SNPSTRs: Empirically Derived, Rapidly Typed, Autosomal Haplotypes for Inference of Population History and Mutational Processes (original) (raw)
Abstract
Each independently evolving segment of the genomes of a sexually reproducing organism has a separate history reflecting part of the evolutionary history of that organism. Uniparentally or clonally inherited DNA segments such as the mitochondrial and chloroplast genomes and the nonrecombining portion of the Y chromosome have provided, to date, most of the known data regarding compound haplotypic variation within and among populations. These comparatively small segments include numerous polymorphic sites and undergo little or no recombination. Recombining autosomes, however, comprise the major repository of genetic variation. Technical challenges and recombination have limited large-scale application of autosomal haplotypes. We have overcome this barrier through development of a general approach to the assessment of short autosomal DNA segments. Each such segment includes one or more single nucleotide polymorphisms (SNPs) and exactly one short tandem repeat (STR) locus. With dramatically different mutation rates, these two types of genetic markers provide complementary evolutionary information. We call the combination of a SNP and a STR polymorphism a SNPSTR, and have developed a simple, rapid method for empirically determining gametic phase for double and triple heterozygotes. Here, we illustrate the approach with two SNPSTR systems. Although even one system provides insight into population history, the power of the approach lies in combining results from multiple SNPSTR systems.
[Supplemental material is available online at http://www.genome.org. The following individual kindly provided reagents, samples, or unpublished information as indicated in this paper: L. Luca Cavelli-Sforza.]
In 1996, researchers reported the global pattern of haplotype frequency variation and linkage disequilibrium (LD) for a pair of linked genetic markers on human chromosome 12 (Tishkoff et al. 1996). The pattern, the authors concluded, provides evidence in support of a common and recent African origin for all non-African human populations. In reaching this conclusion, the authors took advantage of the different mutation rates of the two linked markers. One of the markers is a rapidly evolving short tandem repeat (STR, or microsatellite) locus, whereas the other is a partial deletion of an ancient Alu retroposon insertion, a unique event. That report served to demonstrate the evolutionary information content of autosomal haplotypes composed of different classes of markers.
Other research groups investigating human evolutionary history have studied linked sets of genetic markers with very different mutation rates on the nonrecombining portion of the Y chromosome (NRY) (e.g., Ruiz-Linares et al. 1999; Gresham et al. 2001; Nebel et al. 2001). Compound single nucleotide polymorphism (SNP) and STR haplotypes on the NRY provide powerful tools for inferring the histories of populations (de Knijff 2000). For example, estimates of the age of the SNP have been inferred from the STR diversity of SNP-defined monophyletic groups (clades or haplogroups) of lineages (e.g., Hurles et al. 1999). Conversely, each SNP provides insight into STR diversity as the SNP defines a clade for which STR diversity may then be interpreted free of independently evolved, homoplastic diversity in other clades (Bosch et al. 1999; Makova et al. 2000).
Although the Y chromosome has proven highly informative, that segment of the human genome reflects only a fraction of human history. The mitochondrial genome, although also highly informative, similarly reflects only a fraction of the history of a species. A more complete history requires information from recombining chromosomes that trace to many ancestors.
Evolutionary histories of individual species would be most accurately elucidated from the combined histories of a large number (>50) of DNA regions (Wall 2000). Within most DNA regions short enough for recombination to be rare (on a geographically global scale), however, few informative SNPs exist. A second complication is that with conventional methods, empirical determination of gametic phase for double heterozygotes requires cloning of PCR products. The expense and time involved limit the numbers of samples or genetic systems that may be studied to the point that population studies based on empirical data are precluded. For these reasons, gene histories have been inferred for a relatively small set of autosomal regions. One such region is MS205 (Rogers et al. 2000), wherein 10 SNPs were identified within 2 kb of a minisatellite array. The relatively large number of SNPs combined with inference of phase through typing of the linked microsatellite made possible inference of the history of the MS205 region. Given current estimates of the average frequency of polymorphism across the genome (∼1 per 1000 np), regions with these characteristics are exceedingly rare. An alternative with wide potential application is to consider a large number of short DNA regions with at least two polymorphisms, one of which is a rapidly evolving STR.
With this goal in mind, we recently undertook development of a set of independent, compound haplotypic systems. We chose to focus on SNPs linked tightly to STR polymorphisms. Such combinations of polymorphisms, which we refer to as SNPSTRs (Fig. 1), satisfy the following three requirements: (1) close physical linkage of two or more polymorphisms; (2) significant difference in mutation rate between polymorphisms; and, (3) potential for a large number of independent compound haplotypic systems.
Figure 1.

Schematic of SNPSTR system depicting double heterozygote autosomal genohaplotype for a diploid organism. In this example, one homolog has a C allele at the SNP and an STR of 21 repeat units. The other homolog has a T allele at the SNP and an STR of 23 repeat units. The C allele is amplified via PCR with an allele-specific primer, with C at the 3′ terminus, and is labeled with the fluorescent dye 6-FAM. The T allele is amplified with an allele-specific primer, with T at the 3′ terminus, and is labeled with the fluorescent dye HEX. Both labeled PCR products are produced with the same reverse primer “r.” As the length of the PCR product varies depending solely upon the repeat number of the STR, and length is determined by fluorescent detection of electrophoretic mobility, the allelic states of the SNP and STR, and gametic phase, are all determined simultaneously by fluorescent electrophoretic fragment analysis.
Close Physical Linkage
The Alu and STR originally studied by Tishkoff et al. (1996) are 9.8 kb apart, and the PLAT locus (Tishkoff et al. 2000b) spans 22 kb. We have considered SNPs and STRs fewer than 400 bases apart in order to facilitate data generation. For such tightly linked markers, we have developed a method to empirically determine the homologous SNP and STR allelic states of each individual, including gametic phase for double heterozygotes, using conventional fluorescent fragment analysis. The result is an individual's genohaplotype, in which a genohaplotype is one diploid individual's pair of haplotypes at a given SNPSTR system. The method described below enables us to accomplish these goals rapidly and cost effectively with the potential to be scaled up via automation. Closely linked markers also simplify interpretation of data by minimizing the number of parameters.
Different Mutation Rates
Each independent SNPSTR system combines a slowly evolving polymorphic locus and a more rapidly evolving polymorphic locus. SNPs mutate at a rate on the order of 2.0–2.5 × 10−8 mutations per nucleotide position per generation (Nachman and Crowell 2000). Although estimates of autosomal STR mutation rates vary widely on the basis of motif type (Chakraborty et al. 1997), an effective mutation rate of ∼1.5 × 10−3 per STR per generation has been obtained for dinucleotide repeat polymorphisms (Zhivotovsky 2001). These different rates enable us to take advantage of the different time scales for which each type of marker is informative. Potentially, each SNP provides information into the history of the linked STR, and each STR provides insight into the history of the linked SNP(s). In the context of molecular evolutionary and population genetic models, we can draw inferences regarding both molecular events and processes (e.g., mutation and recombination) and population history.
Large Number of Independent Systems
We consider a class of linked-marker systems that is large and broadly distributed in the genomes of sexually reproducing species. SNPs and STR polymorphisms are frequent in the genomes of many species. Numerous SNPSTR systems exist whenever recombination decouples the histories of short DNA regions that include SNPs and a STR polymorphism. The independent histories of these short genetic regions can be combined to infer population histories. For any given research question, a particular number of these compound haplotypic systems are required to draw a robust conclusion.
Here, we report our method for developing and screening SNPSTRs. Data obtained for two SNPSTR systems, one on human chromosome 22 and one on human chromosome 5, demonstrate the utility of the method and provide additional evidence of the information content inherent in such compound haplotypes.
RESULTS
Development of New SNPSTR Systems
The Protocol for development of a SNPSTR system consists of a sequence of three steps, summarized as follows.
Step 1
Locate a STR locus and its flanking regions via GenLink (http://mapper.wustl.edu/), GenBank (http://www.ncbi.nlm.nih.gov/), or another database. STRs with a larger number of repeat units are more likely to be informative of evolutionary history; STRs with fewer than 10 repeat units often reveal little variation. STRs with perfect repeats (relatively common in the human genome) are optimal as the evolution (specifically, mutational mechanisms and rates) of imperfect STRs is less well understood (Macaubas et al. 1997).
Step 2
Determine whether at least one SNP is present within ∼400 bp of the STR on either or both upstream and downstream flanking regions. We design PCR primers from the STR-flanking regions available in GenBank clones. These primers are designed to encompass a target from as close as possible to the STR (within constraints of primer design) to ∼400 bp away from the STR, for both upstream and downstream flanking regions. The amplified flanking regions are then examined for SNPs using a rapid, inexpensive screening method such as denaturing high-performance liquid chromatography (DHPLC, Oefner and Underhill 1998) or SSCP analysis (Orita et al. 1989). The appropriate set of samples used for SNP discovery (the screening set) depends on the nature of the scientific questions to be addressed. To minimize ascertainment bias (Mountain et al. 1994; Wakely et al. 2001) and to maximize the applicability of SNPSTR systems, we use a global screening set composed of individual samples representing geographically and linguistically diverse human groups. Putative polymorphic samples are sequenced to determine the basis of apparent polymorphism.
Step 3
After discovering a SNP, design allele-specific primers, each labeled with a different fluorescent dye (Fig. 1). Design an unlabeled reverse primer across the STR region to produce labeled PCR product with either of the fluorescent allele-specific primers. The fluorescently labeled PCR product then encompasses the SNP at one end and the STR at the other. The length of the PCR product varies among chromosomes, depending solely on the copy number of repeats at the STR. Once these primers and corresponding PCR parameters are optimized, determine the genohaplotypes of each individual by electrophoresis on a genetic analysis instrument using fluorescent detection (Fig. 2). As with any electrophoretic fragment analysis, for each SNPSTR locus at least one individual PCR product, homozygous at the STR, must also be sequenced for comparison with the fragment analysis to ensure accurate determination of repeat number of STR alleles by fragment mobility.
Figure 2.

GeneScan (Applied Biosystems) analysis of chromosome 22 SNPSTR system 22SR1, a system with a C/T SNP and a CA STR. Detection was performed using a 310 Genetic Analyzer. As in Fig. 1, the C SNP was labeled with 6-FAM (blue). The T SNP was labeled with HEX (green). Size standard HD400 ROX is red. (A) Genohaplotype that is homozygous C at the SNP locus and heterozygous at the STR locus, with repeat numbers of 21 and 23. (B) Doubly homozygous T genohaplotype with a CA repeat number of 22. (C) Double heterozygous genohaplotype depicted schematically in Figure 1. The chromosome with the C SNP has an STR with 21 repeat units. The chromosome with the T allele has an STR with 23 repeat units.
Generation of Labeled Fragments
We developed two procedures for generation of allele-specific fluorescently labeled fragments for electrophoretic determination of SNPSTR genohaplotypes. The first approach involves exponential PCR amplification of labeled fragments starting with genomic sample DNA. Using this exponential approach, for each SNPSTR locus, each individual sample DNA is amplified separately with each allele-specific, fluorescently labeled primer and the same reverse primer (two separate PCR reactions per individual are carried out). Incorporation of a deliberate mismatch one base from the 3′ terminus of allele-specific primers increases specificity. Optimal annealing temperature must be determined experimentally. For some loci, complete specificity is difficult to achieve. If low-frequency amplification of the alternative allele occurs during early cycles, these nonspecific amplicons increase exponentially, so that detectable levels are produced by later cycles. A build-up of nonspecific, labeled fragments then prevents determination of the genohaplotype.
A second approach that circumvents the problem of nonspecific amplification involves linear production of labeled fragments and requires two steps. Consider the case of a SNP upstream of a STR. First, genomic DNA is amplified via PCR with primers that target a region from a forward primer upstream of the SNP to a reverse primer downstream of the STR. Both primers are unlabeled. Successful amplification is confirmed by visualization of an aliquot on an agarose gel. Next, an aliquot (typically 10 μL) of the completed PCR reaction is added as template to an equal volume of a new, second-step reaction mixture. The second-step mixture contains the same PCR reagents as the first reaction, except that only a single, labeled, allele-specific primer is included. No reverse primer is included. Residual reverse primer will not interfere with this step, so no clean-up is needed. With this linear procedure, we have found no need to introduce a deliberate mismatch to enhance specificity; each allele-specific primer is a perfect match to its respective SNP allele. The second step reaction mixture is then subjected to a single cycle of denaturing, annealing, and extension. This linear procedure produces ample labeled allele-specific fragments without exponential build-up of nonspecific fragments. A second advantage is that the method is not sensitive to annealing temperature. A single, stringent annealing temperature will work for most primers. As exemplified below, we have successfully used both the exponential and linear methods to determine SNPSTR genohaplotypes.
SNPSTR System 22SR1
The SNPSTR system 22SR1 includes the CA STR spanning np 112278 to 112323 in Homo sapiens clone CTA-390C10 on chromosome 22q11.21–22q12.1, GenBank Accession Number AL008721 (see supplemental data for further details). We amplified the flanking regions of the STR and screened for SNPs via DHPLC. No heteroduplexes were apparent in the downstream flanking region. In the upstream flanking region, we observed a heteroduplex and verified a C/T SNP at 112227 by sequencing (update submitted to GenBank). We synthesized allele-specific primers spanning np 112207 to the SNP. One allele-specific primer terminated in C and was labeled with 6-FAM. The other allele-specific primer terminated in T and was labeled with HEX. We incorporated a deliberate mismatch one base from the 3′ terminus to increase specificity (C and T, respectively). We obtained consistent allele specificity at 58°C annealing for 15 sec, using Taq DNA polymerase (Promega) in the presence of 2.5 mM MgCl2, by use of the exponential procedure described above. Using this system, we obtained 22SR1 SNPSTR genohaplotypes for 52 individuals representing globally diverse populations, about half from across the African continent (see supplemental data). Outgroup comparison using chimpanzee (Pan troglodytes) provided evidence that the ancestral SNP allele is T, whereas C is derived. Table 1 provides absolute frequencies of the 19 different SNPSTR haplotypes observed in the sample. A total of 21 of 52 individuals (40%) are doubly heterozygous. Highly significant levels of LD were detected for the non-African (P <0.001), but not for the African (P = 0.838) segment of the sample.
Table 1.
22SR1 and 5SR1 SNPSTR Haplotype Frequencies Observed in a Sample of 52 Individuals (104 Haplotypes) Representing Globally Diverse Populations
| System | Origin | SNP | Linked STR CA repeat number | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | ||||
| 22SR1 | global | C | 1 | 1 | 1 | 0 | 3 | 6 | 6 | 18 | 18 | 4 | 1 | 59 |
| T | 0 | 0 | 1 | 3 | 14 | 5 | 3 | 7 | 4 | 1 | 7 | 45 | ||
| African | C | 1 | 1 | 1 | 0 | 3 | 5 | 5 | 10 | 6 | 3 | 1 | 36 | |
| T | 0 | 0 | 1 | 1 | 1 | 4 | 2 | 3 | 2 | 0 | 0 | 14 | ||
| non-African | C | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 8 | 12 | 1 | 0 | 23 | |
| T | 0 | 0 | 0 | 2 | 13 | 1 | 1 | 4 | 2 | 1 | 7 | 31 | ||
| 9 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | ||||
| 5SR1 | global | T | 0 | 53 | 4 | 8 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 67 |
| G | 4 | 0 | 0 | 0 | 0 | 3 | 4 | 13 | 5 | 7 | 1 | 37 | ||
| African | T | 0 | 19 | 0 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 23 | |
| G | 4 | 0 | 0 | 0 | 0 | 3 | 4 | 7 | 2 | 7 | 0 | 27 | ||
| non-African | T | 0 | 34 | 4 | 5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 44 | |
| G | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 3 | 0 | 1 | 10 |
SNPSTR System 5SR1
The SNPSTR system 5SR1 includes the GT (CA) STR spanning np 147561 to 147594 in Homo sapiens chromosome 5 clone RP11–121L11, GenBank Accession Number AC026743 (Table 1). We amplified the flanking regions of the STR and screened for SNPs via DHPLC. No heteroduplexes were apparent in the downstream flanking region. In the upstream flanking region we observed a heteroduplex and verified a G/T SNP at np 147511 by sequencing (update submitted to GenBank). We synthesized allele-specific primers spanning np 147488 to the SNP. One allele-specific primer terminated in G and was labeled with 6-FAM. The other allele-specific primer terminated in T and was labeled with HEX. Each primer was a perfect match to its respective allele. We obtained consistent allele specificity at 58°C annealing for 15 sec, using Taq DNA polymerase (Promega) in the presence of 2.5 mM MgCl2, using the two-step linear procedure (above). We obtained 5SR1 SNPSTR genohaplotypes for 52 individuals (see Supplemental Data). Table 1 provides haplotype frequencies for the global, African, and non-African segments of the overall sample. A total of 17 of 52 individuals (33%) are doubly heterozygous. Highly significant levels of LD were detected for both the African (P <0.001) and non-African (P <0.001) segments of the sample.
DISCUSSION
The 22SR1 and 5SR1 SNPSTR systems described above meet the three goals stated in the introduction. (1) Within each system the SNP and STR are physically linked; (2) mutation rates of the SNP and STR differ significantly; and (3) the two systems, located on different chromosomes, evolve independently. In addition, typing is rapid and cost effective now that the systems have been developed and optimized. With genetic marker systems that meet these goals, we have flexible, powerful tools suitable for a variety of applications. SNPSTR systems may be used wherever genetic markers such as restriction fragment-length polymorphisms (RFLPs), SNP, and STR markers have been applied. As demonstrated by the example of Tishkoff et al. (1996), pairs of such markers provide more information than any single polymorphism system, or than a pair of polymorphisms considered without empirical determination of gametic phase. For instance, by comparing the STR alleles on the multiple SNP backgrounds, we gain information regarding the extent of homoplasy at the STR locus. The additional information derives from the resetting of the STR at the time of the SNP-generating mutation. The derived SNP allele arises on a single chromosome with, by necessity, zero STR variation.
One informative component of SNPSTR data is empirically determined haplotypic phase. One-third or more of the individuals tested for the 22SR1 and 5SR1 systems were doubly heterozygous and, therefore, required phase determination. In the absence of family data, the physical association of an allele at one locus with an allele at a second locus is typically inferred via estimation methods (Hawley and Kidd 1995). Such estimation methods may underestimate the frequency of rare haplotypes (Tishkoff et al. 2000a). As is true for rare alleles, however, rare haplotypes are likely to be particularly informative regarding gene flow between populations (Barton and Slatkin 1986) and mutation rates (Chakraborty 1981).
Given the extensive number of STRs discovered in the genomes of a wide array of species during recent years, numerous potential SNPSTR systems exist. Because SNPs and STRs are frequent in the nuclear genomes of most species, closely linked pairs are quite common (e.g., Makova et al. 2000). According to the dbSNP summary (http://www.ncbi.nlm.nih.gov/SNP/snp_summary.cgi), >2.5 million SNPs have already been identified using human samples. This total corresponds to an average of ∼1 SNP every 1–2 kb. Consistent with this estimate, in our experience, at least one SNP is found within 400 bp of roughly 50% of STRs.
Recent studies (Daly et al. 2001; Jeffreys et al. 2001; Reich et al. 2001) of the patterns of LD and distribution of recombination hotspots within the human genome have implications for SNPSTR application. If the two polymorphisms (SNP and STR) are located at a recombination hotspot, haplotype frequencies may reflect reciprocal recombination as well as other evolutionary forces. If current estimates of the distribution of such hotspots (Daly et al. 2001) are roughly accurate, such cases are expected to be relatively rare. Consider a simplified model of one hotspot of length Z every X nucleotides. If a SNPSTR system (from the SNP through the STR) spans Y nucleotides, the chance of the SNPSTR system overlapping the hotspot is (Y + Z)/X. For instance, for SNPSTR systems of length Y = 400 nucleotides, hotspots of length Z = 1000 nucleotides, and inter-hotspot (LD block) distances of X = 50,000 nucleotides, overlap is expected <3% of the time. Within LD blocks, we may be able to develop compound-linked SNPSTR systems that extend beyond the typical 500 bp and still assume that recombination is essentially absent.
Gene conversion has probably had greater influence on haplotype frequencies at most SNPSTR systems than has reciprocal recombination. Recent comparisons of LD and levels of polymorphism indicate that gene conversion has played a significant role in reducing LD at closely linked sites (Ardlie et al. 2001; Frisse et al. 2001). Whereas interpretation of SNPSTR haplotype frequencies must include the possibility of gene conversion, STR mutation (on the order of 10−3 per generation) plays a much larger role than gene conversion in SNPSTR evolution. A large set of SNPSTR systems, however, may provide insights into the rate of gene conversion across the genome. The 5SR1 system, for instance, lacks evidence of gene conversion, whereas the 22SR1 system is less informative in this regard.
The set of applications for which a given SNPSTR system is particularly suited depends in part on the date of the mutation that gave rise to the SNP. When information regarding the age of an allele (at a SNP or a STR polymorphism) is valuable, a sufficiently polymorphic SNPSTR system will be informative. Two obvious areas of application are in evolutionary genetics and medical genetics. Other areas of potential application include agriculture, entomology, wildlife management, and forensics. Within evolutionary genetics, we expect SNPSTRs to be informative regarding the evolutionary histories of humans and many other species. SNPSTRs may also be informative regarding molecular evolutionary processes such as gene conversion. Information resides in the extent of variation and of LD within populations and in differences in haplotype frequencies and LD among populations (Tishkoff et al. 1996; Ardlie et al. 2002). SNPSTR haplotype frequency differences arise largely through the molecular process of mutation and the population genetic processes of genetic drift, migration, and natural selection.
In the case of humans, our data demonstrate that even single systems provide insights into history. Whereas only a limited number of individuals have been screened for SNPSTR systems 5SR1 and 22SR1, the resulting haplotype frequencies reveal patterns consistent with patterns of previously generated genetic data. Both systems reveal greater genetic diversity within Africa than outside of Africa, particularly in terms of overall numbers of STR alleles. Considering the number of STR alleles on each SNP background, however, reveals a more complex pattern. Wheres for 22SR1, the set of African samples reveals 11 STR alleles, 10 are found on the C background and 7 are found on the T background. The set of non-African samples, with 8 STR alleles total, reveals only 5 on the C background, but 8 on the T background. The set of non-African samples, therefore, exhibits greater (in absolute terms) diversity on the T background than does the set of African samples. Further typing of this SNPSTR system in both African and non-African populations may clarify whether the T diversity in non-Africans reflects population expansion outside of Africa or the diverse nature of the groups that have migrated out of Africa at various points in human history.
The 5SR1 system is striking with respect to the level of LD both within and outside of Africa. There is no overlap in the STR alleles found on each of the SNP backgrounds. This system is unusual for a SNPSTR in that we detect no evidence of homoplasy at the STR locus. This pattern suggests that the SNP arose much earlier than the initial spread of modern humans from Africa. In the context of a model of recent African origin of anatomically modern humans, the overall STR frequencies on the SNP backgrounds of the 5SR1 and 22SR1 systems indicate that when modern humans migrated out of Africa, they carried with them only a subset of the total African diversity. Furthermore, the diversity on each of the SNP backgrounds in the Americas and Oceania is lower than the diversity in Eurasia, which, in turn, is lower than that in Africa (see supplemental data).
Within medical genetics, we expect SNPSTRs to be informative regarding the influence of genetic regions on disease susceptibility. If the precise location of a gene is unknown, association or linkage studies might take advantage of the information contained within SNPSTR systems distributed throughout the genome. As noted by Akey et al. (2001) and Ardlie et al. (2002), among others, haplotype information significantly improves the power to map disease genes. Where a predisposing or candidate gene has been identified, nearby SNPSTRs may be informative regarding the ages of mutations or haplotypes associated with disease. Whereas SNPSTRs not known to be influenced by natural selection are ideal for inference of population history, SNPSTRs located in or near genes may be developed to recover the evolutionary history of those genes.
The number and characteristics of a set of SNPSTR systems required to address a given research question depend on the nature of that question. As with SNP and STR discovery, many laboratories might develop SNPSTR systems and compile a shared resource suitable for a range of applications. Although the contributions of several laboratories would lead to a valuable resource, even research groups focusing on less-frequently studied species might take advantage of SNPSTR technology. A single research group can develop a small number of systems and obtain insights into the evolutionary history of that species. Once developed, these systems can be typed in a large sample very rapidly. Because typing of SNPSTR systems is carried out using fluorescently labeled primers, only very small quantities of DNA are needed (∼50 ng/PCR).
We are in the process of developing a number of human SNPSTR systems using a global screening set. Some of these will be informative at the global level, as are 22SR1 and 5SR1, with geographically broad distributions of the SNP alleles. Such systems make possible more precise inferences regarding the initial spread of our human ancestors throughout the world. Other SNPSTR systems, however, will be regionally informative, say, for the peopling of the Americas. Regionally informative systems are characterized by restricted distribution of one of the two SNP alleles, or low STR diversity within one region relative to other regions. Screening sets may be tailored (by including many individuals from a subset of regions) to generate SNPSTR systems relevant to particular regions of the world. Each SNPSTR system, when unlinked to others on recombining chromosomes, provides independent information regarding the evolutionary history of a species. As with other sets of polymorphisms (Pamilo and Nei 1988), the combination of information from several SNPSTR systems dramatically increases the power to distinguish between alternative hypotheses regarding molecular processes and population history.
METHODS
Primer Design
PCR primers were designed on the basis of sequences surrounding SNPSTR loci retrieved from GenBank. Within the constraints of the region, design considerations included melting temperature, thermal profile, secondary structure, dimer formation, and 3′ terminal base. For allele-specific primers, as they must terminate at the SNP, there is little one may do to influence some of these factors, other than adjusting primer length. Certain SNPs are not amenable to allele-specific PCR (and therefore SNPSTR analysis) due to the nature of the sequences adjacent to the SNP.
PCR
All PCR was performed using Taq DNA polymerase (Promega) and supplied 10× buffer, at 2.5 mM MgCl2, 0.8 μM each primer, for 35 cycles. Reaction reagents and concentrations were standard (Sambrook et al. 1989). Reactions were prepared on ice and transferred to a preheated thermal cycler block at 94°C for 15 sec, followed by 35 cycles of 94°C for 15 sec, 58°C for 15 sec, and 72°C for 40 sec.
Allele-Specific Amplification
For exponential amplification of SNPSTR system 22SR1, a range of annealing temperatures was tested. Almost perfect specificity was found at 58°C. This was improved by addition of Perfect Match PCR Enhancer (Stratagene) used according to the manufacturer's instructions. For linear amplification of labeled 5SR1, both initial and second step (fluorescent) PCR was performed at 58°C. Addition of Perfect Match was unnecessary, as sufficient allele specificity was obtained with the linear, two-step procedure. Primer concentration was 0.8 μM.
Fluorescent Fragment Analysis
For fragment-length determination, fluorescently labeled PCR products were separated by capillary electrophoresis on a 310 Genetic Analyzer in POP-4 (Applied Biosystems). Individual samples were prepared as follows: 2.0 μL of labeled PCR product and 0.7 μL of HD400 ROX size standard (Applied Biosystems) were added to 15.0 μL of formamide, heated at 94°C for 2 min, and snap cooled in an ice bath.
Significance of Linkage Disequilibrium
For each population, for each pair of alleles at the two linked polymorphisms, we test the significance of linkage disequilibrium. LD for each SNPSTR system is measured by a Monte Carlo implementation of a χ2 test of the D statistic. D is calculated (Weir 1996) for each allele in a SNPSTR as follows:
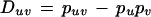
The χ2 statistic to test the hypothesis that none of the Duv's is significantly different from zero is:
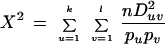
The χ2 statistic is calculated given the haplotype frequencies observed for a SNPSTR system. To test significance of the statistic, we generate 1000 random SNPSTR haplotype frequency distributions with the same row and column sums. The fraction of occurrences in which the randomized SNPSTR's χ2 statistic exceeds the observed SNPSTR's statistic is taken to be the probability that the observed SNPSTR's LD occurred by chance. The chance of falsely rejecting the null hypothesis is dependent on the number of randomizations in the test (Roff and Bentzen 1989).
WEB SITE REFERENCES
http://mapper.wustl.edu/; GenLink Home Page
http://www.ncbi.nlm.nih.gov/; National Center for Biotechnology Information
http://www.stanford.edu/group/mountainlab/; Mountain Laboratory
Acknowledgments
We thank NIH (GM 28428) and NSF (BCS-9905574) for partially funding this research. We thank Prescott L. Deininger for suggesting the linear method and for technical advice, Todd Ward for technical advice, and L. Luca Cavalli-Sforza for providing DNA samples for the screening set as well as laboratory facilities during the initial phase of the project.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mountain@stanford.edu; FAX (650) 725-9996.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.238602.
REFERENCES
- Akey J, Jin L, Xiong M. Haplotypes vs single marker linkage disequilibrium tests: What do we gain? Eur J Hum Genet. 2001;9:291–300. doi: 10.1038/sj.ejhg.5200619. [DOI] [PubMed] [Google Scholar]
- Ardlie K, Liu-Cordero SN, Eberle MA, Daly M, Barrett J, Winchester E, Lander ES, Kruglyak L. Lower-than-expected linkage disequilibrium between tightly linked markers in humans suggests a role for gene conversion. Am J Hum Genet. 2001;69:582–589. doi: 10.1086/323251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nature Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- Barton NH, Slatkin M. A quasi-equilibrium theory for the distribution of rare alleles in a subdivided population. Heredity. 1986;56:409–415. doi: 10.1038/hdy.1986.63. [DOI] [PubMed] [Google Scholar]
- Bosch E, Calafell F, Santos FR, Pérez-Lezaun A, Comas D, Benchemsi N, Tyler-Smith C, Bertranpetit J. Variation in short tandem repeats is deeply structured by genetic background on the human Y chromosome. Am J Hum Genet. 1999;65:1623–1638. doi: 10.1086/302676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R. Estimation of mutation rates from the number of rare alleles in a sample. Ann Hum Biol. 1981;8:221–230. doi: 10.1080/03014468100004991. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Kimmel M, Stivers DN, Davison LJ, Deka R. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc Natl Acad Sci. 1997;94:1041–1046. doi: 10.1073/pnas.94.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nature Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- de Knijff P. Messages through bottlenecks: On the combined use of slow and fast evolving polymorphic markers on the human Y chromosome. Am J Hum Genet. 2000;67:1055–1061. doi: 10.1016/s0002-9297(07)62935-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A. Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet. 2001;69:831–843. doi: 10.1086/323612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Morar B, Underhill PA, Passarino G, Lin AA, Wise C, Angelicheva D, Calafell F, Oefner PJ, Shen P, et al. Origins and divergence of the Roma (Gypsies) Am J Hum Genet. 2001;69:1314–1331. doi: 10.1086/324681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley ME, Kidd KK. HAPLO: A program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered. 1995;86:409–411. doi: 10.1093/oxfordjournals.jhered.a111613. [DOI] [PubMed] [Google Scholar]
- Hurles ME, Veitia R, Arroyo E, Armenteros M, Bertranpetit J, Pérez-Lezaun A, Bosch E, Shlumukova M, Cambon-Thomsen A, McElreavey K, et al. Recent male-mediated gene flow over a linguistic barrier in Iberia, suggested by analysis of a Y-chromosomal DNA polymorphism. Am J Hum Genet. 1999;65:1437–1448. doi: 10.1086/302617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nature Genet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- Macaubas C, Jin L, Hallmayer J, Kimura A, Mignot E. The complex mutation pattern of a microsatellite. Genome Res. 1997;7:635–641. doi: 10.1101/gr.7.6.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova KD, Nekrutenko A, Baker RJ. Evolution of microsatellite alleles in four species of mice (genus Apodemus) J Mol Evol. 2000;51:166–172. doi: 10.1007/s002390010077. [DOI] [PubMed] [Google Scholar]
- Mountain JL, Cavalli-Sforza LL. Inference of human evolution through cladistic analysis of nuclear DNA restriction polymorphisms. Proc Natl Acad Sci. 1994;91:6515–6519. doi: 10.1073/pnas.91.14.6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimation of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel A, Filon D, Brinkmann B, Majumder PP, Faerman M, Oppenheim A. The Y chromosome pool of Jews as part of the genetic landscape of the Middle East. Am J Hum Genet. 2001;69:1095–1112. doi: 10.1086/324070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oefner PJ, Underhill PA. Current Protocols in Human Genetics. (Suppl. 19) New York: Wiley and Sons; 1998. DNA mutation detection using denaturing high-performance liquid chromotography; pp. 7.10.1–7.10.12. [DOI] [PubMed] [Google Scholar]
- Orita M, Suzuki Y, Sekiya T, Hayashi K. Rapid and sensitive detection of point mutations and SNA polymorphisms using the polymerase chain reaction. Genomics. 1989;5:874–879. doi: 10.1016/0888-7543(89)90129-8. [DOI] [PubMed] [Google Scholar]
- Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol. 1988;5:568–583. doi: 10.1093/oxfordjournals.molbev.a040517. [DOI] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, et al. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- Roff DA, Bentzen P. The statistical analysis of mitochondrial DNA polymorphisms: χ 2 and the problem of small samples. Mol Biol Evol. 1989;6:539–545. doi: 10.1093/oxfordjournals.molbev.a040568. [DOI] [PubMed] [Google Scholar]
- Rogers EJ, Shone AC, Alonso S, May CA, Armour JAL. Integrated analysis of sequence evolution and population history using hypervariable compound haplotypes. Hum Mol Genet. 2000;9:2675–2681. doi: 10.1093/hmg/9.18.2675. [DOI] [PubMed] [Google Scholar]
- Ruiz-Linares A, Ortiz-Barrientos D, Figueroa M, Mesa N, Múnera JG, Bedoya G, Vélez ID, García LF, Pérez-Lezaun A, Bertranpetit J, et al. Microsatellites provide evidence for Y chromosome diversity among the founders of the New World. Proc Natl Acad Sci. 1999;96:6312–6317. doi: 10.1073/pnas.96.11.6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd edition. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonné-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M, et al. Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science. 1996;271:1380–1387. doi: 10.1126/science.271.5254.1380. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Pakstis AJ, Ruano G, Kidd KK. The accuracy of statistical methods for estimation of haplotype frequencies: An example from the CD4 locus. Am J Hum Genet. 2000a;67:518–522. doi: 10.1086/303000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Pakstis AJ, Stoneking M, Kidd JR, Destro-Bisol G, Sanjantila A, Lu R-b, Deinard AS, Sirugo G, Jenkins T, et al. Short tandem-repeat polymorphism/Alu haplotype variation at the PLAT locus: Implications for modern human origins. Am J Hum Genet. 2000b;67:901–925. doi: 10.1086/303068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J, Nielsen R, Ardlie K, Liu-Cordero SN. The discovery of single nucleotide polymorphisms and inferences about human demographic history. Am J Hum Genet. 2001;69:1332–1347. doi: 10.1086/324521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD. Detecting ancient admixture in humans using sequence polymorphism data. Genetics. 2000;154:1271–1279. doi: 10.1093/genetics/154.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS. Genetic Data Anlaysis II. Sunderland, MA: Sinauer Associates; 1996. [Google Scholar]
- Zhivotovsky LA. Estimating divergence time with the use of microsatellite genetic distances: Impacts of population growth and gene flow. Mol Biol Evol. 2001;18:700–709. doi: 10.1093/oxfordjournals.molbev.a003852. [DOI] [PubMed] [Google Scholar]