Ligand Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) Inhibits Cell Growth of Human N/TERT-1 Keratinocytes (original) (raw)
. Author manuscript; available in PMC: 2008 Jun 1.
Abstract
The functional role of peroxisome proliferator-activated receptor-β (PPARβ; also referred to as PPARδ) in epidermal cell growth remains controversial. Recent evidence suggests that ligand activation of PPARβ/δ increases cell growth and inhibits apoptosis in epidermal cells. In contrast, other reports suggest that ligand activation of PPARβ/δ leads to the induction of terminal differentiation and inhibition of cell growth. In the present study, the effect of the highly specific PPARβ/δ ligand GW0742 on cell growth was examined using a human keratinocyte cell line (N/TERT-1) and mouse primary keratinocytes. Ligand activation of PPARβ/δ with GW0742 prevented cell cycle progression from G1 to S phase and attenuated cell proliferation in N/TERT-1 cells. Despite specifically activating PPARβ/δ as revealed by target gene induction, no changes in PTEN, PDK and ILK expression or downstream phosphorylation of Akt were found in either N/TERT-1 cells or primary keratinocytes. Further, altered cell growth resulting from serum withdrawal and the induction of caspase-3 activity by ultraviolet radiation were unchanged in the absence of PPARβ/δ expression and/or the presence of GW0742. While no changes in the expression of mRNAs encoding cell cycle control proteins were found in response to GW0742, a significant decrease in the level of ERK phosphorylation was observed. Results from these studies demonstrate that ligand activation of PPARβ/δ does not lead to an anti-apoptotic effect in either human or mouse keratinocytes, but rather, leads to inhibition of cell growth likely through the induction of terminal differentiation.
Keywords: peroxisome proliferator-activated receptor-β/δ, PPARβ/δ, keratinocytes, N/TERT-1 human keratinocytes, differentiation, cell proliferation, apoptosis
1. Introduction
PPARs are ligand activated transcription factors belonging to the nuclear receptor superfamily. In the past decade, the functional role(s) of PPARs have been delineated and range from modulation of glucose and lipid homeostasis to regulation of cell growth and differentiation (reviewed in [1-5]). There is great potential for agonists of PPARβ/δ for the treatment and prevention of a number of diseases. Highly specific PPARβ/δ ligands can significantly increase serum HDL cholesterol [6-8], increase skeletal muscle fatty acid catabolism [9, 10] and decrease serum glucose in a diabetic mouse model [11]. The observed changes in HDL cholesterol levels, cholesterol efflux, increased skeletal muscle fatty acid catabolism and reductions in serum glucose induced by PPARβ/δ ligands support the hypothesis that PPARβ/δ represents a good molecular target to prevent/treat dyslipidemia, obesity, diabetes and/or atherosclerosis [12, 13].
While there is good reason to develop high affinity ligands for PPARβ/δ for the prevention/treatment of human disease, there is considerable controversy regarding the safe use of high affinity PPARβ/δ ligands, especially for indications requiring long-term, chronic treatment regimen. This concern is due in large part to the effects of PPARβ/δ and its ligands on cell growth and carcinogenesis. Indeed, some reports suggest that ligand activation of PPARβ/δ potentiates cell growth, while other reports suggest that ligand activation of PPARβ/δ attenuates cell growth (reviewed in [3]). For example, culturing human breast and prostate cancer cell lines in the presence of a PPARβ/δ ligand causes an increase in cell proliferation [14]. Similarly, colon cancer cell lines cultured in the presence of the PPARβ/δ ligand GW501516 exhibit inhibited levels of apoptosis [15, 16]. It has been postulated that apoptosis is inhibited by PPARβ/δ-dependent down-regulation of the tumor suppressor phosphatase and tensin homologue deleted on chromosome ten (PTEN) and up-regulation of the oncogenes 3-phosphoinositide-dependent kinase-1 (PDK1) and integrin-linked kinase-1 (ILK1) [17]. The net effect of this change in activity would be increased phosphorylation of Akt and inhibition of apoptosis, and these changes were shown in cultured primary keratinocytes [17]. More recently it was suggested that ligand activation of PPARβ/δ induces expression of COX2, which could theoretically promote cell growth and inhibit apoptosis through mechanisms that involve the production of prostaglandins and/or inflammation-dependent signaling [18]. However, there are several observations that are inconsistent with the idea that PPARβ/δ and/or ligands of PPARβ/δ potentiate cell growth. For example, inhibition of cell growth is observed in a variety of different cells and cell lines cultured in the presence of highly specific PPARβ/δ ligands including, human colonocytes [19], a human lung adenocarcinoma cell line [20], mouse lung fibroblasts [21], rat cardiomyocytes [22] a human keratinocyte cell line [23], normal human keratinocytes [24] and mouse primary keratinocytes [25]. The mechanisms underlying the inhibitory effect of PPARβ/δ ligands on cell growth likely include PPARβ/δ-dependent modulation of terminal differentiation. Indeed, there is strong evidence that ligand activation of PPARβ/δ induces terminal differentiation of keratinocytes [24-27] and it has also been shown that differentiation of breast and colon cancer cell lines is associated with increased expression of PPARβ/δ [28]. Additionally, PPARβ/δ ligands can significantly inhibit the expression of proinflammatory molecules such as interleukins and TNFα in immune cells [29, 30], cardiomyocytes [31], a human endothelial cell line [32], C2C12 mouse myoblasts [33] and liver [34]. Thus, while there is great potential for PPARβ/δ ligands as therapeutic agents, this potential is negatively affected by the controversy regarding the biological role(s) of PPARβ/δ in cell proliferation and carcinogenesis. For this reason, the present study examined the effect of ligand activation of PPARβ/δ on cell growth in a human keratinocyte cell line and mouse primary keratinocytes from wild-type and PPARβ/δ-null mice.
2. Materials and methods
2.1. Cell culture
N/TERT-1 keratinocytes are an hTERT-immortalized human keratinocyte cell line that maintains the ability to differentiate ([35], kindly provided by J. Rheinwald). N/TERT-1 cells are maintained at low density in keratinocyte serum-free media (Ker-SFM, Invitrogen) supplemented with 0.4 mM Ca2+, 0.2 ng/mL EGF, 25 μg/mL BPE, 5 mM L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin. To achieve healthy confluent cultures, medium was changed to a 1:1 mixture of Ker-SFM and DF-K medium (DF-K medium consists of equal volumes of Ham's F-12 and DMEM supplemented with 0.4 mM Ca2+, 0.2 ng/mL EGF, 25 μg/mL BPE, 5 mM L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin). Mouse primary keratinocytes were obtained from wild-type or PPARβ/δ-null neonatal pups, and cultured as previously described [25, 36].
2.2. Transient transfections
Forty-eight hours after plating, subconfluent N/TERT-1 cells were transfected with 0.5 μg 3X-PPRE luciferase and 0.25 μg β-galactosidase reporter plasmids per well using Lipofectamine-Plus (Invitrogen) for five hours in DF-K medium lacking antibiotics and growth factors. Cells were allowed to recover overnight in complete Ker-SFM/DF-K medium, and the following day, treated in triplicate for 24 hours with 0.1% DMSO, 0.2 μM GW0742 or 1.0 μM GW0742. Cells were lysed and the lysate used to measure luciferase activity with a Turner 20/20 luminometer and luciferase assay reagents (Promega). Luciferase activity was normalized to β-galactosidase activity and total cellular protein (Pierce-BCA).
2.3. Protein analysis
To verify expression of PPARβ/δ in N/TERT-1 cells, cells were cultured as described above and treated for 24 hours with 0.1% DMSO, 0.2 μM GW0742 or 1.0 μM GW0742. Following treatment, cells were washed twice in ice-cold PBS, and then lysed in cold lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM PMSF, 1X protease inhibitor cocktail). Insoluble material was removed by centrifugation and protein concentration quantified from the supernatants (Pierce-BCA). Equal amounts of protein were separated using SDS-PAGE, and transferred to a PVDF membrane. Transfer efficiency and equal protein loading was verified with Ponceau-S staining. Following blocking for one hour in 5% non-fat dry milk in Tris-buffered saline with 0.1% Tween-20, immunoblots were incubated in primary antibody against PPARβ/δ (Santa Cruz). Detection was performed using an HRP-conjugated secondary antibody and enzyme-linked chemiluminescence. Positive and negative control lysates were generated by transfecting COS-1 cells with pSG5-mPPARβ, pSG5-mPPARα, pSG5-mPPARγ or pSG5.
To examine expression of phosphorylated proteins involved in cell proliferative signaling, triplicate samples of N/TERT-1 cells were cultured as described above in either 0.1% DMSO or 1.0 μM GW0742 for either 6 or 24 hours. Cell lysates were prepared as above from the triplicate samples and equal amounts of protein were pooled for screening analysis by Kinexus on the Kinetworks Cell Cycle Status Screen [KPSS-10.0]. Confirmatory western blot analysis was performed for proteins that were potentially influenced by GW0742. Primary antibodies against phospho-Akt (Ser473), Akt, phospho-ERK1/2 (Thr202/Tyr204), ERK, PTEN, retinoblastoma (Rb), phospho-Rb (S780, S807/S811), Elk-1, phospho-Elk-1 (S383) were obtained from Cell Signaling (Beverly, MA). Primary antibodies against PDK1 and ILK1 were from Upstate Biotechnologies (Lake Placid, NY) and the antibody for lactic dehydrogenase (LDH) was from Jackson Immunoresearch (West Grove, PA). For quantitative immunoblots, membranes were probed with primary antibodies and then incubated with biotinylated secondary antibodies (Jackson Immunoresearch, West Grove, PA), washed, incubated in 125I-strepavidin, and exposed to phosphorimager plates. Hybridization signals were normalized to the signal detected for LDH, which was used as a loading control.
2.4. Analysis of cell growth, cell cycle progression and apoptosis
N/TERT-1 keratinocytes were seeded onto 6-well tissue culture dishes at 30,000 cells per well in Ker-SFM. Twenty-four hours later, cell number was measured with a Z1 coulter particle counter® (Beckman Counter, Inc., Hialeah, FL) to determine plating efficiency (Day 0). For the remaining cells, medium was changed to Ker-SFM/DF-K, and cells were treated in triplicate with 0.1% DMSO, 0.1 μM or 1 μM GW0742. Cell number was determined at daily intervals, and the remaining cells were retreated with fresh media and treatment each day for up to 6 days. For flow cytometry analysis, N/TERT-1 cells were treated in triplicate as described above. Every 24 h, cells were suspended with trypsin, washed in ice-cold PBS, and then fixed in ice-cold ethanol. DNA content was determined with propidium iodide staining and flow cytometry (Beckman EPICS XL) using standard techniques. The percentage of cells at each phase of the cell cycle ± S.D. was determined with MultiCycle® analysis software on at least 10,000 gated events. To examine the role of PPARβ/δ on apoptosis, primary keratinocytes from wild-type and PPARβ/δ-null mice were cultured as described above. Cells were maintained in low calcium medium (0.05 mM) with or without 8% serum in the presence or absence of 0.5 μM GW0742. The number of cells was quantified 24 and 72 hours after serum removal using a Z1 coulter particle counter® (Beckman Counter, Inc., Hialeah, FL). This approach allowed for examination of serum withdrawal-induced apoptosis, and whether ligand activation of PPARβ/δ modified this effect through a PPARβ/δ-dependent mechanism. As an alternative to this approach, primary keratinocytes from wild-type and PPARβ/δ-null mice were also irradiated with 20,000 μJ/cm2 UV light using the CL-1000 Ultra Violet Crosslinker, and caspase 3 activity was measured 9 or 18 hours post-irradiation using the Caspase 3 Glo reagent (Promega) following manufacturer’s recommended procedures.
2.5. Analysis of mRNA expression
N/TERT-1 cells were cultured as described above in Ker-SFM/DF-K media. Cells in the log-growth phase were treated in triplicate with 0.1% DMSO or 1 μM GW0742 for 1, 2, 4, 8, 12 or 24 hours. Following each timepoint, cells were washed twice with PBS, and RNA was purified with Trizol® reagent according to the manufacturer's instructions. Quantitative real-time PCR was carried out on equal amounts of cDNA (10-50 ng) as previously described [37] to quantify the level of expression of mRNA encoding the known PPARβ/δ target gene ADRP [37, 38], as well as mRNA encoding PDK1, ILK1, transglutaminase-I (TG-I), involucrin, small proline-rich protein 1A (SPR1A), and keratin 10 (K10). Relative mRNA expression was determined with a standard curve created from various amounts (0-200 ng) of untreated N/TERT-1 cDNA and normalized to GAPDH mRNA levels. Similar analysis for changes in gene expression induced by ligand activation of PPARβ/δ was performed using cultured primary keratinocytes from wild-type and PPARβ/δ-null mice, except that samples were obtained after 8 or 12 hours of treatment with 0.2 μM GW0742. Additionally, mRNA analysis of the primary keratinocytes was performed using northern blotting as previously described [36] and quantitative western blotting as described above.
For RNase protection assays (RPAs), subconfluent N/TERT-1 cells were cultured in the presence or absence of 1.0 μM GW0742 for 24, 48 or 72 hours as described above. RNA was isolated from these cells and used for analysis by RPA (BD Biosciences, San Diego, CA). RPAs were carried out essentially as described [39]. Unlabeled sense RNA encoding p130, Rb, p107, p53, p57, p21, p19, p18, p16, p14/15, ribosomal protein L32 (L32), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from BD Biosciences (San Diego, CA). For synthesis of radiolabeled antisense RNA, the final reaction mixture (10 μl) contained 70 μCi of [α-32P]UTP (3,000 Ci/mmol; NEN, Cambridge, MA); 10 μmol of UTP; 500 μmol each of GTP, ATP, and CTP; 10 μmol of dithiothreitol; 1X transcription buffer; 12 U of RNasin; 8 U of T7 polymerase and 1 μL of the template probe. After 1 h at 37° C, the mixture was treated with RNase-free DNase (2 U; Pharmingen) for 30 min at 37°C, and the probes were purified by extractions with phenol-chloroform and chloroform, followed by precipitation with ethanol. Dried probes were dissolved (3 × 105 cpm/μl) in hybridization buffer (80% formamide, 0.4 M NaCl, 1 mM EDTA, and 40 mM PIPES, pH 6.6) and added (2 μl; 3 × 105 cpm/μl) to tubes containing sample RNA (10 μg) dissolved in 8 μl of hybridization buffer. The samples were heated at 80°C for 3 min and incubated at 56°C for 16 h. The single stranded RNA was subsequently digested by the addition of a solution (100 μl) of RNase A (0.2 μg/ml; Sigma) and RNase T1 (600 U/ml; GIBCO BRL) in 10 mM Tris, 300 mM NaCl, and 5 mM EDTA, pH 7.5. After incubation (30 min at 37°C), the samples were treated with 18 μl of a mixture of proteinase K (1 mg/ml; GIBCO BRL), SDS (5%), and yeast tRNA (200 μg/ml). The RNA duplexes were isolated by extraction and precipitation as above, dissolved in 5 μl of gel loading buffer (65% formamide, 5.5 mM EDTA, and dyes), and electrophoresed in standard 5% acrylamide-8 M urea sequencing gels. Dried gels were placed on XAR film (Kodak, Rochester, NY) with intensifying screens and were developed at -80 °C for 18 h. For quantification, dried gels were developed on phosphorimager screens, and gel band intensity was analyzed by filmless autoradiographic analysis.
3. Results
3.1. GW0742 inhibits growth of N/TERT-1 keratinocytes
N/TERT-1 cells are an immortalized human keratinocyte cell line that retain growth factor dependence and normal differentiation characteristics [35]. PPARβ/δ is known to be highly expressed in skin and keratinocytes (reviewed in [3]). To determine whether N/TERT-1 keratinocytes express a functional PPARβ/δ, cells were transfected with a PPRE-driven luciferase reporter gene and treated with either 0.2 μM or 1.0 μM GW0742. Consistent with PPARβ/δ activity, a significant increase in reporter activity was observed after treatment with GW0742 (Fig. 1A). Additionally, western blot analysis confirmed that N/TERT-1 keratinocytes express PPARβ/δ and that GW0742 had no effect on PPARβ/δ expression (Fig. 1B). To begin to determine the effect of ligand activation of PPARβ/δ on cell growth of N/TERT-1 keratinocytes, cells were cultured in the presence of either 0.1 μM or 1.0 μM GW0742. No significant differences in cell proliferation were observed in the presence of 0.1 μM GW0742, while significant inhibition in the average number of cells was found after four to six days of culture in 1.0 μM GW0742 (Fig. 2A). To determine if GW0742 altered cell cycle progression in N/TERT-1 cells, parallel cultures were treated as in Fig. 2A, and DNA content determined with propidium iodide staining and analysis by flow cytometry. Activation of PPARβ/δ with GW0742 was associated with an increase in the number of cells in the G1 phase and a decrease in the number of cells in the S phase after two to four days of culture (Fig. 2B).
Fig. 1.
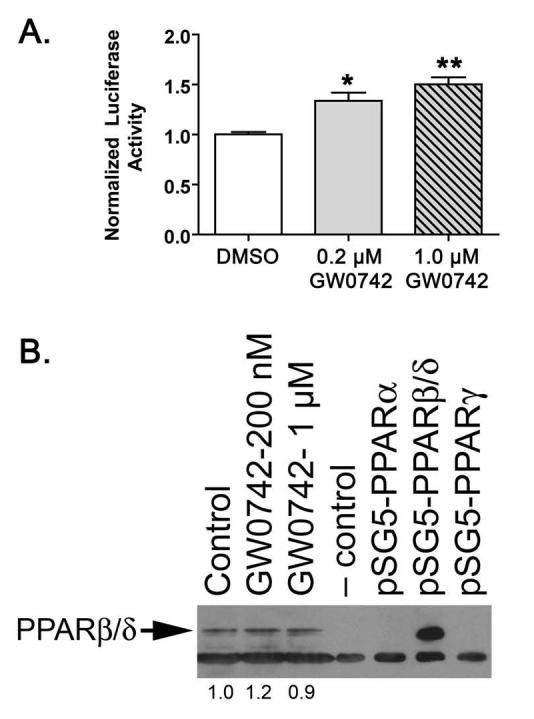
N/TERT-1 keratinocytes express a functional PPARβ/δ. (A) N/TERT-1 keratinocytes were cultured as described and transfected with 3X-PPRE luciferase and β-galactosidase reporter plasmids. Luciferase activity was determined after culture with either 0, 0.2 or 1.0 μM GW0742 and normalized to β-galactosidase and total cellular protein (Pierce-BCA). Values represent the mean ± S.E.M.. *Significantly greater than control DMSO-treated, P ≤ 0.05. (B) N/TERT-1 cells were treated for 24 hours with either 0, 0.2 or 1.0 μM GW0742, and assayed for PPARβ/δ expression by western blotting. Positive and negative control lysates were generated by transfecting COS-1 cells with pSG5-mPPARβ/δ or pSG5, respectively. Image J software (v 1.37) was used to quantify PPARβ/δ expression and values are presented as the fold change relative to control.
Fig. 2.
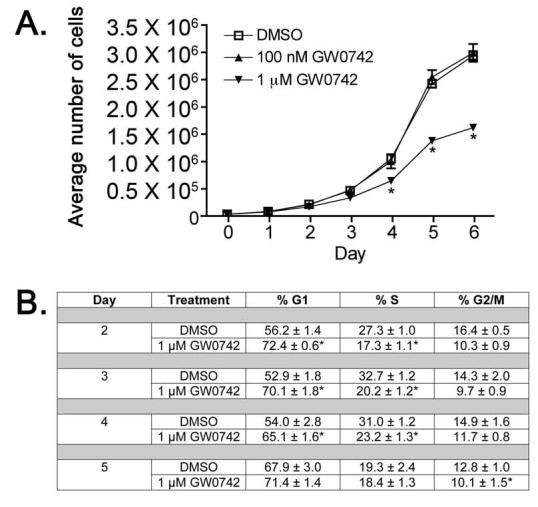
Ligand activation of PPARβ/δ leads to inhibition of cell growth in N/TERT-1 keratinocytes (A) N/TERT-1 keratinocytes were seeded onto 6-well tissue culture dishes and cultured over a six day period in the presence of 0, 0.1, or 1.0 GW0742. Cell number was quantified daily with a Coulter counter. Values represent the mean ± S.E.M. from triplicate, independent samples. *Significantly greater than control DMSO-treated, P ≤ 0.05. (B) Cells were cultured and treated in triplicate as described in Part A. Every 24 h, cells were trypsinized, washed in ice-cold PBS, and fixed in ethanol for the determination of DNA content using flow cytometry. The mean percentage of cells at each phase of the cell cycle ± S.D. was determined as described in Materials and Methods. *Significantly greater than control DMSO-treated, P ≤ 0.05.
3.2. Ligand activation of PPARβ/δ by GW0742 in N/TERT-1 keratinocytes does not alter expression of the PTEN/PDK1/ILK1/Akt pathway
Previous studies by others suggest that ligand activation of PPARβ/δ in mouse primary keratinocytes causes anti-apoptotic signaling mediated by inhibition of PTEN expression and increased expression of the oncogenes PDK1 and ILK1 leading to increased phosphorylation of Akt [17]. To determine whether this pathway functions similarly in N/TERT-1 cells, mRNA expression was examined using quantitative real-time PCR. Analysis of mRNA expression demonstrated that treatment of N/TERT-1 cells with 1.0 μM GW0742 caused a significant increase in the mRNA encoding ADRP (Fig. 3A), a known PPARβ/δ target gene which was evident at 1 hour and reached maximal levels at 24 hours after ligand treatment [38]. Despite activation of PPARβ/δ, no change in the expression of mRNA encoding PDK1 or ILK1 was found (Fig. 3A). Quantitative western blot analysis was consistent with these observations as no differences in the expression of PTEN, PDK1, ILK1, phospho-Akt or Akt protein levels were observed after treatment with 1.0 μM GW0742 in multiple timepoints up to 24 hours post-treatment (Fig. 3B).
Fig. 3.
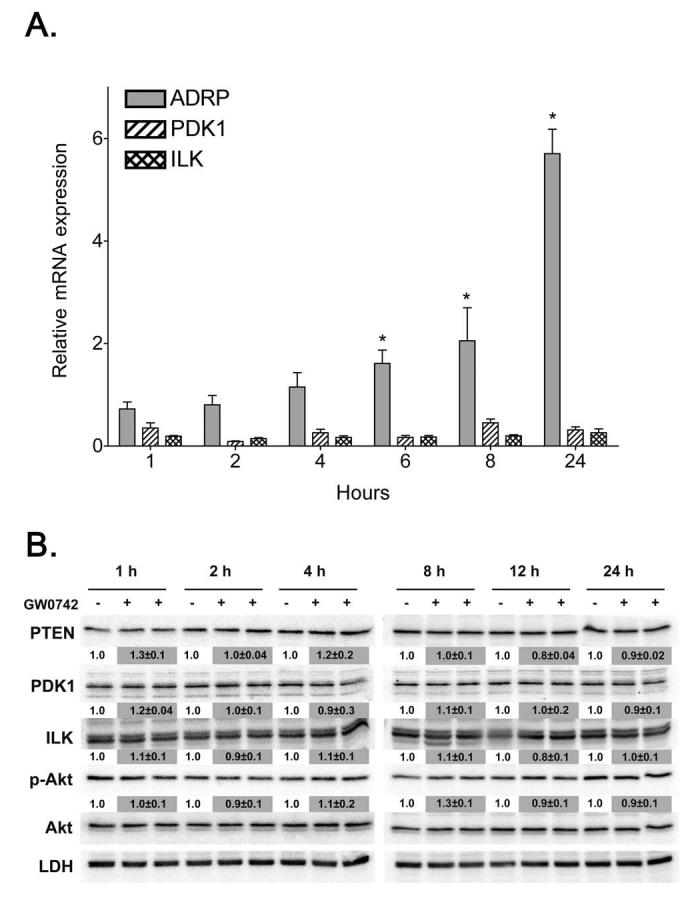
Ligand activation of PPARβ/δ does not modulate the PTEN/PDK1/ILK1/Akt pathway in N/TERT-1 cells (A) Triplicate cultures of N/TERT-1 keratinocytes were treated with either 0.1% DMSO or 1.0 μM GW0742 and quantitative real-time PCR was carried out on equal amounts of cDNA. Results were calculated from a standard curve created from various amounts (0-200 ng) of untreated N/TERT-1 cDNA and normalized to GAPDH expression relative to DMSO controls. Values represent the mean ± S.E.M.. *Significantly different than DMSO control, P ≤ 0.05. (B) N/TERT-1 cells were cultured as described in Materials and methods and treated with either 0.1% DMSO or 1.0 μM GW0742 for 1, 2, 4, 8, 12 or 24 h. Protein was separated by SDS-PAGE, transferred to PVDF membrane, and protein expression determined by western blotting. Normalized hybridization values are presented as the fold change relative to control and represent the mean ± S.E.M..
3.3. Ligand activation of PPARβ/δ by GW0742 in mouse primary keratinocytes does not alter expression of the PTEN/PDK1/ILK1/Akt pathway
The lack of change in the PTEN/PDK1/ILK1/Akt pathway in N/TERT-1 cells (Fig. 3), suggests that there could be a species difference in the response to ligand activation of PPARβ/δ. To examine this hypothesis, primary keratinocytes from wild-type and PPARβ/δ-null mice were cultured in the presence of GW0742 and used for analysis of mRNA expression by northern blotting and protein expression by quantitative western blotting. Consistent with observations made in N/TRET-1 cells, a significant increase in the expression of ADRP mRNA was observed in wild-type keratinocytes after eight and twelve hours of culture in 0.2 μM GW0742, and this effect was not observed in similarly treated PPARβ/δ-null keratinocytes (Fig. 4A). These results demonstrate a PPARβ/δ-dependent increase of ADRP mRNA expression by GW0742. However, in contrast to a previous report [17], ligand activation of PPARβ/δ led to an increase in the mRNA encoding PTEN, and no significant changes in the expression of mRNA encoding PDK1 or ILK1 in wild-type keratinocytes (Fig. 4A). Interestingly, constitutive expression of mRNA encoding PDK1 and ILK1 was significantly higher in PPARβ/δ-null keratinocytes as compared to wild-type keratinocytes (Fig. 4A). Quantitative protein analysis showed no significant differences in the level of PTEN, PDK1, ILK1, phospho-Akt or Akt by GW0742 in wild-type or PPARβ/δ-null keratinocytes (Fig. 4B). However, constitutive expression of ILK1 was significantly higher in PPARβ/δ-null keratinocytes (Fig. 4B), consistent with mRNA analysis (Fig. 4A). These results suggest that ligand activation of PPARβ/δ in keratinocytes would not lead to inhibition of the apoptotic pathway as previously suggested by others [15-17]. Indeed, when apoptosis was induced in primary keratinocytes using serum withdrawal, similar decreases in cell number were observed in both genotypes (Fig. 5A). Further, treatment with 0.5 μM GW0742 did not alter the decrease in average cell number caused by serum withdrawal in either genotype at either timepoint (Fig. 5A). Similar to N/TERT-1 cells, treatment of wild-type keratinocytes with 0.5 μM GW0742 resulted in a significant decrease in the average number of cells after three days of culture in the presence of serum, and this effect was not found in similarly treated PPARβ/δ-null keratinocytes (Fig. 5A). Additionally, average cell number was significantly greater in PPARβ/δ-null keratinocytes cultured in the presence of serum after three days as compared to wild-type keratinocytes (Fig. 5A). These results demonstrate that while ligand activation of PPARβ/δ can inhibit cell growth of primary keratinocytes, GW0742 does not exacerbate or attenuate the decrease in cell number resulting from serum withdrawal. Since serum withdrawal can reduce cell number through mechanisms of growth inhibition including necrosis and apoptosis, the effect of UV irradiation on apoptosis was examined in mouse primary keratinocytes using caspase-3 activity as a marker. Consistent with the analysis of serum withdrawal, UV-induced caspase-3 activity was not different between wild-type or PPARβ/δ-null keratinocytes (Fig. 5B).
Fig. 4.
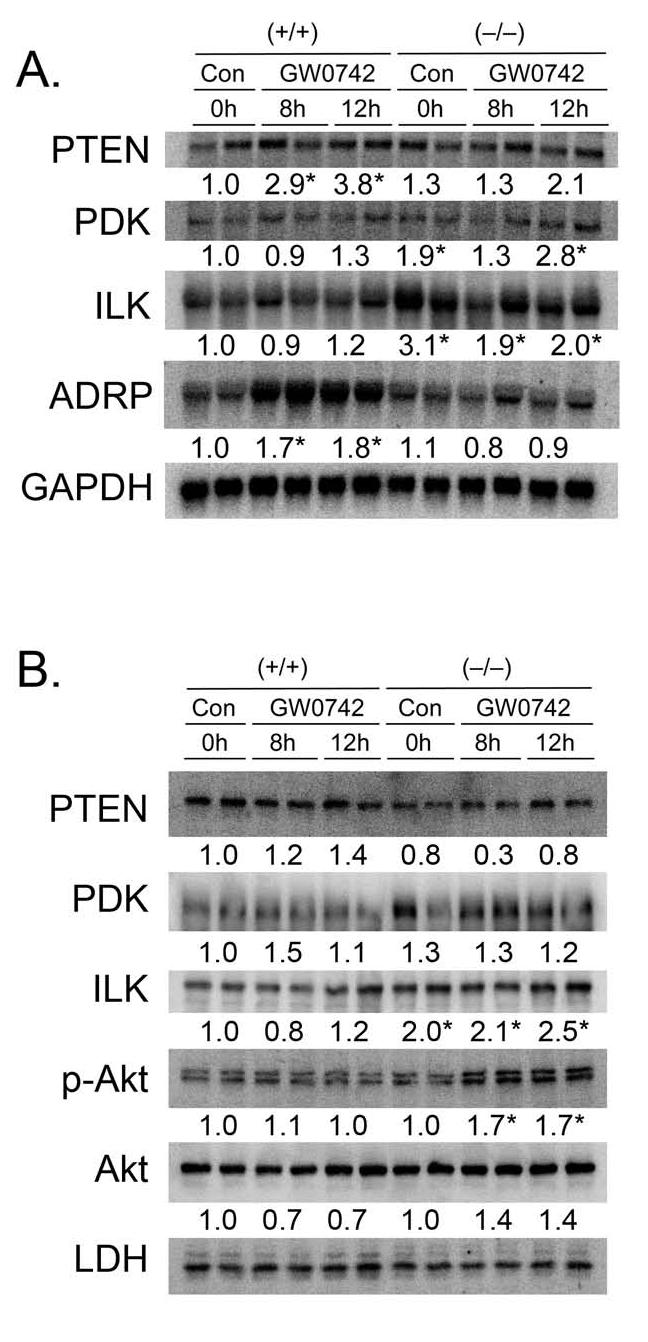
Effect of GW0742 on the PTEN/PDK1/ILK1/Akt pathway in primary mouse keratinocytes from wild-type (+/+) and PPARβ/δ-null (-/-) mice. (A) Expression of mRNA encoding PTEN, PDK1, ILK1 or the PPARβ/δ target gene ADRP was determined by Northern blot analysis as described in Materials and methods. Normalized hybridization values are presented as the fold change relative to control and represent the mean ± S.E.M.. *Significantly different than (+/+) control at 0 hours, P ≤ 0.05. (B) Protein was isolated from (+/+) and (-/-) keratinocytes cultured in the presence of absence of 0.2 μM GW0742 for up to 12 hours and separated by SDS-PAGE. Quantitative western blot analysis was performed using radioactive detection methods as described in Materials and methods. Normalized hybridization values are presented as the fold change relative to control and represent the mean ± S.E.M.. *Significantly different than control (+/+) at 0 hours, P ≤ 0.05.
Fig. 5.
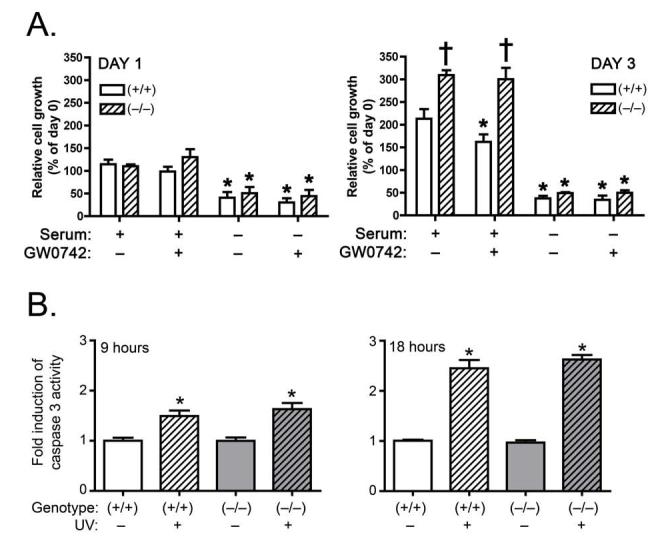
Effect of PPARβ/δ on serum deprivation and UV irradiation changes in cell growth in wild-type (+/+) and PPARβ/δ-null (-/-) primary keratinocytes. (A) Primary keratinocytes from (+/+) and (-/-) mice were treated with and without 8% serum in the presence or absence of 0.5 μM GW0742. Cell number was measured using a Coulter counter after 1 day of culture (left panel) and 3 days of culture (right panel). Values represent the mean ± S.E.M.. *Significantly less than (+/+) control cultured in the presence of 8% serum. †Significantly greater than (+/+) control cultured in the presence of 8% serum. (B) UV-induced caspase 3 activity was measured in (+/+) and (-/-) keratinocytes after 9 hours (left panel) and 18 hours (right panel) Values represent the mean ± S.E.M.. *Significantly different than control, P<0.05.
3.4. Ligand activation of PPARβ/δ by GW0742 in N/TERT-1 keratinocytes does not alter expression of mRNAs encoding cell cycle regulators
The previous findings suggest that the growth inhibition observed in both N/TERT-1 cells and primary keratinocytes is not due to altered expression of the PTEN/PDK1/ILK1/Akt pathway, and that ligand activation of PPARβ/δ does not significantly influence the apoptotic pathway. To determine if the G1 to S-phase block found in the N/TERT-1 cells (Fig. 2B) was associated with changes in the mRNA expression of cell cycle regulators, an RPA was performed. Detectable levels of Rb, p107, p53, p27, p21 and p16 were measured by RPA analysis. However, no changes in the expression of mRNA encoding these genes were found in response to 1.0 μM GW0742 after either 24, 48 or 72 hours of treatment (Fig. 6); when increased G1 phase cells were detected (Fig. 2B). The mRNAs encoding p130, p19, p18 and p14/15 were not detectable by RPA.
Fig. 6.
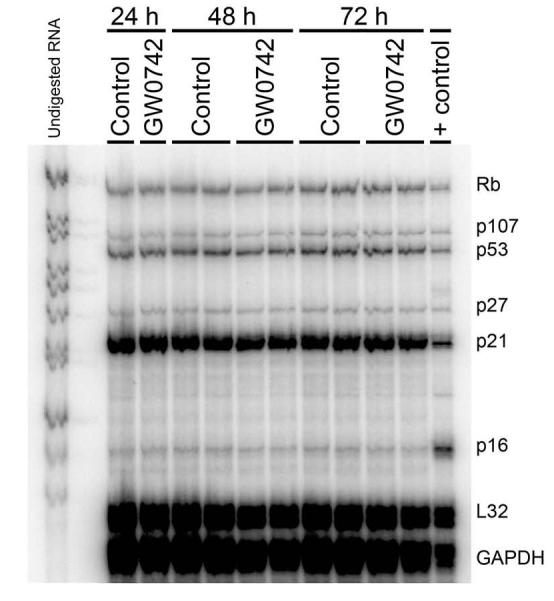
Effect of GW0742 on mRNA encoding cell cycle regulators. An RNase protection assay was performed using RNA from N/TERT-1 keratinocytes cultured in either 0.1% DMSO or 1.0 μM GW0742 after 24, 48 or 72 hours.
3.5. GW0742 inhibits MAPK signaling in N/TERT-1 keratinocytes
In addition to transcriptionally-mediate events, ligands for PPARs can also cause changes in the phosphorylation of proteins that can also modulate cell cycle, and these events can be mediated by both receptor-dependent and receptor-independent mechanisms (reviewed in [40]). To begin to examine the changes in phosphorylation-dependent signaling, global analysis of phosphorylated cell cycle proteins was performed using the Kinexus KPSS 10.0 Screen. Consistent with the analysis performed in mouse primary keratinocytes, no consistent changes in the phosphorylation pattern of PDK1, PTEN, Akt or the Akt substrates mTOR, GSK3, and p27 were detected in N/TERT-1 cells cultured in 1.0 μM GW0742 after either 6 or 24 h of culture (data not shown). No consistent differences were found in the phosphorylation of other known regulators of the cell cycle including p53, Src, RSK or Raf in N/TERT-1 cells cultured in 1.0 μM GW0742 after either 6 or 24 h of culture (data not shown). Significantly reduced phosphorylation of retinoblastoma (Rb) was detected in N/TERT-1 cells cultured in 1.0 μM GW0742 after 24 h, based on the Kinexus screen protein, consistent with inhibited cell growth. However, confirmatory quantitative western blots revealed no significant differences in phosphorylation of Rb at S780 or S870/811 (Fig. 7). In contrast, a significantly lower level of p42/44 ERK (MAPK) in N/TERT-1 cells cultured in 1.0 μM GW0742 after 24 h was confirmed (Fig. 7). Consistent with this observation, phosphorylation of Elk-1 (a known ERK/MAPK substrate) was reduced in N/TERT-1 cells cultured in 1.0 μM GW0742 (Fig. 7).
Fig. 7.

Inhibition of phospho-MAPK (ERK) by GW0742. Confirmatory quantitative western blots were performed using protein from N/TERT-1 cells after preliminary screening by Kinexus, using phospho-specific antibodies as indicated. Normalized hybridization values are presented as the fold change relative to control and represent the mean ± S.E.M.. *Significantly less than DMSO control, P ≤ 0.05.
3.6. GW0742 increases expression of differentiation-related proteins in N/TERT-1 cells
The inhibition of cell proliferation observed in N/TERT-1 cells after treatment with GW0742 could also be due in part to the induction of differentiation, as ligand activation of PPARβ/δ has been shown to cause an increase in terminal differentiation of keratinocytes with a concomitant inhibition of cell growth [24-26]. Indeed, treatment of N/TERT-1 cells with 1.0 μM GW0742 led to an increase in the mRNA encoding a number of known markers of terminal differentiation including TG-I, SPR1A, K10 and involucrin (Fig. 8)
Fig. 8.
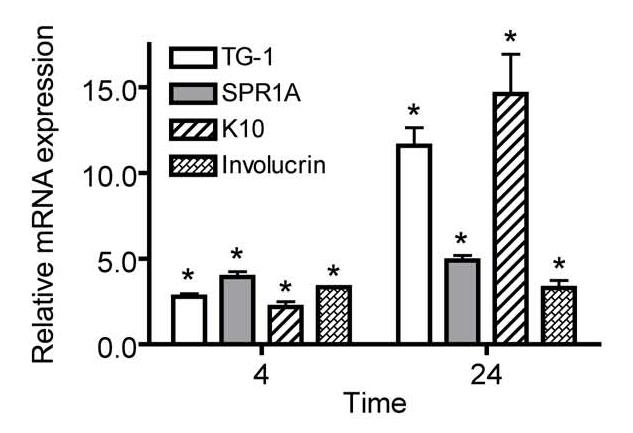
GW0742 induces expression of differentiation marker mRNAs. Triplicate cultures of N/TERT-1 keratinocytes were treated with either 0.1% DMSO or 1.0 μM GW0742 and quantitative real-time PCR was carried out on equal amounts of cDNA to determine the level of mRNA encoding TG-I, SPR1A, involucrin, and K10. Results were calculated from a standard curve created from various amounts (0-200 ng) of untreated N/TERT-1 cDNA and normalized to GAPDH expression relative to the DMSO control. Values represent the mean ± S.E.M.. *Significantly different than DMSO control, P ≤ 0.05.
4. Discussion
There is considerable controversy regarding the effect of ligand activation of PPARβ/δ, as some studies show enhanced cell growth, while others show no influence or inhibited cell growth (reviewed in [3]). There are a number of possible explanations that could account for the reported disparity, including species-specific effects, ligand-specific effects, differences in experimental design and experimental error. Given the potential beneficial use of PPARβ/δ ligands for the treatment of human diseases, it is essential to precisely determine the effect of ligand activation on cell proliferation. This includes differentiating between the effects that are universally related to PPARβ/δ activation versus those that are due to off-target pharmacology and/or species-specific effects. For this reason, the present studies examined the effects of PPARβ/δ ligand activation in a human keratinocyte cell line and in mouse primary keratinocytes.
Results from the present investigation demonstrate that N/TERT-1 cells express a functional PPARβ/δ, and that ligand activation of PPARβ/δ inhibits cell growth in this human keratinocyte cell model. Specifically, the inhibition of cell proliferation correlated with a G1/S phase block in response to treatment with the highly specific PPARβ/δ ligand GW0742. These findings are consistent with previous studies showing inhibition of cell growth in mouse and human keratinocytes [24, 25]. To begin to understand how GW0742 leads to inhibition of cell growth, a number of possible mechanisms were examined.
No differences in the expression of mRNA encoding PDK1 and ILK1 were found in N/TERT-1 keratinocytes cultured in the presence of GW0742. Further, the protein expression of PTEN, PDK1, ILK1 and Akt, and the phosphorylation status of Akt were unchanged in N/TERT-1 keratinoytes following treatment with GW0742. Importantly, under identical conditions, GW0742 increased the expression of the known PPARβ/δ target gene ADRP indicating that GW0742 was pharmacologically active in N/TERT-1 cells. Since work by others has shown repressed expression of PTEN and increased PDK1, ILK1 and Akt activity in passaged mouse keratinocytes [17], the lack of GW0742-induced changes in N/TERT-1 cells suggested that there was a species difference in the response to ligand activation of PPARβ/δ. However, follow up experiments in mouse primary keratinocytes to confirm this hypothesis are inconsistent with this view. Despite the presence of PPARβ/δ-dependent increased ADRP mRNA expression in mouse primary keratinocytes, ligand activation of PPARβ/δ did not lead to repression of mRNA encoding PTEN or increased expression of mRNA encoding PDK1 or ILK1. These findings were verified at the protein level using quantitative western blotting and were similar to the findings in N/TERT-1 keratinocytes. Another consistent finding from this analysis was that constitutively higher expression of ILK1 was found at both the mRNA and protein level in PPARβ/δ-null keratinocytes. The significance of this observation is uncertain but might contribute to increased cell growth found in PPARβ/δ-null keratinocytes [36]. Collectively, these findings also suggest that ligand activation of PPARβ/δ does not negatively influence apoptosis in keratinocytes. To further evaluate this hypothesis, the effect of serum deprivation and UV irradiation was examined in wild-type and PPARβ/δ-null mouse primary keratinocytes. Consistent with the previous observations, decreased cell number was comparable in response to serum deprivation in the presence or absence of PPARβ/δ expression. Additionally, GW0742 did not affect cell number in response to serum withdrawal; although inhibition of cell growth was found in wild-type keratinocytes treated with GW0742, and this effect was consistent with the effect of GW0742 in N/TERT-1 cells. Further, UV-induced apoptosis as measured by caspase-3 activity was similar in both wild-type and PPARβ/δ-null keratinocytes. Combined, this comprehensive analysis reveals that ligand activation by GW0742 does not lead to inhibition of apoptosis by modulating PTEN/PDK1/ILK1/Akt pathway in either N/TERT-1 keratinocytes or mouse primary keratinocytes.
To examine other possible mechanisms underlying the inhibition of cell growth of N/TERT-1 keratinocytes in response to GW0742, the expression pattern of mRNA encoding proteins known to modulate cell cycle transition was examined. While no changes in the expression of Rb, p107, p53, p27, p21 or p16 were found after GW0742 treatment in N/TERT-1 keratinocytes, it remains possible that ligand activation of PPARβ/δ leads to transcriptional changes of other target genes not examined with the RPA probe set that could cause a G1/S phase block. Further work is necessary to test this hypothesis. Interestingly, GW0742 also caused reduced levels of MAPK in N/TERT-1 cells. Whether this effect contributes to the G1/S phase block that resulted from GW0742, or is mediated by a PPARβ/δ-dependent mechanism cannot be determined from the present study, but both possibilities should be examined in future studies.
The most consistent finding from these studies is the induction of differentiation related marker mRNA caused by GW0742 treatment in N/TERT-1 cells. A number of independent laboratories have provided strong evidence that PPARβ/δ can mediate the terminal differentiation of keratinocytes and other cell types [24-28]. Findings from the present studies support these observations as markers of terminal differentiation were also found in N/TERT-1 cells treated with GW0742. This suggests that one mechanism by which ligand activation of PPARβ/δ leads to inhibition of cell growth is through the induction of terminal differentiation, since it is known that the induction of terminal differentiation in keratinocytes is associated with a concomitant decrease in cell growth (reviewed in [3]). However, it is also of interest to note the GW0742 induction of the differentiation marker K10. While it is known that K10 is a functional subunit of the cornified epithelium, there is also evidence that K10 can function to inhibit cell growth of human keratinocytes [41]. Indeed, overexpression of K10 not only inhibits cell growth of human keratinocytes, but this inhibition is associated with a G1/S phase block [41]. These findings are consistent with results from the present studies. Further studies are necessary to determine whether the induction of K10 by GW0742 is mediated by direct regulation by PPARβ/δ, and whether this contributes to the inhibition of cell growth induced by GW0742.
In summary, results from the current studies show that the human keratinocyte cell line N/TERT-1 express a functional PPARβ/δ and that ligand activation of PPARβ/δ in N/TERT-1 and mouse keratinocytes leads to inhibition of cell growth. The inhibition of cell growth induced by GW0742 does not appear to be influenced by PPARβ/δ-dependent modulation of PTEN/PDK1/ILK1/Akt pathway or apoptosis. While the specific target genes that contribute to GW0742-induced inhibition of cell growth are still uncertain, it is likely that this is due in part to the induction of terminal differentiation. Further work is still necessary to definitively determine the mechanisms underlying this effect. However, it is clear from these studies that enhanced cell growth in human and mouse keratinocytes does not occur in response to ligand activation of PPARβ/δ as suggested by others in other cell types. Further work is also necessary to more definitively examine the mechanisms underlying these disparities.
Acknowledgements
The authors kindly acknowledge Dr. J. Rheinwald for providing the N/TERT-1 cells. This work supported by grants from the National Institutes of Health (CA89607, CA97999, CA124533, J.M.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Akiyama TE, Meinke PT, Berger JP. Curr Diab Rep. 2005;5:45–52. doi: 10.1007/s11892-005-0067-3. [DOI] [PubMed] [Google Scholar]
- [2].Beaven SW, Tontonoz P. Annu Rev Med. 2006;57:313–329. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- [3].Burdick AD, Kim DJ, Peraza MA, Gonzalez FJ, Peters JM. Cell Signal. 2006;18:9–20. doi: 10.1016/j.cellsig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- [4].Desvergne B, Michalik L, Wahli W. Physiol Rev. 2006;86:465–514. doi: 10.1152/physrev.00025.2005. [DOI] [PubMed] [Google Scholar]
- [5].Michalik L, Desvergne B, Wahli W. Nat Rev Cancer. 2004;4:61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- [6].Leibowitz MD, Fievet C, Hennuyer N, Peinado-Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, Santini C, Marquis RW, Tolman RL, Smith RG, Moller DE, Auwerx J. FEBS Lett. 2000;473:333–336. doi: 10.1016/s0014-5793(00)01554-4. [DOI] [PubMed] [Google Scholar]
- [7].Oliver WR, Jr., Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. Proc Natl Acad Sci U S A. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wallace JM, Schwarz M, Coward P, Houze J, Sawyer JK, Kelley KL, Chai A, Rudel LL. J Lipid Res. 2005;46:1009–1016. doi: 10.1194/jlr.M500002-JLR200. [DOI] [PubMed] [Google Scholar]
- [9].Holst D, Luquet S, Nogueira V, Kristiansen K, Leverve X, Grimaldi PA. Biochim Biophys Acta. 2003;1633:43–50. doi: 10.1016/s1388-1981(03)00071-4. [DOI] [PubMed] [Google Scholar]
- [10].Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Proc Natl Acad Sci U S A. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM, Evans RM. Proc Natl Acad Sci U S A. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ali FY, Davidson SJ, Moraes LA, Traves SL, Paul-Clark M, Bishop-Bailey D, Warner TD, Mitchell JA. Faseb J. 2006;20:326–328. doi: 10.1096/fj.05-4395fje. [DOI] [PubMed] [Google Scholar]
- [13].Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- [14].Stephen RL, Gustafsson MC, Jarvis M, Tatoud R, Marshall BR, Knight D, Ehrenborg E, Harris AL, Wolf CR, Palmer CN. Cancer Res. 2004;64:3162–3170. doi: 10.1158/0008-5472.can-03-2760. [DOI] [PubMed] [Google Scholar]
- [15].Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Nat Med. 2004;10:245–247. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- [16].Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, Das SK, Dey SK, DuBois RN. Cancer Cell. 2004;6:285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- [17].Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Molecular Cell. 2002;10:721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- [18].Xu L, Han C, Wu T. J Biol Chem. 2006;281:33982–33996. doi: 10.1074/jbc.M600135200. [DOI] [PubMed] [Google Scholar]
- [19].Matthiessen MW, Pedersen G, Albrektsen T, Adamsen S, Fleckner J, Brynskov J. Scand J Gastroenterol. 2005;40:198–205. doi: 10.1080/00365520410009573. [DOI] [PubMed] [Google Scholar]
- [20].Fukumoto K, Yano Y, Virgona N, Hagiwara H, Sato H, Senba H, Suzuki K, Asano R, Yamada K, Yano T. FEBS Lett. 2005;579:3829–3836. doi: 10.1016/j.febslet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- [21].Ali FY, Egan K, Fitzgerald GA, Desvergne B, Wahli W, Bishop-Bailey D, Warner TD, Mitchell JA. Am J Respir Cell Mol Biol. 2005;34:242–246. doi: 10.1165/rcmb.2005-0289OC. [DOI] [PubMed] [Google Scholar]
- [22].Planavila A, Rodriguez-Calvo R, Jove M, Michalik L, Wahli W, Laguna JC, Vazquez-Carrera M. Cardiovasc Res. 2005;65:832–841. doi: 10.1016/j.cardiores.2004.11.011. [DOI] [PubMed] [Google Scholar]
- [23].Martinasso G, Maggiora M, Trombetta A, Canuto RA, Muzio G. J Toxicol Environ Health A. 2006;69:353–365. doi: 10.1080/15287390500227522. [DOI] [PubMed] [Google Scholar]
- [24].Westergaard M, Henningsen J, Svendsen ML, Johansen C, Jensen UB, Schroder HD, Kratchmarova I, Berge RK, Iversen L, Bolund L, Kragballe K, Kristiansen K. J Invest Dermatol. 2001;116:702–712. doi: 10.1046/j.1523-1747.2001.01329.x. [DOI] [PubMed] [Google Scholar]
- [25].Kim DJ, Bility MT, Billin AN, Willson TM, Gonzalez FJ, Peters JM. Cell Death Differ. 2006;13:53–60. doi: 10.1038/sj.cdd.4401713. [DOI] [PubMed] [Google Scholar]
- [26].Schmuth M, Haqq CM, Cairns WJ, Holder JC, Dorsam S, Chang S, Lau P, Fowler AJ, Chuang G, Moser AH, Brown BE, Mao-Qiang M, Uchida Y, Schoonjans K, Auwerx J, Chambon P, Willson TM, Elias PM, Feingold KR. J Invest Dermatol. 2004;122:971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- [27].Tan NS, Michalik L, Noy N, Yasmin R, Pacot C, Heim M, Fluhmann B, Desvergne B, Wahli W. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aung CS, Faddy HM, Lister EJ, Monteith GR, Roberts-Thomson SJ. Biochem Biophys Res Commun. 2006;340:656–660. doi: 10.1016/j.bbrc.2005.12.061. [DOI] [PubMed] [Google Scholar]
- [29].Jakobsen MA, Petersen RK, Kristiansen K, Lange M, Lillevang ST. Scand J Immunol. 2006;63:330–337. doi: 10.1111/j.1365-3083.2006.01745.x. [DOI] [PubMed] [Google Scholar]
- [30].Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. Proc Natl Acad Sci U S A. 2003;100:6712–6717. doi: 10.1073/pnas.1031789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ding G, Cheng L, Qin Q, Frontin S, Yang Q. J Mol Cell Cardiol. 2006;40:821–828. doi: 10.1016/j.yjmcc.2006.03.422. [DOI] [PubMed] [Google Scholar]
- [32].Rival Y, Beneteau N, Taillandier T, Pezet M, Dupont-Passelaigue E, Patoiseau JF, Junquero D, Colpaert FC, Delhon A. Eur J Pharmacol. 2002;435:143–151. doi: 10.1016/s0014-2999(01)01589-8. [DOI] [PubMed] [Google Scholar]
- [33].Woo CH, Massett MP, Shishido T, Itoh S, Ding B, McClain C, Che W, Vulapalli SR, Yan C, Abe JI. J Biol Chem. 2006;281:32164–32174. doi: 10.1074/jbc.M602369200. [DOI] [PubMed] [Google Scholar]
- [34].Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- [35].Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kim DJ, Murray IA, Burns AM, Gonzalez FJ, Perdew GH, Peters JM. J Biol Chem. 2005;280:9519–9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- [37].Marin HE, Peraza MA, Billin AN, Willson TM, Ward JM, Kennett MJ, Gonzalez FJ, Peters JM. Cancer Res. 2006;66:4394–4401. doi: 10.1158/0008-5472.CAN-05-4277. [DOI] [PubMed] [Google Scholar]
- [38].Chawla A, Lee CH, Barak Y, He W, Rosenfeld J, Liao D, Han J, Kang H, Evans RM. Proc Natl Acad Sci U S A. 2003;100:1268–1273. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Barrett EG, Johnston C, Oberdorster G, Finkelstein JN. Am J Physiol. 1998;275:L1110–1119. doi: 10.1152/ajplung.1998.275.6.L1110. [DOI] [PubMed] [Google Scholar]
- [40].Peraza MA, Burdick AD, Marin HE, Gonzalez FJ, Peters JM. Toxicol Sci. 2006;90:269–295. doi: 10.1093/toxsci/kfj062. [DOI] [PubMed] [Google Scholar]
- [41].Paramio JM, Casanova ML, Segrelles C, Mittnacht S, Lane EB, Jorcano JL. Mol Cell Biol. 1999;19:3086–3094. doi: 10.1128/mcb.19.4.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]