Blockade of IL-12 during the induction of collagen-induced arthritis (CIA) markedly attenuates the severity of the arthritis (original) (raw)
Abstract
The effect of blocking IL-12, a potent inducer of interferon-gamma (IFN-γ) and promoter of Th1 cell responses, during the induction phase of CIA was investigated. Arthritis was elicited in male DBA/1 mice by immunizing with type II collagen (CII) in Freund's complete adjuvant. Neutralizing anti-IL-12 antibodies were administered twice weekly from CII immunization. It was found that administration of anti-IL-12 from immunization until the onset of clinical arthritis did not lower the incidence of arthritis, but dramatically attenuated the severity of the disease, both clinically and histopathologically. This regime was associated with reduced IFN-γ levels produced by ex vivo CII-stimulated draining lymph node cells, and with diminished spontaneous ex vivo production of tumour necrosis factor (TNF), IL-6 and IL-10 by freshly isolated synovial cells. Total anti-CII antibody serum levels in these mice were lower than in the controls, but there was no change in the IgG2a/IgG1 ratio. These findings confirm that IL-12 has a major role in the induction of murine CIA and suggests that this disease is propagated, in part, by cells of the Th1 phenotype.
Keywords: IL-12, collagen-induced arthritis, cytokines
INTRODUCTION
IL-12 is a heterodimeric cytokine, consisting of a p40 and a p35 subunit, with potent immunoregulatory properties, primarily released by antigen-presenting cells, dendritic cells, and monocytes/macrophages in response to bacterial products and immune signals (reviewed in [1]). It enhances natural killer (NK)-mediated cytotoxicity and induces interferon-gamma (IFN-γ) production by NK cells and T lymphocytes [2, 3]. IL-12 plays a key role in promoting Th1 immune responses, as demonstrated both in vitro [4] and in vivo [5, 6]. Accordingly, antibodies against IL-12 have been used to beneficial effect in experimental models for autoimmune diseases that are Th1-driven, such as experimental allergic encephalomyelitis (EAE) [7] and 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced chronic intestinal inflammation in mice, a model for human inflammatory bowel disease [8]. In TNBS-treated mice, administration of anti-IL-12 after induction of colitis led to a striking improvement of established disease, clinically and histopathologically, associated with a decrease in IFN-γ production by ex vivo stimulated lamina propria CD4+ cells. Similarly, anti-IL-12 treatment in C3H mice infected with Borrelia burgdorferi significantly reduced the severity of Lyme arthritis, accompanied by a decrease in IFN-γ serum levels [9].
Murine CIA is an experimental model for rheumatoid arthritis (RA) that can be induced in genetically susceptible DBA/1 mice by immunization with heterologous native type II collagen (CII) emulsified in Freund's complete adjuvant (FCA) [10]. The clinical course of the disease is characterized by an acute arthritis affecting several limbs, resulting in joint destruction and deformities. Pathologically, there is massive infiltration of the synovium by immunocompetent cells, formation of an invasive pannus, and subsequent cartilage and bone erosion. The immune response to CII involves both cellular and humoral mechanisms [11] and CD4+ cells have been strongly implicated in the induction phase of the disease [12].
It has been recently demonstrated that IL-12 can replace Mycobacterium tuberculosis when immunizing DBA/1 mice with CII in oil, resulting in severe arthritis, associated with enhanced IFN-γ production by ex vivo CII-stimulated spleen cells, and an increased collagen-specific IgG2a antibody response [13]. Consistent with this observation, a study from our laboratory investigated the role of Th1/Th2-type responses in the development of CIA, and found that IFN-γ production dramatically increased at the time of disease onset and subsequently declined throughout the disease and the remission phase [14].
In view of the association between a Th1 response and the onset of arthritis, we explored the role of IL-12 in the pathogenesis of CIA, by administration of a neutralizing anti-IL-12 MoAb during the induction phase of the disease. We report here that the blockade of IL-12 is not able to prevent disease onset, but dramatically attenuates the severity of the arthritis.
MATERIALS AND METHODS
Induction of arthritis
Bovine CII was purified from hyaline cartilage, as previously described [15]. Male DBA/1 mice (8–12 weeks old) were immunized with 100 μg of CII emulsified in FCA (Difco, Detroit, MI) by intradermal injection at the base of the tail.
Administration of anti-IL-12 antibody
The rat IgG2b anti-mouse IL-12p40 MoAb, designated 10F6 [16], was used to treat mice immediately after immunization with CII. Doses of 500 μg in 100 μl PBS/injection, were given twice weekly until onset of clinical arthritis. At the doses used this antibody has previously been found to have potent neutralizing activity in vitro and in preliminary in vivo studies (Gately and Presky, unpublished results). This MoAb is able to block endotoxin-induced IFN-γ production and antigen (keyhole limpet haemocyanin (KLH))-induced Th1 responses with a potency comparable to that of the polyclonal goat anti-mouse IL-12 IgG described previously [17, 18]. In three experiments, a total of 42 mice were treated with anti-IL-12, and 27 mice were treated with PBS alone, while eight received an equal dose of rat IgG2b isotype control antibody directed against a Chlamydomonas glycoprotein, AFRC MAC1 (European Collection of Animal Cell Cultures, Salisbury, UK).
Monitoring of arthritis
From day 15 after immunization mice were examined daily for onset of disease using two clinical parameters: paw swelling and clinical score [15]. Paw swelling was assessed by measuring the thickness of the first affected hind paw with callipers. For the clinical score, 0 = normal; 1 = slight swelling and erythema; 2 = pronounced oedema; 3 = joint rigidity. Each limb was graded, resulting in a maximal clinical score of 12 per animal. Unless otherwise specified, arthritis was monitored over 10 days, after which the mice were killed. Clinical monitoring was performed in a blinded manner.
Anti-collagen antibody ELISA
Mice were killed after 3 or 10 days of arthritis and bled post mortem. Serum levels of total IgG anti-CII antibodies, and of anti-CII antibodies of the IgG1 and IgG2a subclasses, were measured by modification of an ELISA for the detection of human IgG [15]. Briefly, microtitre plates were coated with 2 μg/ml native bovine CII, blocked, and then incubated with serially diluted test sera. Bound IgG was detected by incubation with alkaline phosphatase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, Luton, UK), or sheep anti-mouse IgG1, or IgG2a (The Binding Site, Birmingham, UK) followed by substrate (dinitrophenyl phosphate). Plates were washed between steps with 0.01% Tween–PBS. Optical density (OD) was read at 405 nm. To obtain anti-CII antibody concentrations, serum samples were titred in parallel to a sample of affinity-purified anti-CII-IgG of known concentration.
Lymph node cell culture
Mice were killed at day 6 after immunization, and at day 3 and day 10 after disease onset. Inguinal lymph nodes were excised, teased apart to make a single-cell suspension, washed and then cultured in 96-well plates at a density of 1 × 106 cells/ml (200 μl/well) in RPMI (Bio-Whittaker, Verviers, Belgium) containing heat-inactivated fetal calf serum (FCS; 10% v/v), 2 mm glutamine, penicillin 100 U/ml, streptomycin 100 μg/ml, and 2-mercaptoethanol (2 × 10−5 m). Cells were cultured alone, or in the presence of bovine CII (50 μg/ml in Tris-buffered saline). Supernatants were collected after 72 h and stored at −20°C until measured for cytokines.
Synovial cell culture
Mice were killed at the aforementioned time points and knee joints were removed. Synovial membranes were excised under a dissecting microscope and digested with collagenase A (1 mg/ml) (Boehringer-Mannheim UK, Lewes, UK) and DNAse type IV (150 μg/ml; Sigma, Poole, UK) at 37°C for 20 min, in the presence of polymyxin B (33 μg/ml). The cells were pooled for a given treatment group, washed extensively and cultured in 96-well plates at a density of 4 × 106 cells/ml (100 μl/well) in RPMI, supplemented with the additives mentioned above. Supernatants were collected after 24 h and stored at −20°C until cytokine measurement.
Cytokine assays
For determination of tumour necrosis factor (TNF) levels, a bioassay was performed using the WEHI 164 cell line [19], as previously described [20]. All other cytokines were detected by ELISA, as described previously [14]. The antibody pairs used were as follows (listed by capture/detection MoAb): IL-6, 20F3/32C11; IL-10, 2 A5/SXC-1; IFN-γ, R4-6 A2/XMG1.2. These antibodies were obtained from the ATCC, courtesy of Dr J. Abrams (DNAX, Palo Alto, CA). The antibody pair for a heterodimer-specific mouse IL-12 ELISA was 9A5/5C3 and the detection limit was 15 pg/ml. The detection limits for all other ELISAs were 40 pg/ml.
Histological analysis
Hind paws were removed post mortem on day 10 of arthritis, fixed in formalin and decalcified in 5% EDTA. Paws were then embedded in paraffin, sectioned and stained with haematoxylin and eosin (H–E). Arthritic changes in the ankle, the metatarsophalangeal joints, the proximal interphalangeal and the distal interphalangeal joints were scored blindly as mild (mild synovial hyperplasia); moderate (pannus formation and erosions limited to the cartilage–pannus junction); or severe (extended bone and cartilage erosions with loss of joint architecture).
Statistical analysis
The Mann–Whitney _U_-test for comparing non-parametric data for statistical significance was applied on all clinical results. The χ2 test was applied for analysis of histological data.
RESULTS
Treatment with anti-IL-12 attenuates the severity of arthritis
Neutralization of IL-12 during the entire induction phase of CIA did not affect the incidence or the time of onset of arthritis, as depicted in Table 1. Figure 1a,b shows the clinical course of arthritis in one representative experiment. The additive value for paw thickness over the course of arthritis (day 1 to day 10) was significantly lower in the treated group than in the controls (P = 0.0005) (Fig. 1a), as was the clinical score (P = 0.0001) (Fig. 1b). Pooled data from the three experiments showed that the arthritis was dramatically attenuated by anti-IL-12 administration, as assessed by maximal paw thickness (P < 0.0001) and by maximal clinical score (P = 0.0001). Inflammation of the front paws did not occur in the treated animals, but was present in 45% of the control mice. No differences in clinical severity were observed between the IgG2b isotype control antibody-treated mice and the PBS-treated mice (not shown).
Table 1.
Incidence and time of onset of CIA after treatment with anti-IL-12
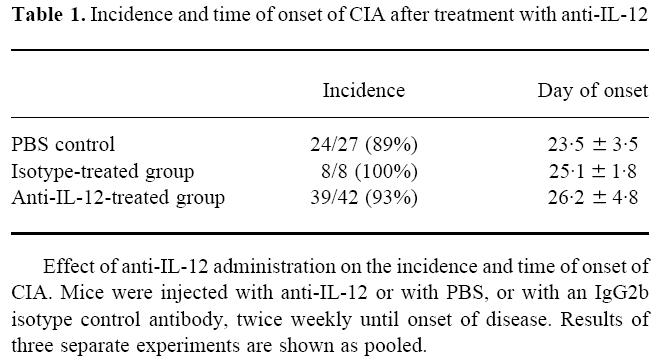
Fig. 1.
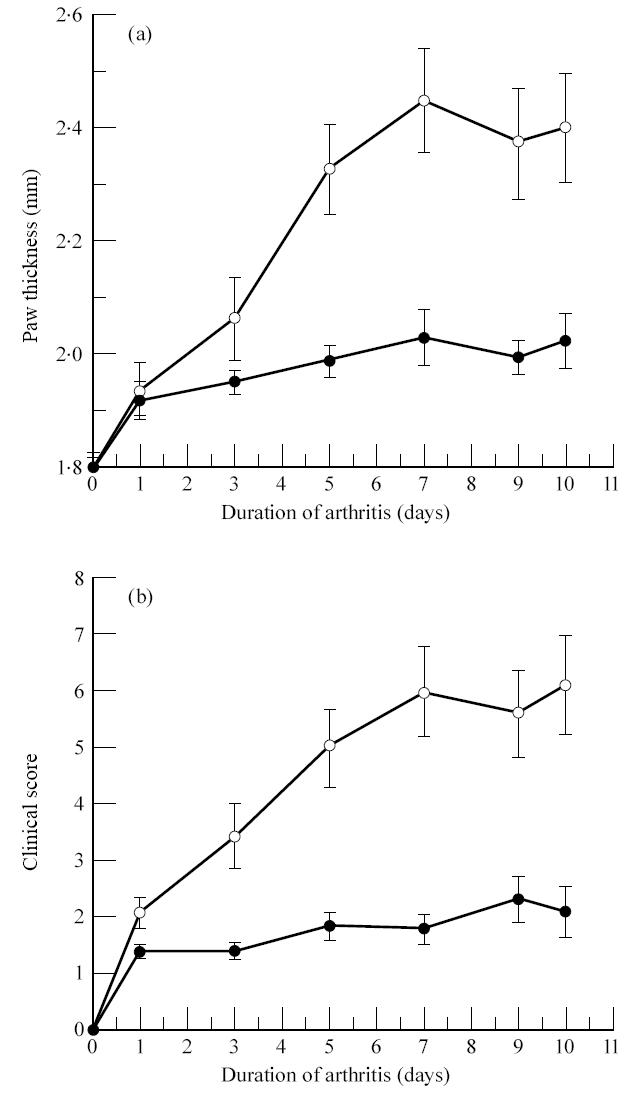
(a) Paw thickness, expressed in mm, of a representative experiment, measured daily over 10 days of arthritis. Mice were injected with anti-IL-12 or with PBS (control) from the time of immunization onwards, twice weekly until onset of arthritis. Controls, n = 11 (○); anti-IL-12-treated animals, n = 21 (•). Results are the mean of n mice ± s.e.m. (b) Clinical score of a representative experiment, measured daily over 10 days of arthritis. Mice were injected with anti-IL-12 or with PBS (control) from the time of immunization onwards, twice weekly until onset of arthritis. Controls, n = 11 (○); anti-IL-12-treated animals, n = 21 (•). Results are the mean of n mice ± s.e.m.
Histological findings confirm the clinical data
Ten days after onset of arthritis, the mice were killed and the hind paws were removed for histological processing. Microscopic analysis of H–E-stained paw sections showed that more joints were only mildly affected in the anti-IL-12-treated group than in the control group (P < 0.05; χ2) (Fig. 2). In most control mice, the joints of the hind paw were completely destroyed, with loss of joint architecture (Fig. 3a). Only 20% of the control animals had joints with mild lesions (Fig. 2). Eighty percent of the anti-IL-12-treated mice showed either mild or moderate joint lesions (Fig. 2). Typical lesions found in these mice consisted of small erosions limited to the cartilage–pannus junction (Fig. 3b).
Fig. 2.
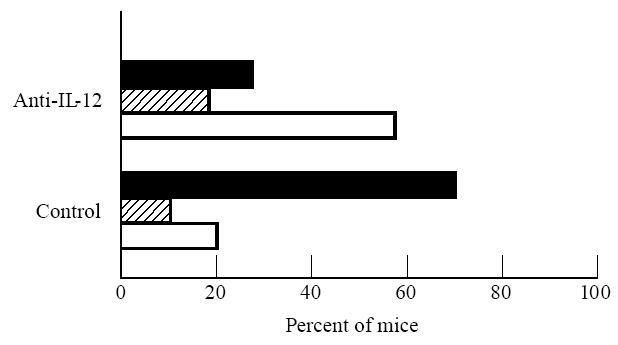
Histological findings after administration of PBS controls (n = 17 mice) or anti-IL-12 (n = 34 mice). Arthritis in the joints from hind paw sections was assessed histologically 10 days after the onset of clinical arthritis and was scored as mild (□), moderate ( ), or severe (▪), as described in Materials and Methods.
), or severe (▪), as described in Materials and Methods.
Fig. 3.
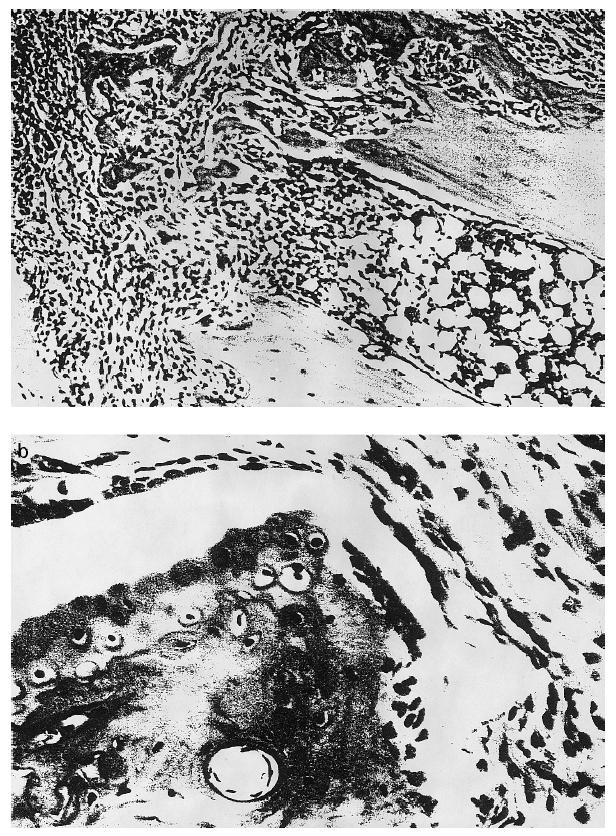
(a) Haematoxylin and eosin staining of the metatarsophalangeal joint of a DBA/1 mouse, killed after 10 days of clinical arthritis (× 100). Infiltration by inflammatory cells is extensive (left), with total destruction and loss of joint architecture. (b) In mice treated with anti-IL-12, severely damaged lesions were rarely seen, as shown in this figure of the most extensively damaged joints of this treatment group, which has only mild synovitis and erosions of the cartilage–pannus junction. (× 250).
The effect of anti-IL-12 on serum levels of anti-CII antibodies
Total IgG anti-CII antibody levels in the serum after 10 days of arthritis were slightly lower in the anti-IL-12-treated mice than in controls (Fig. 4). IL-12 affects the development of Th1 cells, and hence the Th1-dependent IgG subclass, IgG2a, might have diminished by anti-IL-12 therapy. However, the ratio of serum IgG2a/IgG1 subclasses varied widely between 1 and 6 in all the experiments, and no effect of anti-IL-12 administration on this ratio was found.
Fig. 4.
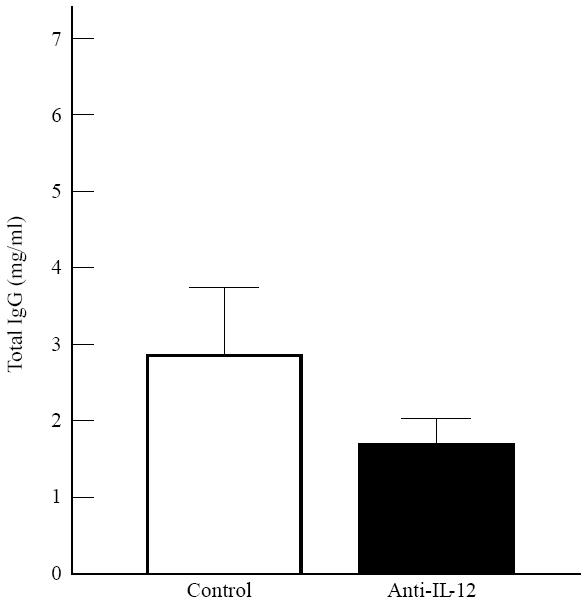
Serum levels of total IgG anti-collagen type II (CII) antibodies, 10 days after onset of clinical arthritis, in control animals (n = 11) and anti-IL-12-treated animals (n = 23). Error bars represent 1 s.d.
The effect of anti-IL-12 on spontaneous ex vivo cytokine production by synovial cells
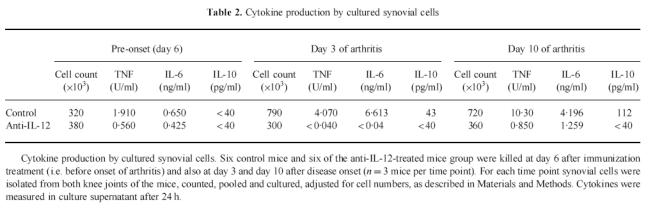
The most critical site of cytokine expression in arthritis is within the joint itself, and in particular within the synovium [21]. We evaluated the effect of anti-IL-12 treatment on the balance between production of pro- and anti-inflammatory cytokines in the synovium and compared this with the severity of clinical disease. For this purpose, knee joints were removed before disease onset (6 days after immunization), 3 days after onset of arthritis and 10 days after onset of arthritis, and synovial cells were counted and cultured as described. To obtain sufficient material for cytokine study synovium from mice of a given treatment group was pooled. Synovial hyperplasia was not observed in the anti-IL-12-treated animals, whereas the knee synovial cell count increased two- to three-fold in the controls (Table 2). This was reflected in the spontaneous production of cytokines in vitro(Table 2), with control animals producing some bioactive TNF before disease onset (1.9 U/ml) and increased levels after onset of clinical arthritis (4 U/ml at day 3 and 10 U/ml at day 10 of arthritis). TNF secretion was markedly reduced by synovial membrane cells from mice treated with anti-IL-12 at day 6 after immunization (0.5 U/ml), and TNF production was virtually absent after onset of disease. A similar pattern of response was found for IL-6 production. IL-10 was produced by synovial membrane cells from control animals after disease onset (43 pg/ml at day 3 of arthritis and 112 pg/ml at day 10), but not by synovial cells from treated animals (< 40 pg/ml). IL-12 could not be detected in the controls, or in any of the treated mice, using an ELISA for murine IL-12p70, with a sensitivity of 15 pg/ml (data not shown). The results in Table 2 are from culture supernatants derived from cultures of standardized cell numbers, thus the potential for the in vivo local cytokine concentrations in the synovium of the control mice may be far greater due to the increased number of leucocytes in these tissues.
Table 2.
Cytokine production by cultured synovial cells
The effect of anti-IL-12 on CII-stimulated ex vivo IFN-γ production by draining lymph node cells
Inguinal lymph nodes were excised 6 days after immunization, and at days 3 and 10 after disease onset. Cell counts increased three-fold by day 3 of arthritis in the control animals, while treated mice with mild arthritis showed only a limited increase in lymph node cell counts (Table 3). IFN-γ was produced by inguinal lymph node cells only when stimulated with CII in vitro(Table 3). Control mice produced IFN-γ 6 days after immunization, reaching peak levels at day 3 of arthritis. By day 10 of arthritis, IFN-γ secretion had fallen below detection limits (< 40 pg/ml). Anti-IL-12 administration effectively prevented IFN-γ production before disease onset and IFN-γ was undetectable at any stage of disease in the mice that received anti-IL-12. We have previously shown that IFN-γ production in this system can be abrogated by anti-CD4 [22].
Table 3.
IFN-γ production (pg/ml) by cultured lymph node cells

DISCUSSION
The data presented in this study suggest an important role for IL-12 in the development of CIA, as neutralization by MoAb during the induction phase of arthritis caused dramatic effects on the severity of the disease. This effect is most likely to be due to the suppression of the generation of CII-specific Th1 cells, a hypothesis which is supported by the reduction, or absence, of IFN-γ production by CII-stimulated lymph node cells in vitro. The enhancement of Th2 cytokines, such as IL-4 or IL-10, was not found (data not shown).
Humoral responses to CII are required for the manifestation of CIA [11]. Successful anti-IL-12 treatment was associated with lower serum levels of total anti-IgG anti-CII antibody levels (Fig. 4), but a reproducible effect on the ratio of IgG2a/IgG1 subclasses was not observed. Humoral responses developing under Th1 or Th2 conditions are characterized by IgG2a/2b/3 and IgG1/IgE antibodies, respectively [23]. IL-12 has been shown to promote the synthesis of antigen-specific IgG2a antibodies in vivo [24], and up-regulates IgG2a anti-CII antibodies when used to induce CIA with Freund's incomplete adjuvant (FIA) [13]. Nevertheless, we could not demonstrate the selective down-regulation of IgG2a antibodies to CII following IL-12 blockade. The reason for this variability in IgG subclass expression is not clear. Similar findings have been obtained in IL-12-deficient DBA/1 mice, where the anti-CII-antibody titres were reduced in comparison with the wild-type controls, although there was no shift in the IgG2a/IgG1 ratio [25].
Anti-IL-12 did not reduce the incidence of arthritis. This could be the result of an incomplete neutralization of IL-12, but it has to be noted that in a recent study with IL-12p40 knock-out mice on the DBA/1 background a proportion of mice still developed arthritis (20%) upon CII immunization [25]. These two observations suggest that although IL-12 plays an important role in the development of CIA, there are other possible pathways for the induction of CIA. The arthritis that developed after anti-IL-12 administration was not only clinically mild, with limited oedema and inflammation of the affected paws, and restricted to one or two hind limbs, but the histological damage was also less extensive than in the controls. Concordantly, synovial hyperplasia was limited. FACS analysis of synovial cell suspensions showed that there was a reduced influx of leucocytes, in particular of neutrophils (data not shown), a finding which is consistent with reports that IL-12 is chemotactic for polymorphonuclear cells [26]. Spontaneous production of the proinflammatory cytokines TNF and IL-6 by cultured synovial cells was low throughout the course of the mild disease, compared with control arthritis. Interestingly, detectable IL-10 was not found in these synovia, whereas the controls, which had more severe arthritis, released considerable amounts of this cytokine. This is compatible with evidence that proinflammatory cytokines (IL-1β, TNF) are involved in the up-regulation of IL-10 production [27, 28]. IL-10 inhibits the synthesis of IL-1β, TNF, IL-6, IL-8 and granulocyte-macrophage colony-stimulating factor (GM-CSF) by monocytes [29, 30] and is produced in abundance in rheumatoid synovium [31]. IL-10 has been postulated to be part of a homeostatic regulatory mechanism that partially controls the inflammatory response [32]. In this context it is interesting to note that IL-12 has been reported to be important in the production of IL-10 [33]. Diminished IL-12 may thus lead to less IL-10, which may reduce the benefit of IL-12 blockade, thus explaining the arthritic lesions observed in the anti-IL-12-treated mice.
The present study shows that anti-IL-12 blocks one of the prominent Th1 responses to CII and thus dramatically attenuates CIA. This attenuation did not appear to be caused by a shift in the Th1/Th2 balance, as the up-regulation of Th2 responses was not observed with the treated mice. We conclude that IL-12 has a major role in the induction of CIA and is involved in mediating a vigorous Th1 immune and inflammatory response to CII.
Acknowledgments
This study was funded by the Arthritis and Rheumatism Council, the Medical Research Council of Great Britain and the European Union. The authors would like to thank Paul Warden and the staff of our Biological Services Unit; Philip Connolly for preparation of the histological sections; and Maurice Gately (Hoffmann-La Roche, Nutley, NJ) for advice.
References
- 1.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13:251–76. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 2.Wolf SF, Temple PA, Kobayashi M, et al. Cloning of cDNA for natural kill cell stimulatory factor, a heterodimeric cytokine with multiple biological effects on T and natural killer cells. J Immunol. 1991;146:3074–81. [PubMed] [Google Scholar]
- 3.Chan SH, Perussia B, Gupta JW, et al. Induction of interferon γ production by natural killer stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med. 1991;173:869–79. doi: 10.1084/jem.173.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manetti R, Parronchi P, Giudizi MG, Piccinni M-P, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor (interleukin 12) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177:1199–204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sypek JP, Chung CL, Mayor SEH, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG. Resolution of cutaneous leishmaniasis: interleukin-12 initiates a protective T helper type 1 immune response. J Exp Med. 1993;177:1797–802. doi: 10.1084/jem.177.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heinzel FP, Schoenhaut DS, Rerko RM, Rosser LE, Gately MK. Recombinant interleukin 12 cures mice infected with Leishmania major. J Exp Med. 1993;177:1505–9. doi: 10.1084/jem.177.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med. 1995;181:381–6. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–90. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anguita J, Persing DH, Rincon H, Barthold SW, Fikrig E. Effect of anti-interleukin 12 treatment on murine Lyme Borreliosis. J Clin Invest. 1996;97:1028–34. doi: 10.1172/JCI118494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Courtenay JS, Dallman MJ, Dayan AD, Martin A, Mosedale B. Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283:666–8. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- 11.Seki N, Sudo Y, Yoshioka T, et al. Type II collagen-induced arthritis. I. Induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. J Immunol. 1988;140:1477–84. [PubMed] [Google Scholar]
- 12.Ranges GE, Sriram S, Cooper SM. Prevention of type II collagen-induced arthritis by in vivo treatment with anti-L3T4. J Exp Med. 1985;162:1105–10. doi: 10.1084/jem.162.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Germann T, Szeliga J, Hess H, Storkel S, Podlaski FJ, Gately MJ, Schmitt E, Rude E. Administration of interleukin 12 in combination with type II collagen induces severe arthritis in DBA/1 mice. Proc Natl Acad Sci USA. 1995;92:4823–7. doi: 10.1073/pnas.92.11.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mauri C, Williams RO, Walmsley M, Feldmann M. Relationship between Th1/Th2 cytokine patterns and the arthritogenic response in collagen-induced arthritis. Eur J Immunol. 1996;26:1511–4. doi: 10.1002/eji.1830260716. [DOI] [PubMed] [Google Scholar]
- 15.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–8. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkinson VL, Warrier RR, Truitt TP, Nunes P, Gately MK, Presky DH. Characterization of anti-mouse IL-12 monoclonal antibodies and measurement of mouse IL-12 by ELISA. J Immunol Methods. 1996;189:15–24. doi: 10.1016/0022-1759(95)00223-5. [DOI] [PubMed] [Google Scholar]
- 17.Tripp CS, Gately MK, Hakimi J, Ling P, Unanue ER. Neutralization of IL-12 decreases resistance to Listeria in SCID and C.B-17 mice. Reversal by IFNγ. J Immunol. 1994;152:1883–7. [PubMed] [Google Scholar]
- 18.Heinzel FP, Rerko RM, Ling P, Hakimi J, Schoenhaut DS. Interleukin 12 is produced in vivo during endotoxemia and stimulates synthesis of gamma interferon. Immunity. 1994;62:4244–9. doi: 10.1128/iai.62.10.4244-4249.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espevik T, Nissen-Meyer J. A highly sensitive cell line, WEHI 164 clone 13, for measuring cytotoxic factor/tumor necrosis factor from human monocytes. J Immunol Methods. 1986;95:99–105. doi: 10.1016/0022-1759(86)90322-4. [DOI] [PubMed] [Google Scholar]
- 20.Baker D, Butler D, Scallon BJ, O'Neill JK, Turk JL, Feldmann M. Control of established experimental allergic encephalomyelitis by inhibition of tumor necrosis factor (TNF) activity within the central nervous system using monoclonal antibodies and TNF-receptor-immunoglobulin fusion proteins. Eur J Immunol. 1994;24:2040–8. doi: 10.1002/eji.1830240916. [DOI] [PubMed] [Google Scholar]
- 21.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 22.Ross SE, Williams RO, Mason LJ, Mauri C, Marinova-Mutafchieva L, Malfait A-M, Maini RN, Feldmann M. Suppression of TNFα expression, inhibition of Th1 activity and amelioration of collagen-induced arthritis by rolipram. J Immunol. 1997 in press. [PubMed] [Google Scholar]
- 23.Finkelman FD, Holmes J, Katona IM, et al. Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol. 1990;8:303–34. doi: 10.1146/annurev.iy.08.040190.001511. [DOI] [PubMed] [Google Scholar]
- 24.Germann T, Bongartz M, Dlugonska H, et al. Interleukin-12 profoundly upregulates the synthesis of antigen-specific complement-fixing IgG2a, IgG2b and IgG3 antibody subclasses in vivo. Eur J Immunol. 1995;25:823–9. doi: 10.1002/eji.1830250329. [DOI] [PubMed] [Google Scholar]
- 25.McIntyre KW, Shuster DJ, Gillooly KM, et al. Reduced incidence and severity of collagen-induced arthritis in interleukin-12 deficient mice. Eur J Immunol. 1996;26:2933–8. doi: 10.1002/eji.1830261219. [DOI] [PubMed] [Google Scholar]
- 26.Allavena P, Paganin C, Zhou D, Bianchi G, Sozzani S, Mantovani A. Interleukin-12 is chemotactic for natural killer cells and stimulates their interaction with vascular endothelium. Blood. 1994;84:2261–8. [PubMed] [Google Scholar]
- 27.Tilg H, Atkins MB, Dinarello CA, Mier JW. Induction of circulating interleukin 10 by interleukin 1 and interleukin 2, but not by interleukin 6 immunotherapy. Cytokine. 1995;7:734–9. doi: 10.1006/cyto.1995.0087. [DOI] [PubMed] [Google Scholar]
- 28.Wanidworanum C, Strober W. Predominant role of tumor necrosis factor-α in human monocyte IL-10 synthesis. J Immunol. 1993;151:6853–61. [PubMed] [Google Scholar]
- 29.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–22. [PubMed] [Google Scholar]
- 30.deWaal-Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;74:1209–20. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katsikis P, Chu CQ, Brennan FM, Maini RN, Feldmann M. Immunoregulatory role of interleukin 10 (IL-10) in rheumatoid arthritis. J Exp Med. 1994;179:1517–27. doi: 10.1084/jem.179.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldmann M, Brennan FM, Walmsley M, Cohen S, Katsikis P, Maini RN. Homeostatic regulatory mechanisms in rheumatoid arthritis synovium: interleukin 10 is the major anti-inflammatory cytokine. In: Lightman S, editor. Horizons in medicine. London: Blackwell Science; 1996. pp. 343–51. No. 7. [Google Scholar]
- 33.Morris SC, Madden KB, Adamovicz JJ, Gause WC, Hubbard BR, Gately MK, Finkelman FD. Effects of IL-12 on in vivo cytokine gene expression and Ig isotype selection. J Immunol. 1994;152:1047–56. [PubMed] [Google Scholar]