer, followed by a mild treatment with Proteinase K (10 μg/ml for 10 minutes). The tissue morphology was significantly better preserved than after Proteinase K treatment alone. DNA was denatured by 0.4 mol/L of NaOH at 37°C followed by an overnight incubation with 3 mol/L of sodium bisulfite at 55°C in Gene Frames (Advanced Biotechnologies, Surrey, UK), and the modification was completed by a 5-minute incubation with 0.4 mol/L of NaOH. The amplification solution contained 1× polymerase chain reaction (PCR) buffer II (Applied Biosystems, Foster City, CA); 3.5 mmol/L MgCl; 200 μmol/L dNTP; 1 μmol/L U-, M-, or W-primers; 0.1 U/μl Amplitac Gold polymerase (Applied Biosystems, Foster City, CA) and 0.1% bovine serum albumin was applied in Gene Frames of 65 μl. After denaturation at 94°C for 7 minutes, 40 cycles of PCR were performed on the Omnislide Thermal Cycler (Hybaid, Ashford, UK): annealing at 55°C for 2 minutes and denaturing at 94°C for 1 minute.
Digoxygenin-labeled PCR products (U, M, and W) from the solution-phase PCR were used as probes for in situ hybridization. A 100-μl probe cocktail contained 50 μl formamide, 30 μl 25% dextran sulfate, 10 μl 20× standard saline citrate, and 10 μl probes. Target DNA and probes were co-denatured at 95°C for 9 minutes, incubated overnight at 37°C under coverslides, and washed for 10 minutes in 1× standard saline citrate (pH 7.0) at 52°C. The probes were visualized by the DIG Nucleic Acid Detection Kit (Roche, Mannheim, Germany) showing a dark-blue signal in positive cases, and using nuclear fast red as counterstain.
Sections treated with DNase, cases incubated without primers, or cases incubated without probes served as negative controls. All cases were analyzed with the U-, M-, and W-primers and -probes, and the W-probes served as control for the efficiency of the bisulfite treatment.
PCR-SSCP and LOH
Tumor tissue and surrounding normal tissues were dissected from 185 primary tumors with sufficient material left in the tissue blocks. The DNA was isolated separately and screened for polymorphisms at the 3′-UTR of the CDKN2A gene as recently described. 20
In addition, 50 cases were screened for exon 1 to 3 mutations of the CDKN2A gene as reported. 10 The primers used are given in Table 1 ▶ . All shifts detected by single-stranded conformational polymorphism (SSCP) were confirmed by direct sequencing of both the forward and the reverse strand. The analyses were repeated, starting with PCR amplification of DNA. Direct sequencing was performed using a rhodamine dye terminator cycle sequencing kit (Big Dye, Applied Biosystems), and the sequencing reaction products were electrophoresed on a denaturing polyacrylamide gel in an automated sequencer (ABI 377, Applied Biosystems).
Table 1.
Primers Used in PCR Amplification of the Three Exons of the CDKN2A Gene
| Exon | Primer sequences | Annealing temperature | Product size (bp) | |
|---|---|---|---|---|
| 1α | Forward | 5′CGG CTG CGG AGA GGG GGA GAC | 66 | 246 |
| Reverse | 5′CAG CGC CCG CAC CTC CTC TA | |||
| 2 (5′ end) | Forward | 5′GGG CTC TAC ACA AGC TTC CTT | 63 | 277 |
| Reverse | 5′AGC CAG GTC CAC GGG CAG AC | |||
| 2 (3′ end) | Forward | GGG AGG GCT TCC TGG ACA C | 63 | 243 |
| Reverse | TTT GGA AGC TCT CAG GGT ACA | |||
| 3 | Forward | GCC TGT TTT CTT TCT GCC CTC TG | 57 | 144 |
| Reverse | CGA AAG CGG GGT GGG TTG T |
In the same 50 cases, four microsatellite markers on chromosome 9p21, namely D9S736, D9S974, D9S171, and D9S1870, were amplified using primer sequences described in the Genome Data Base. One primer in each set was labeled at the 5′ end with Cy-5 or FAM fluorescent dyes, and PCR products were electrophoresed on 6% denaturing polyacrylamide gel and detected in an automated sequencer (ABI 377). In case of informative markers LOH was scored when a reduction of at least 50% was seen in one allele in DNA isolated from tumor tissue compared with DNA isolated from the corresponding normal tissue.
Statistics
Analyses were performed using the statistical package SPSS. 32 Associations between different categorical variables were assessed by Pearson’s chi-square test. Continuous variables not following the normal distribution were compared between two or more groups using the Mann-Whitney U or Kruskal-Wallis H tests. Univariate analyses of time to death because of malignant melanoma were performed using the product-limit procedure (Kaplan-Meier method), with date of histological diagnosis as the starting point. Differences between categories were tested by the log-rank test. The influence of covariates on patient survival was analyzed by the proportional hazards method. 33 Model assumptions were tested by log-minus-log plots.
Results
p16 Protein Expression
The results of the p16 protein expression analysis, as well as the proliferative rate by Ki-67 expression, are previously reported. 21 Briefly, the immunohistochemical staining pattern for p16 protein was homogeneous throughout the tumor in 75% of the positive cases. Some cases showed a more heterogeneous staining pattern (mosaic type), with groups of positive and negative tumor cells side by side, whereas other cases showed positive and negative areas, such as the case in Figure 1g ▶ ). Of all 190 primary tumors, 45% showed no or minimal nuclear staining of p16 protein (staining index, 0 to 1), compared with 77% for the metastases (Wilcoxon signed rank test, P = 0.001).
Figure 1.
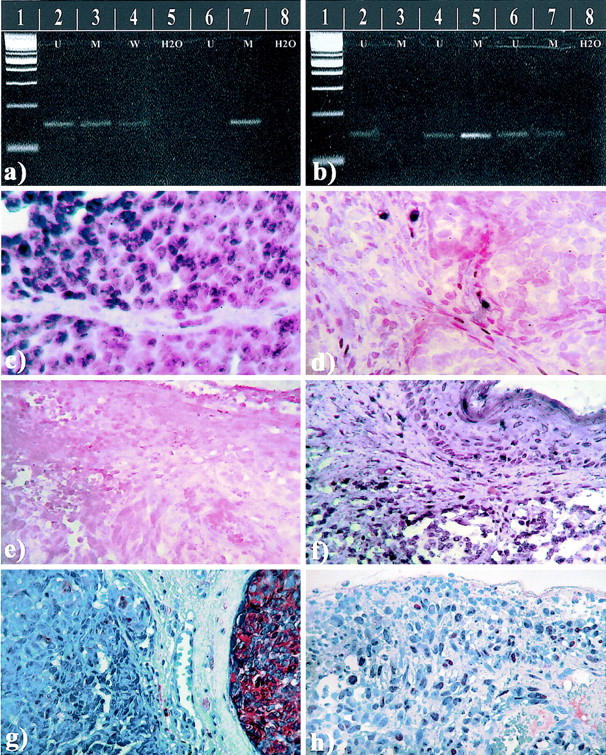
a and b: Solution-phase MSP of the CDKN2A promoter region. a: Lane 1, 100-bp marker; lane 2, unmethylated control; lane 3, methylated control; lane 4, unmodified DNA wild-type control; lane 5, blank; lanes 6 and 7, case 274 positive with the M-primers; lane 8, blank. b: Lane 1, 100-bp marker; lanes 2 and 3, case 276 positive with the U-primers; lanes 4 and 5, case 393 positive with both the U- and M-primers; lanes 6 and 7, case 396 positive with both the U- and M-primers; lane 8, H2O. In situ MSP. c: Case 393 shows nuclear blue and granular positivity with the M-primers/probe. d: The same case with the U-primers/probe, note the positive signals in some stromal cells. e: Case 396 showed no positivity with the M-primers/probe, in contrast to the positive reaction for U-primers/probe shown for tumor tissue and overlying epidermis in f. g: Case 393 shows tumor cell heterogeneity with respect to immunohistochemical staining of the p16 protein. h: Positive nuclear staining of the p16 protein in case 396.
Mean proliferative rate, assessed by Ki-67 expression, was 35% in cases with absent or minimal nuclear p16 staining, compared with 24% in the remaining cases with moderate/strong nuclear p16 expression (_t_-test, P < 0.0001).
Solution-Phase MSP
Fifty-nine primary tumors and all 12 metastases showed reproducible amplification with the U- and/or the M-primers. Forty-three primary tumors (73%) were unmethylated, 11 cases (19%) showed bands with both the U- and M-primers, whereas 5 cases (8%) had positive M-bands only (Figure 1) ▶ . Eight metastases (67%) were unmethylated and four (33%) were methylated at the p16 promoter region (Wilcoxon signed rank test, P = 0.25). All five methylated primary tumors (100%) showed complete loss of p16 protein expression, compared with 24 of the unmethylated (55%) and 5 of the mixed cases (45%) (chi square, P = 0.05). None of the two mutated cases had promoter hypermethylation.
In Situ MSP and Hybridization
To interpret the 11 cases being positive after both methylation- and unmethylation-specific solution-phase PCR, in situ MSP was performed. Apparently, the bisulfite modification was a critical step, and incubations impairing the tissue morphology (NaOH and proteinase K) were necessary to get sufficient DNA modification. However, all mixed cases gave evaluable staining results with blue nuclear grains contrasting the red counterstain (Figure 1) ▶ . Cases positive with the M-probe/primers in tumor cells and the U-probe/primers only in tumor stroma were recorded as methylated (n = 4). Cases with tumor cell positivity for both the U- and M-probe/primers were interpreted as heterogeneous (partial methylation, n = 7) and recorded as predominantly methylated if the M-probe/primer positivity was dominant (n = 2), and predominantly unmethylated if the U-probe/primer positivity was dominant (n = 5). Thus, the cases were regrouped, without knowledge of patient outcome or clinicopathological data, into methylated (n = 11, 19%) and unmethylated (n = 48, 81%) tumors (Figure 2) ▶ .
Figure 2.

Flow chart illustrating the reclassification of cases with bands for both unmethylated and methylated p16 promoter (mixed cases) after in soluti MSP. The mixed cases were subjected to in situ MSP and subsequently reclassified into predominantly unmethylated (U) and predominantly methylated (M). Ht, heterogeneous cases; Hm, homogeneous cases.
After the in situ MSP, there was no significant association between promoter methylation (n = 11) and loss of nuclear p16 staining (P = 0.30). Four of the seven cases with heterogeneous methylation (partial methylation) showed presence of p16 protein expression, and the protein-staining pattern was heterogeneous in these cases as illustrated in Figure 1g ▶ . Methylated cases (n = 11) showed significantly increased proliferative rate by Ki-67 expression (Mann-Whitney test, P = 0.01), and these cases were significantly thicker (Mann-Whitney test, P = 0.004). As illustrated in Figure 1g ▶ , histological heterogeneity was observed in some of the cases with heterogeneous methylation. Still, no particular feature was exclusive to these cases, and four of seven cases did not show histological heterogeneity.
The number of different genetic alterations (promoter methylation, point mutations, LOH, and polymorphisms) was recorded for each case. Two or more alterations, found in 25% of the cases, were significantly associated with loss of p16 protein expression (P < 0.001).
Mutations in CDKN2A
In total, 50 primary melanomas were screened, and sequence changes were detected in two cases (4%). Case 364: codon 99 (exon 2) CGG>CGA (Arg>Arg) for p16, and codon 114 (exon 2) GGC>AGC (Gly>Ser) for _p14_ARF. Case 453: codon 55 (exon 2) GGC>CGC (Gly>Arg) for p16, and codon 69 (exon 2) GGG>GCG (Gly>Ala) for _p14_ARF. The mutation for case 453 in p16 is a novel mutation and is at the residue which is conserved in all four CDK inhibitors. 34
LOH
LOH was determined at four different polymorphic markers on chromosome 9p21 in 50 primary melanomas. These markers cover the entire INK4 locus as well as flanking centromeric and telomeric loci on chromosome 9p21. Our data show that 45 cases (90%) had lost an allele at one or more loci, and 3 cases showed loss at all four loci. LOH at the chromosome 9p21 markers ranged from 34 to 56% of informative cases.
Both mutated cases had LOH at D9S974, D9S736, and D9S1870, whereas one had LOH at D9S171. Further, D9S171 was significantly less frequent in cases with Clark’s level 5 of invasion (P = 0.02). None of these cases with LOH at this locus were hypermethylated at the promoter region (P = 0.07), and the same trend was seen for LOH at D9S736 (P = 0.06). LOH at D9S1870 were more often found in ulcerated cases (P = 0.02) and in cases with strong p53 staining (P = 0.02). No significant associations were present between LOH at any of the four markers and p16 protein expression, proliferative rate by Ki-67, tumor thickness, or patient survival.
Polymorphisms in CDKN2A
In total, 185 primary tumors had sufficient material left in the paraffin blocks for the study of polymorphisms in the 3′-UTR of the CDKN2A gene. The frequency of cases that were heterozygous for the T allele in the 540 C>T polymorphism was 23% (n = 42), whereas one case was homozygous. Forty-seven cases (25%) were heterozygous, and three (1.6%) were homozygous for the G allele at the 500 C>G polymorphism.
There was no significant association between either of the polymorphisms and methylation status, LOH, or p16 protein expression. Both mutated cases had the 540 C>T and one had the 500 C>G polymorphism. Further, there was no association with the other clinicopathological variables studied.
Survival Analyses
Significantly reduced survival was present for cases showing absent or minimal nuclear p16 protein expression (log rank test, P = 0.0003), and recurrence-free survival was also significantly decreased in this subgroup of patients (log rank test, P = 0.007).
Estimated 10-year survival for cases regarded as predominantly methylated after in situ MSP (n = 11) was 31%, compared with 74% for unmethylated cases (log rank test_, P_ = 0.025; Figure 3 ▶ ). No significant associations were present between LOH at any of the four markers and patient survival. However, presence of the 540 C>T polymorphism was significantly associated with improved patient survival (log rank test, P = 0.03; Figure 2 ▶ ). No significant survival differences were found with respect to number of genetic alterations (methylation, mutations, LOH, polymorphisms).
Figure 3.
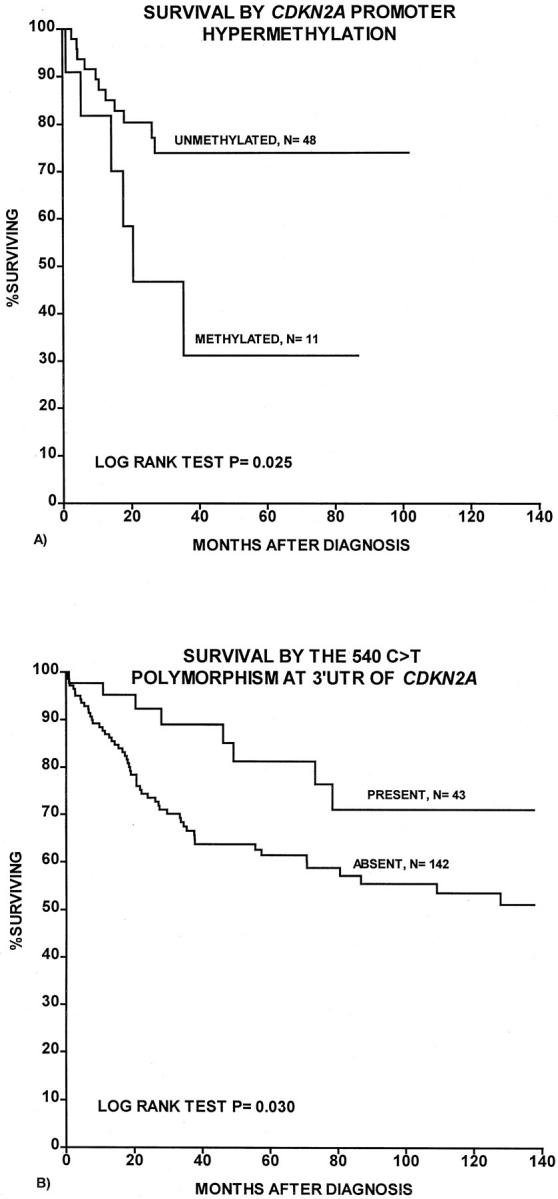
Survival curves were estimated according to the Kaplan-Meier method with death as the result of melanoma as end point. A: Survival by CDKN2A promoter methylation. B: Survival by presence of the 540 C>T polymorphism at 3′-UTR of CDKN2A.
In multivariate analysis according to the proportional hazards method, the positive impact on survival of the 540 C>T polymorphism was still significant (Table 2) ▶ , with a Hazard ratio of 2.6 (L ratio test, P = 0.02) for cases without this polymorphism, whereas promoter methylation of CDKN2A had no independent prognostic importance, possibly because of the low number of cases included in this subset analysis.
Table 2.
Multivariate Survival Analysis According to the Proportional Hazards Method, for Patients with Vertical Growth Phase Melanoma, Using Death from Melanoma as End Point
| Variable | Categories | n | HR* | P value† |
|---|---|---|---|---|
| Level of invasion (Clark) | II, III, IV | 143 | 1 | |
| V | 34 | 5.0 | <0.0001 | |
| Anatomic site | Trunk | 49 | 3.3 | |
| Other | 128 | 1 | <0.0001 | |
| Vascular invasion | Absent | 141 | 1 | |
| Present | 36 | 1.96 | 0.034 | |
| Proliferative rate by Ki-67 expression | Low‡ | 46 | 1 | |
| High | 131 | 4.69 | 0.001 | |
| Microvessel density | Low§ | 120 | 1 | |
| High | 57 | 2.12 | 0.015 | |
| p53 expression | Absent¶ | 27 | 1 | |
| Present | 150 | 19.5 | 0.003 | |
| p16 expression | Absent/low∥ | 80 | 2.31 | |
| Present | 97 | 1 | 0.003 | |
| 540 C > T polymorphism | Absent | 137 | 2.61 | |
| Present | 40 | 1 | 0.019 |
Discussion
Previous studies indicate a major role for CDKN2A (p16) as a tumor suppressor gene in a variety of human cancers, being inactivated by point mutations, promoter methylation, and homozygous deletions. 9 Recent publications have also demonstrated that p16 knockout mice, as well as mice with _p16-_specific mutations or deletions, show an increased incidence of spontaneous and carcinogen-induced cancers including melanoma. 35,36 We previously showed that loss of p16 protein expression was associated with increased tumor cell proliferation and reduced survival in vertical growth phase cutaneous melanoma, 21 supporting that p16 is an important cell cycle regulator in these tumors. However, the mechanisms of inactivation are not completely understood.
In this series of cutaneous melanoma, we found that 19% of the cases were hypermethylated at the promoter region of CDKN2A. This frequency is higher than what has been indicated in a few small studies, 13-16 but lower than recently reported for uveal melanoma (32%). 37 The frequency tended to be increased in corresponding metastases (33%), indicating that p16 hypermethylation might also occur in late tumor progression. Some of the cases were heterogeneous, as also described by others, 38 showing both methylated and unmethylated tumor cells by in situ PCR, similar to the mixed pattern of p16 protein expression observed by immunohistochemistry. 21
The importance of p16 promoter hypermethylation in vertical growth phase melanoma is supported by a significant correlation with increased tumor cell proliferation by Ki-67 expression, indicating loss of cell cycle control. 39 Further, there was a significantly reduced patient survival in methylated cases, suggesting a functional importance for disease progression in these tumors. This is in line with studies of p16 promoter methylation and survival impact in other tumors. 40-43
There was no clear association between promoter methylation and reduced p16 protein expression after in situ MSP. This may, in part, be because of the low number of cases analyzed for methylation. Another reason might be partial or monoallelic methylation and heterozygous tumor cells with varying penetrance of the wild-type allele, similar to what has been described for p27 in melanoma. 44 Also, other mechanisms of inactivation may act in concert with methylation and explain the loss of p16 protein expression. An association between p16 promoter methylation and reduced protein expression has been reported in other cancer types. 12,14,37,45-48
Only two point mutations (4%) were detected in this series, supporting most previous studies that mutational inactivation is not a major mechanism of gene silencing in sporadic primary melanoma. The frequency of mutations varies from 0 to 26% according to recent reports, 10,13,17,49,50 but differences in case selection exist. Still, our data show that 90% of the cases had LOH at one or more markers within the INK4 locus, suggesting that monoallelic loss at 9p21 is frequent and might represent one of two or more events causing reduced expression of p16 and also that of p14ARF. 19 Although LOH has been correlated to reduced p16 protein expression, 15 we found no significant association between LOH at any marker and p16 staining, indicating that monoallelic loss alone may not be sufficient for genetic silencing.
Recent evidence strongly suggests that the 3′-untranslated region of mRNA is involved in regulation of gene expression by controlling nuclear export, polyadenylation status, subcellular targeting, translation rates, and mRNA degradation. 51 We recently found a significant overrepresentation of the heterozygote frequency of the 540 C>T polymorphism of CDKN2A when compared to controls. 20 In our present study, the presence of this polymorphism provided independent prognostic information in multivariate survival analysis, and patients positive for this marker had significantly improved survival. To our knowledge, only Sauroja and colleagues 52 have reported on possible survival differences with respect to this polymorphism, although the analysis was performed on melanoma metastases and not primary tumors. Our present results strongly suggest a role for the 540 C>T polymorphism of CDKN2A in the initiation or early progression of a subset of cutaneous melanomas with less aggressive behavior.
In conclusion, point mutations of CDKN2A seem to be rare in cutaneous melanoma of the vertical growth phase, whereas promoter hypermethylation was present in a significant subgroup of the cases (19%), being associated with increased tumor cell proliferation and significantly reduced patient survival. Further, the presence of one specific polymorphism (540 C>T) at 3′-UTR, which previously was found to be increased in melanoma cases when compared with the general population, was associated with favorable outcome in multivariate analysis, pointing to the possibility that this might represent a distinct subgroup of low-grade vertical growth phase melanoma. Thus, our findings support a significant role of the CDKN2A gene for melanoma progression.
Acknowledgments
We thank Mrs. Gerd Lillian Hallseth and Mr. Bendik Nordanger for excellent technical assistance.
Footnotes
Address reprint requests to Lars A. Akslen, M.D., Ph.D., Department of Pathology, The Gade Institute, Haukeland University Hospital, N-5021 Bergen, Norway. E-mail: lars.akslen@gades.uib.no.
Supported by the Norwegian Cancer Society (contract grant no. D94070) and the Swedish Cancer Society.
References
- 1.Serrano M, Hannon GJ, Beach D: A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366:704-707 [DOI] [PubMed] [Google Scholar]
- 2.Mao L, Merlo A, Bedi G, Shapiro GI, Edwards CD, Rollins BJ, Sidransky D: A novel p16INK4A transcript. Cancer Res 1995, 55:2995-2997 [PubMed] [Google Scholar]
- 3.Kamb A, Gruis NA, Weaver Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, III, Johnson BE, Skolnick MH: A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264:436-440 [DOI] [PubMed] [Google Scholar]
- 4.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA: Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368:753-756 [DOI] [PubMed] [Google Scholar]
- 5.Liggett WH, Jr, Sidransky D: Role of the p16 tumor suppressor gene in cancer. J Clin Oncol 1998, 16:1197-1206 [DOI] [PubMed] [Google Scholar]
- 6.Greene MH: The genetics of hereditary melanoma and nevi. 1998 update. Cancer 1999, 86:2464-2477 [DOI] [PubMed] [Google Scholar]
- 7.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH, Jr, Tucker MA, Dracopoli NC: Germline p16 mutations in familial melanoma. Nat Genet 1994, 8:15-21 [DOI] [PubMed] [Google Scholar]
- 8.Piepkorn MW: Genetic basis of susceptibility to melanoma. J Am Acad Dermatol 1994, 31:1022-1039 [DOI] [PubMed] [Google Scholar]
- 9.Rocco JW, Sidransky D: p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp Cell Res 2001, 264:42-55 [DOI] [PubMed] [Google Scholar]
- 10.Kumar R, Lundh Rozell B, Louhelainen J, Hemminki K: Mutations in the CDKN2A (p16INK4a) gene in microdissected sporadic primary melanomas. Int J Cancer 1998, 75:193-198 [DOI] [PubMed] [Google Scholar]
- 11.Herman JG: Hypermethylation of tumor suppressor genes in cancer. Semin Cancer Biol 1999, 9:359-367 [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA: Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 1995, 55:4531-4535 [PubMed] [Google Scholar]
- 13.Fujimoto A, Morita R, Hatta N, Takehara K, Takata M: p16INK4a inactivation is not frequent in uncultured sporadic primary cutaneous melanoma. Oncogene 1999, 18:2527-2532 [DOI] [PubMed] [Google Scholar]
- 14.Gonzalgo ML, Bender CM, You EH, Glendening JM, Flores JF, Walker GJ, Hayward NK, Jones PA, Fountain JW: Low frequency of p16/CDKN2A methylation in sporadic melanoma: comparative approaches for methylation analysis of primary tumors. Cancer Res 1997, 57:5336-5347 [PubMed] [Google Scholar]
- 15.Funk JO, Schiller PI, Barrett MT, Wong DJ, Kind P, Sander CA: p16INK4a expression is frequently decreased and associated with 9p21 loss of heterozygosity in sporadic melanoma. J Cutan Pathol 1998, 25:291-296 [DOI] [PubMed] [Google Scholar]
- 16.von Eggeling F, Werner G, Theuer C, Riese U, Dahse R, Fiedler W, Schimmel B, Ernst G, Karte K, Claussen U, Wollina U: Analysis of the tumor suppressor gene p16(INK4A) in microdissected melanoma metastases by sequencing, and microsatellite and methylation screening. Arch Dermatol Res 1999, 291:474-477 [DOI] [PubMed] [Google Scholar]
- 17.Ohta M, Berd D, Shimizu M, Nagai H, Cotticelli MG, Mastrangelo M, Shields JA, Shields CL, Croce CM, Huebner K: Deletion mapping of chromosome region 9p21–p22 surrounding the CDKN2 locus in melanoma. Int J Cancer 1996, 65:762-767 [DOI] [PubMed] [Google Scholar]
- 18.Healy E, Rehman I, Angus B, Rees JL: Loss of heterozygosity in sporadic primary cutaneous melanoma. Genes Chromosom Cancer 1995, 12:152-156 [DOI] [PubMed] [Google Scholar]
- 19.Kumar R, Smeds J, Lundh Rozell B, Hemminki K: Loss of heterozygosity at chromosome 9p21 (INK4–p14ARF locus): homozygous deletions and mutations in the p16 and p14ARF genes in sporadic primary melanomas. Melanoma Res 1999, 9:138-147 [DOI] [PubMed] [Google Scholar]
- 20.Kumar R, Smeds J, Berggren P, Straume O, Rozell BL, Akslen LA, Hemminki K: A single nucleotide polymorphism in the 3′ untranslated region of the CDKN2A gene is common in sporadic primary melanomas but mutations in the CDKN2B, CDKN2C, CDK4 and p53 genes are rare. Int J Cancer 2001, 95:388-393 [DOI] [PubMed] [Google Scholar]
- 21.Straume O, Sviland L, Akslen LA: Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin Cancer Res 2000, 6:1845-1853 [PubMed] [Google Scholar]
- 22.Bartsch D, Shevlin DW, Callery MP, Norton JA, Wells SA, Jr, Goodfellow PJ: Reduced survival in patients with ductal pancreatic adenocarcinoma associated with CDKN2 mutation. J Natl Cancer Inst 1996, 88:680-682 [DOI] [PubMed] [Google Scholar]
- 23.Takeuchi H, Ozawa S, Ando N, Shih CH, Koyanagi K, Ueda M, Kitajima M: Altered p16/MTS1/CDKN2 and cyclin D1/PRAD-1 gene expression is associated with the prognosis of squamous cell carcinoma of the esophagus. Clin Cancer Res 1997, 3:2229-2236 [PubMed] [Google Scholar]
- 24.Kawabuchi B, Moriyama S, Hironaka M, Fujii T, Koike M, Moriyama H, Nishimura Y, Mizuno S, Fukayama M: p16 inactivation in small-sized lung adenocarcinoma: its association with poor prognosis. Int J Cancer 1999, 84:49-53 [DOI] [PubMed] [Google Scholar]
- 25.Yamada Y, Hatta Y, Murata K, Sugawara K, Ikeda S, Mine M, Maeda T, Hirakata Y, Kamihira S, Tsukasaki K, Ogawa S, Hirai H, Koeffler HP, Tomonaga M: Deletions of p15 and/or p16 genes as a poor-prognosis factor in adult T-cell leukemia. J Clin Oncol 1997, 15:1778-1785 [DOI] [PubMed] [Google Scholar]
- 26.Elder DE, Murphy GF: Melanocytic tumors of the skin. Rosai J Sobin LH eds. Atlas of Tumor Pathology. 1991:pp 119-131 AFIP, Washington DC
- 27.Breslow A: Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann Surg 1970, 172:902-908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark WJ, From L, Bernardino E: The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res 1969, 29:705-727 [PubMed] [Google Scholar]
- 29.Weidner N, Moore DH, Jr, Vartanian R: Correlation of Ki-67 antigen expression with mitotic figure index and tumor grade in breast carcinomas using the novel “paraffin”-reactive MIB1 antibody [see comments]. Hum Pathol 1994, 25:337-342 [DOI] [PubMed] [Google Scholar]
- 30.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nuovo GJ, Plaia TW, Belinsky SA, Baylin SB, Herman JG: In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci USA 1999, 96:12754-12759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norusis M: SPSS Advanced Statistics 6.1. 1994. Chicago, IL, SPSS Inc.
- 33.Cox DR: Regression models and life-tables. J R Stat Soc 1972, 34:187-222 [Google Scholar]
- 34.Ruas M, Peters G: The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta 1998, 1378:115-177 [DOI] [PubMed] [Google Scholar]
- 35.Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A: Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413:83-86 [DOI] [PubMed] [Google Scholar]
- 36.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA: Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413:86-91 [DOI] [PubMed] [Google Scholar]
- 37.van der Velden PA, Metzelaar-Blok JA, Bergman W, Monique H, Hurks H, Frants RR, Gruis NA, Jager MJ: Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res 2001, 61:5303-5306 [PubMed] [Google Scholar]
- 38.Suh SI, Cho JW, Baek WK, Suh MH, Carson DA: Lack of mutation at p16INK4A gene but expression of aberrant p16INK4A RNA transcripts in human ovarian carcinoma. Cancer Lett 2000, 153:175-182 [DOI] [PubMed] [Google Scholar]
- 39.Foster SA, Wong DJ, Barrett MT, Galloway DA: Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol 1998, 18:1793-1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim DH, Nelson HH, Wiencke JK, Zheng S, Christiani DC, Wain JC, Mark EJ, Kelsey KT: p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res 2001, 61:3419-3424 [PubMed] [Google Scholar]
- 41.Esteller M, Gonzalez S, Risques RA, Marcuello E, Mangues R, Germa JR, Herman JG, Capella G, Peinado MA: K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J Clin Oncol 2001, 19:299-304 [DOI] [PubMed] [Google Scholar]
- 42.Liang JT, Chang KJ, Chen JC, Lee CC, Cheng YM, Hsu HC, Wu MS, Wang SM, Lin JT, Cheng AL: Hypermethylation of the p16 gene in sporadic T3N0M0 stage colorectal cancers: association with DNA replication error and shorter survival. Oncology 1999, 57:149-156 [DOI] [PubMed] [Google Scholar]
- 43.Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ, Baylin SB: Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res 1997, 57:837-841 [PubMed] [Google Scholar]
- 44.Worm J, Bartkova J, Kirkin AF, Straten P, Zeuthen J, Bartek J, Guldberg P: Aberrant p27Kip1 promoter methylation in malignant melanoma. Oncogene 2000, 19:5111-5115 [DOI] [PubMed] [Google Scholar]
- 45.Tanaka H, Fujii Y, Hirabayashi H, Miyoshi S, Sakaguchi M, Yoon HE, Matsuda H: Disruption of the RB pathway and cell-proliferative activity in non-small-cell lung cancers. Int J Cancer 1998, 79:111-115 [DOI] [PubMed] [Google Scholar]
- 46.Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD: Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res 2001, 61:249-255 [PubMed] [Google Scholar]
- 47.Garcia JF, Villuendas R, Algara P, Saez AI, Sanchez-Verde L, Martinez-Montero JC, Martinez P, Piris MA: Loss of p16 protein expression associated with methylation of the p16INK4A gene is a frequent finding in Hodgkin’s disease. Lab Invest 1999, 79:1453-1459 [PubMed] [Google Scholar]
- 48.Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W, Ahrendt S, Eby Y, Sewell D, Nawroz H, Bartek J, Sidransky D: High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 1996, 56:3630-3633 [PubMed] [Google Scholar]
- 49.Healy E, Sikkink S, Rees JL: Infrequent mutation of p16INK4 in sporadic melanoma [see comments]. J Invest Dermatol 1996, 107:318-321 [DOI] [PubMed] [Google Scholar]
- 50.Piccinin S, Doglioni C, Maestro R, Vukosavljevic T, Gasparotto D, D’Orazi C, Boiocchi M: p16/CDKN2 and CDK4 gene mutations in sporadic melanoma development and progression. Int J Cancer 1997, 74:26-30 [DOI] [PubMed] [Google Scholar]
- 51.Conne B, Stutz A, Vassalli JD: The 3′ untranslated region of messenger RNA: a molecular ‘hotspot’ for pathology? Nat Med 2000, 6:637-641 [DOI] [PubMed] [Google Scholar]
- 52.Sauroja I, Smeds J, Vlaykova T, Kumar R, Talve L, Hahka-Kemppinen M, Punnonen K, Jansen CT, Hemminki K, Pyrhonen S: Analysis of G(1)/S checkpoint regulators in metastatic melanoma. Genes Chromosom Cancer 2000, 28:404-414 [DOI] [PubMed] [Google Scholar]