Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts (original) (raw)
Abstract
Human diploid fibroblasts (HDFs) can be grown in culture for a finite number of population doublings before they cease proliferation and enter a growth-arrest state termed replicative senescence. The retinoblastoma gene product, Rb, expressed in these cells is hypophosphorylated. To determine a possible mechanism by which senescent human fibroblasts maintain a hypophosphorylated Rb, we examined the expression levels and interaction of the Rb kinases, CDK4 and CDK6, and the cyclin-dependent kinase inhibitors p21 and p16 in senescent HDFs. Cellular p21 protein expression increased dramatically during the final two to three passages when the majority of cells lost their growth potential and neared senescence but p21 levels declined in senescent HDFs. During this period, p16 mRNA and cellular protein levels gradually rose with the protein levels in senescent HDFs reaching nearly 40-fold higher than early passage cells. In senescent HDFs, p16 was shown to be complexed to both CDK4 and CDK6. Immunodepletion analysis of p21 and p16 from the senescent cell extracts revealed that p16 is the major CDK inhibitor for both CDK4 and CDK6 kinases. Immunoprecipitation of CDK4 and CDK6 and their associated proteins from radiolabeled extracts from senescent HDFs showed no other CDK inhibitors. Based upon these results, we propose that senescence is a multistep process requiring the expression of both p21 and p16. p16 up-regulation is a key event in the terminal stages of growth arrest in senescence, which may explain why p16 but not p21 is commonly mutated in immortal cells and human tumors.
Growth and cell division of human diploid fibroblasts (HDFs) in culture eventually generates a metabolically active but nondividing population of senescent cells. During replicative senescence, as described by Hayflick over three decades ago (1), human embryonic fibroblasts will undergo a total of 60–80 cumulative population doublings. Two tumor suppressor genes, the retinoblastoma gene product (Rb) and p53, have been implicated in the induction of the senescent state. Inactivation of p53 and Rb function by infection with simian virus 40 (SV40), expression of human papilloma viral proteins, E6 and E7, (2) or down-regulation of protein expression with anti-sense oligomers extends the life span of HDFs (3). Rb is regulated by phosphorylation, and in senescent cells it is found in its growth-suppressing hypophosphorylated state even in the presence of growth factors (4). Rb inactivation leads to expression of E2F-dependent genes such as thymidine kinase, DNA polymerase-α, cdc2, and cyclin A (5), which are not expressed in senescent cells (6), indicating that the failure to phosphorylate Rb is important in the growth arrest of senescent cells.
Three cyclin-dependent kinases, CDK2, CDK4, and CDK6, are involved in the phosphorylation of the Rb protein (reviewed in ref. 5). In senescent fibroblasts, CDK2 is catalytically inactive and the protein down-regulated (7). CDK4 is also reported to be down-regulated in senescent cells (8), while the status of CDK6 has not been previously addressed. The activating cyclins for these CDKs, cyclins D1 and E, are present in senescent cells at similar or elevated levels relative to early passage cells (8). A role of the CDK inhibitors in senescence was revealed by the isolation of a cDNA of a highly expressed message in senescent cells that encoded the CDK inhibitor, p21 (9).
In mammalian cells, two distinct families of CDK inhibitors have been characterized, represented by two prototype CDK inhibitors, p21 and p16. The p21 family currently includes two related proteins, p27Kip1 and p57Kip2, and the p16 family currently includes four related proteins: p16INK4a (also variously known as MTS1, CDK4I and CDKN2), p15INK4b (also known as MTS2), p18INK4c, and p19INK4d (reviewed in ref. 10). p16 was the first member of the INK4 family characterized and was isolated based upon its interaction with and inhibition of CDK4 (11). Subsequently, p16 was identified as the MTS1 gene representing the melanoma susceptibility locus (12). Homozygous deletion of p16 gene expression in mice produces normal offspring but shows an increased incidence of lymphomas and sarcomas (13) unlike similarly p21 expression-deleted mice, which show no increased risk for tumor formation (14) although mice similarly deleted for p27Kip1 expression have multiorgan hyperplasias (15, 16, 17).
Concurrent work has recently shown an increase in p16 protein and mRNA in senescent human fibroblasts (18, 19) but it was not determined if this up-regulation resulted in significant CDK binding. In the present study, high cellular expression of p16 protein was found in multiple strains of senescent HDFs. Further, in a detailed analysis of the senescent process in MRC-5 fibroblasts, elevated p16 expression followed an increase in p21 expression. p16 was found in both CDK4 and CDK6 complexes and was bound to the majority of the CDK4 and nearly all of the CDK6 expressed in senescent MRC-5 fibroblasts. Immunoprecipitation of CDK4 and CDK6 and their associated proteins from radiolabeled extracts from senescent MRC-5 fibroblasts showed no other CDK inhibitors present, suggesting p21 and p16 are the molecules responsible for CDK4/6 inhibition. Finally, these results suggest that the growth arrest in senescence fibroblasts results from a multistep process with an increase in p21 expression as cells enter senescence followed by a sustained elevation in the level of p16 mRNA and protein.
MATERIALS AND METHODS
Cells and Culture Conditions.
HDFs used in this study were a strain of HDF from newborn foreskin, NHF1, and three strains from embryonic lung tissue, MRC-5, IMR-90, and WI-38, obtained from M. Cordiero-Stone (University of North Carolina, Chapel Hill), American Type Culture Collection, Olivia Pereira-Smith (Baylor College of Medicine, Houston) and American Type Culture Collection, respectively. GM639, an SV40 immortalized human fibroblast line, was also obtained from O. Pereira-Smith. Cells were serially passaged twice weekly at early passage and at decreasing dilution with increasing age. Medium was changed weekly for senescent cultures. Proliferative potential of the culture was determined as the labeling index (LI) measured by the percentage of cells incorporating BrdUrd following a 48-hr incubation in complete medium containing 10 μm BrdUrd and was detected by immunocytochemistry using anti-BrdUrd antibody (Becton Dickinson catalog no. 347580). Cultures with a LI of <5% were considered senescent. Early passage cells were growth inhibited by three means: (i) growth to high density (HD) with continued cultivation for at least 3 additional days, (ii) incubation in medium containing 0.2% fetal bovine serum for 48 hr (low serum, LS), or (iii) exposure to 4 Gy of gamma-irradiation from a cesium source (Irrad).
Extract Preparation and Protein Analysis.
Total cellular protein extracts were prepared by rinsing cultures with cold PBS, then scraping the cells directly into either 1× Laemmli sample buffer or a small volume of cold PBS with 1 mM sodium vanadate, and a protease cocktail consisting of 170 μg/ml phenylmethylsulfanyl fluoride, 25 μg/ml leupeptin, 25 μg/ml aprotinin, 150 μg/ml benzamidine, and 10 μg/ml trypsin inhibitor. For cells scraped in PBS, concentrated SDS was added (final concentration of 1%) and the proteins were rapidly denatured by boiling. Viscosity was reduced by sonication. For extracts in PBS/SDS, protein concentrations were determined using the DC protein assay (Bio-Rad) and diluted with sample buffer. The total number of cells per ml in the final sample was determined by trypsinizing a companion plate and counting on a Coulter counter. Except where noted, all experiments represent analysis based on an equal number of cells.
Western blots were prepared by separation of proteins using the Laemmli discontinuous gel system, and transferred overnight to supported nitrocellulose (Schleicher & Schuell). Proteins were visualized by standard Enhanced Chemiluminescence protocols (Pierce). Except where noted, antibodies were generated as polyclonal rabbit sera against peptides or bacterially produced glutathione _S_-transferase (GST) fusion products and are as follows: human GST–p16, human GST–p21, rat mitogen-activated protein (MAP) kinase (the generous gift of John Blenis, Harvard University, Boston), human CDK4, and human CDK6. The following commercial antibodies were used from Santa Cruz Biotechnology (Santa Cruz, CA); cyclin D1 (Prad1), and CDK6 (C21). Films were scanned on a Silverscanner DTP scanner (La Cie Unlimited, Beaverton, OR).
Immunoprecipitation and Immunodepletion of Native CDK Complexes.
Native CDK complexes were prepared as detailed (20). Supernatants were incubated overnight with anti-CDK4 or anti-CDK6 antibodies that had been preincubated with PBS or blocking peptide (1 μg/μl sera) for 1 hr. Antibody-bound protein A pellets were washed four times at room temperature and resuspended in 1× Laemmli sample buffer for analysis. For radiolabeled extracts, cells were incubated for 4–6 hr with 50 mM Hepes (pH 7.4) buffered Selectamine labeling medium (GIBCO/BRL) without added cysteine or methionine but supplemented with 10% dialyzed fetal bovine serum, 2 mM glutamine, and 300 μCi (1 Ci = 37 GBq) per milliliter of [35S]cysteine/methionine (Express labeling mix, NEN).
For immunodepletion of complexes, nondenatured lysates as described (20) were prepared from senescent MRC-5 cells, and an aliquot was removed for analysis as the initial extract then centrifuged. The protein pellet was resuspended in the same volume of 1× Laemmli sample buffer. The clarified lysate was divided and incubated for several hours with 30 μl of protein A-agarose beads prebound with an equal volume of either anti-p16 or anti-p21 sera, and centrifuged. The supernatant was immunodepleted a total of three times. The adsorbed protein A-agarose pellets were washed once with lysis buffer and resuspended in 1× Laemmli buffer. Protein concentrations for the initial and final p16 and p21 immunodepleted lysates were determined and a constant amount of protein was analyzed by Western blot for p16, p21, CDK4, CDK6, and MAP kinase protein levels.
RNA Expression Analysis.
Total RNA was isolated from cells using TriReagent (Molecular Research Center, Cincinnati). RNA (20 μg) was separated on a 1.1% formaldehyde agarose gel. The gel was transferred onto Nytran (Schleicher & Schuell) as per standard protocols. RNA was crosslinked to the Nytran using a Stratalinker (Stratagene). Probes were generated using isolated coding sequence DNA from human glyceraldehyde phosphate dehydrogenase, p16α specific fragment (5′ end to _Rca_I), and p21 (kindly provided by James Smith, Baylor College of Medicine, Houston). Blots were quantitated using a PhosphorImager (Molecular Dynamics).
RESULTS
p16 and p21 Expression in Senescence HDFs.
Based upon previous reports of increased expression of the CDK inhibitor p21 in senescent cells (9), the expression of additional inhibitors was examined in early passage and senescent fibroblasts of several strains of HDFs, NHF1, MRC-5, IMR-90, and WI-38. Total cellular protein increased 3- to 5-fold in senescent cells, which increase significantly in size (21). Therefore, we compared young vs. senescent cells on the amount of a given protein per cell (termed cellular level) based on the fact that subcellular localization by immunofluorescent staining showed p21 and p16 proteins primarily within the nucleus (22, 23), which would therefore effectively concentrate their action.
While no increase was seen for p27Kip1(data not shown), an increase in p21 and p16 protein expression was seen in senescent cultures relative to logarithmically growing early passage fibroblasts (Fig. 1 A and B). The p16 protein was at nearly undetectable levels in log phase cells but increased dramatically upon senescence (Fig. 1A). Senescent MRC-5 cells express nearly 40-fold higher cellular p16 protein levels per cell than early passage cells, which translates into an 10-fold increase in protein expression based upon a per-microgram protein analysis (see Fig. 4A). The precise increase is difficult as the basal levels are low. The magnitude of p16 expression was variable from culture to culture of senescent cells, but senescent cells that had been maintained for extended periods of time expressed the highest levels of p16. Further, the relative expression of p21 vs. p16 varied between the strains analyzed with less dramatic but significant increases in p16 levels were seen in senescent IMR-90 and WI-38.
Figure 1.
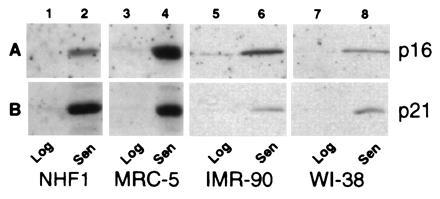
p16 and p21 protein expression in proliferating and senescent HDFs. Total extracts from the same number of cells from NHF1 (lanes 1 and 2), MRC-5 (lanes 3 and 4), IMR-90 (lanes 5 and 6), and WI-38 (lanes 7 and 8) strains of HDFs were analyzed for expression of p16 (A) and p21 (B) under conditions of log phase growth (lanes 1, 3, 5, and 7) and senescence (lanes 2, 4, 6, and 8).
Figure 4.
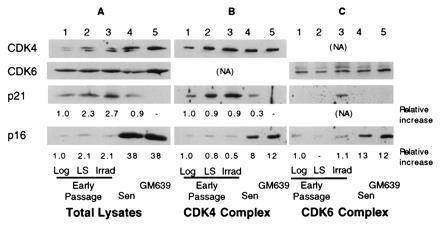
Relative levels of CDK inhibitors, p21 and p16 in total extracts and in CDK complexes in human fibroblasts. Extracts from early passage MRC-5 fibroblasts logarithmically growing (lane 1), arrested by growth in LS (lane 2), or 20 hr after gamma irradiation (Irrad) (lane 3), and senescent MRC-5 cells (Sen) (lanes 4) or a SV40-transformed immortal human fibroblast line, GM639 (lane 5) were either analyzed directly (Total lysates) (A) or immunoprecipitated under nondenaturing conditions with anti-CDK4 (B) or anti-CDK6 (C) antibodies. Total and immunoprecipitated lysates were analyzed for the levels of CDK4, CDK6, p21, and p16 present. The approximate relative levels in total extracts were determined directly by densitometry. For immunoprecipitated extracts, relative levels were normalized for the amount of the appropriate CDK immunoprecipitated.
A more detailed examination of p21 and p16 mRNA and protein expression under different conditions of growth inhibition and at increasing passage in culture was performed on MRC-5 fibroblasts (Fig.2). For p21, cellular protein levels increased in HD cultures and following incubation with reduced or LS. Upon serial propagation, cellular p21 levels reached a maximum during the period in which the majority of the cells lost their growth potential (LI ≈20%) as shown by the decrease in the LI and decreasing cumulative population doublings (Remaining CPD). The p21 level then decreased as the cells reached a LI of <5%, although its cellular level remained several-fold above the levels found in log phase cells (Figs. 1, 2, and5B). p16 protein increased modestly upon growth to HD and upon passage to near senescence, but reached maximal levels when p21 levels had declined and the cells showed no remaining growth potential (Fig. 2). p16 protein levels in senescent cells reached similar cellular levels as those expressed in an SV40-immortalized human fibroblast line, GM639 cells (Fig. 4A, lanes 4 and 5). No significant changes in the cellular levels of either CDK4 or CDK6 were observed in MRC-5 fibroblasts of any age or growth-arrest state (Fig. 2). A several-fold decrease in cellular CDK2 was observed in senescent and serum-starved fibroblasts (data not shown).
Figure 2.
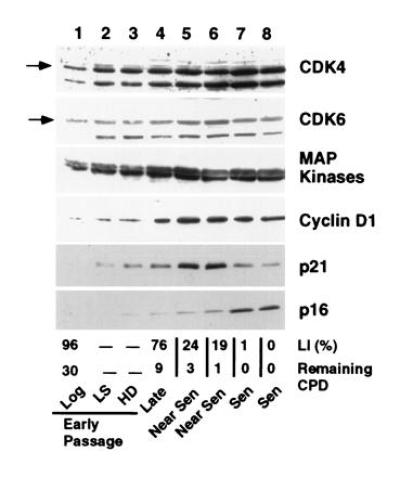
Growth and age related expression of cell cycle related proteins in MRC-5 fibroblasts. Extracts representing the same number of cells from early passage MRC-5 fibroblasts in log phase (lane 1), arrested by growth in reduced serum (LS) (lane 2) or to HD (lane 3) and from MRC-5 fibroblasts at increasing age in culture (lanes 4–8) were analyzed for expression of CDK4 and CDK6, MAP kinase, cyclin D1, the cyclin-dependent inhibitors, p21 and p16. The percentage of the population actively undergoing DNA synthesis during the 48-hr period before the extracts were made is shown as the LI. The degree of senescence is shown as the number of cumulative population doublings remaining before loss of proliferative potential (Remaining CPD) and is denoted as late, near senescent (Near Sen) and senescent (Sen) populations. Lanes 1–8 represent 25, 42, 43, 53, 95, 105, 115, and 100 μg of protein, respectively.
Figure 5.
Relative amounts of CDK4 and CDK6 complexed with p16 and p21 in senescent MRC-5. Extracts of senescent MRC-5 fibroblasts were immunodepleted for either p16 or p21 by three serial precipitations with p16 and p21 antisera. The protein levels of CDK4, CDK6 and MAP kinase (A) or p16 and p21 (B) were examined. (A) Lanes 1 and 2 represent the same number of early passage and senescent cells while lanes 3–6 represent 150 μg of the untreated (lane 3), p16-depleted (lane 4), and p21-depleted extracts (lane 5). (B) Protein (150 μg) was analyzed for total extracts of early passage (lane 1), senescent cells (lane 2), and untreated (lane 4), p16-depleted (lane 5) and p21-depleted (lane 6) extracts. The extracted cell pellet was resuspended in a similar volume of Laemmli sample buffer and a fraction representing the same fraction as loaded for the untreated extract representing an undefined amount of protein (lane 3) was analyzed.
p16 and p21 in CDK4 and CDK6 Complexes of Senescent MRC-5.
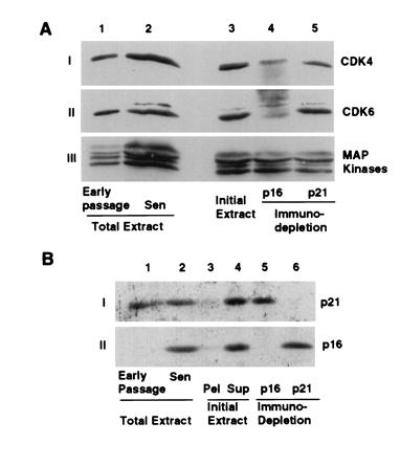
CDK inhibitors regulate CDK activity by forming physical complexes with CDK or CDK-cyclin complexes. To determine which specific CDK inhibitor(s) were involved in senescent cells, we examined the CDK4 and CDK6 complexes. The proteins associated with CDK4 and CDK6 were determined in MRC-5 HDF extracts from growing and growth-arrested early passage cultures and from senescent cultures. When CDK4 was immunoprecipitated from extracts of radiolabeled senescent cells, proteins of 36, 33, 21, and 16 kDa were specifically coprecipitated (Fig.3A, lane 7) representing cyclin D1, CDK4, and the CDK inhibitors p16 and p21, respectively (Fig.4A, data not shown for cyclin D1). The specificity of the immunoprecipitations was confirmed by parallel precipitations with sera blocked by competing peptides. While moderate levels of p21 protein were seen in the CDK4 complexes of growth factor deprived and gamma-irradiated cells, p16 was identified as a significantly labeled protein in growth-factor deprived early passage cells, in senescent MRC-5 cells and in GM639 cells (Fig.3A). In CDK6 complexes, proteins of ≈90, 40, 33, and 16 kDa specifically coprecipitate with CDK6 (Fig. 3B, data not shown). The identity of the p40 and p16 bands are CDK6 and the CDK inhibitor p16, respectively, while the identities of the other proteins are unknown. Anti-CDK6 immunoprecipitations from radiolabeled lysates revealed no p21 protein present under any conditions analyzed although Western analysis of the immune complex of the unlabeled extracts prepared at the same time showed p21 in CDK6 complexes from irradiated cells (Fig. 4C). Because proliferating-cell nuclear antigen is down-regulated during senescence (ref. 6 and data not shown), its absence in either CDK4 or CDK6 complexes is expected for senescent MRC-5, but we have no explanation for its absence in the early passage extracts. No other antibody specific proteins representing CDK4/6 inhibitors of either the INK4 or the p21 family were seen in either the CDK4 or CDK6 radiolabeled complexes, suggesting they play little or no role in CDK4 or CDK6 inhibition in senescent human fibroblasts.
Figure 3.
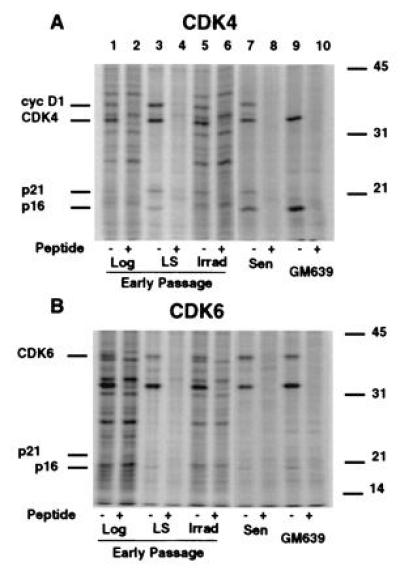
Radiolabeled proteins in immunoprecipitated native CDK4 and CDK6 complexes from MRC-5 extracts under different growth states. Extracts from radiolabeled early passage MRC-5 fibroblasts logarithmically growing (lanes 1 and 2), arrested by growth in LS (lanes 3 and 4), 20 hr after gamma irradiation (Irrad) (lanes 5 and 6), and senescent MRC-5 cells (Sen) (lanes 7 and 8) or a SV40-transformed immortal human fibroblast line, GM639 (lanes 9 and 10) were immunoprecipitated under nondenaturing conditions with anti-CDK4 (A) or anti-CDK6 (B). Specificity of the precipitations was shown by competing the antibody with either a buffered saline solution (−) or peptides used to generate the antibodies in PBS (+). Extracts from the same number of cells were immunoprecipitated. Lanes 7–10 for both A and_B_ were exposed three times longer to compensate for differences in radiolabel incorporation between extracts.
Comparison of relative levels of inhibitors in the complexes by Western blot analysis of unlabeled CDK complexes for p16, p21, and the respective CDK revealed no significant differences in p21 levels associated with CDK4 between log, growth factor-deprived, and in cells 20 hr after irradiation when normalized to the amount of CDK4 precipitated, whereas CDK4 complexes from MRC-5 senescent contained 75% less p21 as compared with complexes from logarithmically growing cells (Fig. 4). Interestingly, significant levels of p21 accumulated in the CDK6 complex from irradiated cells at a time in which no change in levels of p21 was found in the CDK4 complexes from log phase cells, reduced serum-treated and irradiated MRC-5 fibroblasts.
Senescent CDK4 and CDK6 complexes contain 8- and 13-fold, respectively, higher amounts of p16 protein as compared with logarithmically growing MRC-5s (Fig. 4). These levels of complex-bound p16 were similar to the amounts found in GM639 cells. No significant change of p16 levels in complexes from either growth factor deprived or irradiated cells was observed, although a modest increase in cellular p16 and p16/CDK4 complex was found upon growth to HD (data not shown).
Amount of CDK4 and CDK6 Complexed with p16 and p21.
The relative contribution of p16 and p21 in inhibiting CDK4 and CDK6 in senescence was determined by quantitation of the amounts of CDK4 and CDK6 remaining following immunodepletion of p21 and p16 from nondenatured extracts. p16 depletion removed the majority of CDK4 and nearly all of the CDK6, while p21 depletion removed some of the CDK4 and little of the CDK6 (Fig. 5 A and_B_, lanes 4 and 5). When normalized to the amount of MAP kinase present in the immunodepleted extracts, ≈70% of the CDK4 and 85% of the CDK6, were associated with p16, while complexes containing p21 represented the remaining percentages. Removal of either p16 or p21 did not decrease the amount of the other inhibitor from the final extract (Fig. 5B, lanes 4–6). Further, all of the p21, p16, CDK4, and CDK6 was solubilized by the extraction procedure (Fig.5B, lanes 3 and 4, data not shown for CDKs).
Expression of p16 and p21 mRNAs.
Northern blot analysis of total RNA prepared from MRC-5 cells of increasing age revealed that p16 and p21 mRNA levels generally correlated with the magnitude of their protein expression (Fig. 6). As two promoters generate two p16 mRNAs (24), we used an exon 1α specific p16 probe to detect the p16 protein specific message (p16α mRNA). This probe hybridized primarily to a 1.1-kb mRNA and in young cultures, p16α mRNA levels increased ≈4-fold in HD cultures but not under growth factor limiting conditions (Fig. 6A, lanes 2 and 3). Further, expression increased with increasing age, reaching 17- and 12-fold higher levels in two senescent cultures with an average of a 12-fold increase in four independent samples (Fig.6A, lanes 6 and 7, data not shown). Hybridization with the full-length cDNA for human p21 revealed a 2.1-kb mRNA that showed 2- to 3-fold increases under limiting growth factor and HD conditions as well as with increasing age but upon senescence decreased significantly to an average of only a 1.2-fold increase in the four independent samples, which reflects the changes seen in p21 protein expression (Figs. 2 and 6B, data not shown).
Figure 6.
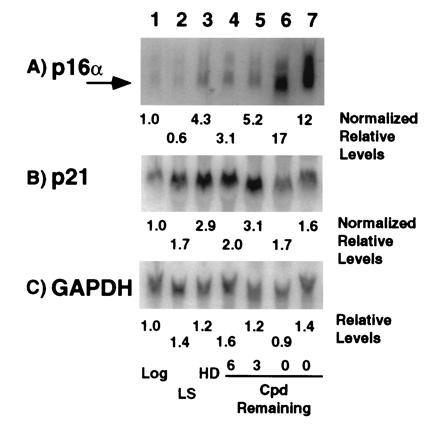
Expression of p16, p21 and glyceraldehyde phosphate dehydrogenase mRNAs in MRC-5 cells. RNA from cells in log phase (lane 1, log), growth arrested by incubation in reduced serum (lane 2, LS), growth arrested by growth to HD (lane 3) or from cells approaching and in senescence (lanes 4 to 7, CPD Remaining) were probed for levels of p16α (A), p21 (B), and glyceraldehyde phosphate dehydrogenase (C) mRNAs. The LIs for the log and increasing age (lanes 1, 4–7) were 72, 49, 39, 33, and 5%, respectively. Increases are expressed relative log phase cell expression based upon normalization to glyceraldehyde phosphate dehydrogenase expression.
DISCUSSION
Isolation and characterization of the CDK inhibitor p21 as a gene with increased expression in senescent human fibroblasts (9), and its characterization as a protein that interacted with and inhibited the CDKs (25, 26) initially suggested a mechanism for why Rb was hypophosphorylated in senescent fibroblasts. Further, the finding that p21 transcription was activated by p53 (27), suggested a mechanism through which SV40 Tag inactivation of p53, and therefore suppression of p21 protein levels, could extend the life span of HDFs in culture (2). In this study we confirm that p21 expression is increased in old MRC-5 fibroblasts, leading to a dramatic decrease in the proliferative potential of the population. However, upon senescence, p21 protein levels decreased and expression of the INK4 family CDK inhibitor, p16, dramatically increased, suggesting that p16 becomes the primary inhibitor of the Rb kinases, CDK4 and CDK6. While Hara et al. (18), have shown an increase in p16 mRNA and protein upon senescence, no direct interaction between the inhibitor and the CDKs or its relative contribution to CDK inhibition was shown. We report here that the majority of the CDK4 and the CDK6 kinases are complexed with p16 in senescent MRC-5 HDFs. p16 accumulation after the cells have exited the cycle suggests that additional cellular changes are ongoing during increasing time in senescence and that cells may exist in one of multiple stages of senescence. These results suggest an alternate model for SV40 Tag-mediated extension of HDF lifespan in which Tag inactivation of Rb disrupts the p16-CDK4/6-Rb growth inhibitory pathway.
The mechanism for the senescence-induced p16 increases is not known. In immortalized cells, inactivation of Rb by deletion or viral inactivation (18, 28) or by hyperphosphorylation as found at the G1/S boundary (29, 30) are reported to increase p16 although the cell cycle increases are disputed (18, 24). Inactivation through hyperphosphorylation cannot explain the p16 increases, as Rb is hypophosphorylated in senescent HDFs and expressed at similar levels relative to early passage cells (4). One possibility is that with the inactivation of the Rb kinases, CDK2/4/6, Rb is dephosphorylated and may no longer interact with transcription factors that may require partial Rb (hypo)phosphorylation for binding. Dephosphorylated Rb may allow the activation of p16 transcription. Alternatively, in senescent cells, p16 increases may result from Rb inactivation through a yet undefined pathway involving the activity of additional proteins interacting with Rb, as suggested by the formation of unique senescent-specific Rb/E2F and p107/E2F complexes containing p21, cyclins, and CDK2 (31). Further, suppression of p16 expression may require a specific Rb-E2F complex such as E2F1 or require Rb in conjunction with another transcriptional corepressor that may be down-regulated upon extended growth suppression. E2F1 expression has been shown to be down-regulated (31) or not expressed in senescent cells (32). An additional possibility is that the p16 promoter may contain additional regions that are Rb-independent and are activated in senescent cells possibly through a p21-initiated mechanism. Finally, increased stability of p16 mRNA in senescence has been reported (18) and may explain the increased p16 mRNA levels, although this mechanism would have to be selective for p16.
p16 may function as a tumor suppressor through its role as a senescence gene. The p16 gene corresponds to the melanoma cancer gene, MTS1, on chromosome 9 (12, 33) and p16 mutations have been found in multiple tumor types (34), unlike the p21 gene where no mutations have been found in screens of a wide range of tumor types (35, 36). Ablation of p16 expression by homozygous deletion in mice produces viable offspring but leads to dramatically increased spontaneous tumor formation and enhanced susceptibility to carcinogens. The cultured embryonic fibroblasts of these mice lack the crisis event before immortalization and are transformed by the expression of a single oncogene (13). However, deletion of the p16 gene also prevents the expression of an alternatively spliced mRNA containing exons 2 and 3 in common with p16. This mRNA produces an unrelated, 19-kDa protein that also suppresses growth (37). In humans, a similar mRNA may generate a 13 kDa protein (24) whose protein expression and activity have yet to be determined. Deletion of p21 in mice also produces viable normal offspring without any apparent phenotype except cells from these mice have a reduced growth-arrest response following gamma-irradiation (14). These results suggest that both p16 and p21 are not developmental growth regulators but checkpoint proteins, with p16 playing a critical role in cellular immortalization and tumor prevention.
In conclusion, we have shown that replicative cellular senescence of HDFs is a multistep process involving the sequential expression of p21 and p16 resulting in p16 becoming the major CDK4/CDK6 inhibitor in senescent MRC-5 fibroblasts. Current models explain a possible mechanism for the initiation of p21 expression, while the mechanism of p21 down-regulation and increased p16 expression have not been delineated. Current work is focusing upon defining these pathways and toward defining the relative roles p21 and p16 may play in the growth arrest found in senescent fibroblasts.
Acknowledgments
We thank Ms. L. Annab for her excellent technical assistance and Dr. David Franklin for advice on the immunodepletion experiments; Ms. R. Montague and Drs. G. Preston, J. Preston, B. Harvat, P. Vojta, C. Afshari, and R. Paules for their critical reading of this manuscript; and Drs. O. Pereira–Smith, J. Smith, and J. Blenis for providing valuable reagents.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “_advertisement_” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: HDF, human diploid fibroblast; CDK, cyclin-dependent kinase; LI, labeling index; SV40, simian virus 40; Rb, retinoblastoma gene product; LS, low serum; HD, high density; MAP, mitogen-activated protein.
References
- 1.Hayflick L. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 2.Shay J W, Pereira-Smith O M, Wright W E. Exp Cell Res. 1991;196:33–39. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- 3.Hara E, Tsurui H, Shinozaki A, Nakada S, Oda K. Biochem Biophys Res Commun. 1991;179:528–534. doi: 10.1016/0006-291x(91)91403-y. [DOI] [PubMed] [Google Scholar]
- 4.Stein G H, Beeson M, Gordon L. Science. 1990;249:666–669. doi: 10.1126/science.2166342. [DOI] [PubMed] [Google Scholar]
- 5.Weinberg R A. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 6.Cristofalo V J, Pignolo R J, Cianciarulo F L, DiPaolo B R, Rotenberg M O. Exp Gerontol. 1992;27:429–432. doi: 10.1016/0531-5565(92)90077-d. [DOI] [PubMed] [Google Scholar]
- 7.Dulic V, Drullinger L F, Lees E, Reed S I, Stein G H. Proc Natl Acad Sci USA. 1993;90:11034–11038. doi: 10.1073/pnas.90.23.11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lucibello F C, Sewing A, Brusselbach S, Burger C, Muller R. J Cell Sci. 1993;105:123–133. doi: 10.1242/jcs.105.1.123. [DOI] [PubMed] [Google Scholar]
- 9.Noda A, Ning Y, Venable S F, Pereira-Smith O M, Smith J R. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 10.Sherr C J, Roberts J M. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 11.Serrano M, Hannon G J, Beach D. Nature (London) 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 12.Kamb A, Gruis N A, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian S V, Stockert E, Day R R, Johnson B E, Skolnick M H. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 13.Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, DePinho R A. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 14.Brugarolas J, Chandrasekaran C, Gordon J I, Beach D, Jacks T, Hannon G J. Nature (London) 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 15.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh D Y, Nakayama K-I. Cell. 1996;86:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 16.Fero M L, Rivkin M, Tasch T, Porter P, Carow C E, Firpo E, Polyak K, Tsai L-H, Broudy V, Perlmutter R M, Kaushansky K, Roberts J M. Cell. 1996;86:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 17.Kiyokawa H, Kineman R D, Manova-Todorova K O, Soares V C, Hoffman E S, Ono M, Khanam D, Hayday A C, Frohman L A, Koff A. Cell. 1996;86:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 18.Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong H, Riabowol K. Exp Gerontol. 1996;31:311–325. doi: 10.1016/0531-5565(95)00025-9. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins C W, Xiong Y. In: Cell Cycle: Material and Methods. Pagano M, editor. New York: Springer; 1995. pp. 250–263. [Google Scholar]
- 21.Cristofalo V J, Pignolo R J. Physiol Rev. 1993;73:617–638. doi: 10.1152/physrev.1993.73.3.617. [DOI] [PubMed] [Google Scholar]
- 22.Bond J A, Blaydes J P, Rowson J, Haughton M F, Smith J R, Wynford Thomas D, Wyllie F S. Cancer Res. 1995;55:2404–2409. [PubMed] [Google Scholar]
- 23.Lukas J, Parry D, Aagaard L, Mann D J, Bartkova J, Strauss M, Peters G, Bartek J. Nature (London) 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- 24.Stone S, Jiang P, Dayananth P, Tavtigian S V, Katcher H, Parry D, Peters G, Kamb A. Cancer Res. 1995;55:2988–2994. [PubMed] [Google Scholar]
- 25.Xiong Y, Hannon G J, Zhang H, Casso D, Kobayashi R, Beach D. Nature (London) 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 26.Harper J W, Adami G R, Wei N, Keyomarsi K, Elledge S J. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 27.el-Deiry W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer W E, Kinzler K W, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Nichols M A, Shay J W, Xiong Y. Cancer Res. 1994;54:6078–6082. [PubMed] [Google Scholar]
- 29.Tam S W, Shay J W, Pagano M. Cancer Res. 1994;54:5816–5820. [PubMed] [Google Scholar]
- 30.Soucek T, Pusch O, Hengstschlager-Ottnad E, Wawra E, Bernaschek G, Hengstschlager M. FEBS Lett. 1995;373:164–169. doi: 10.1016/0014-5793(95)01033-b. [DOI] [PubMed] [Google Scholar]
- 31.Afshari C A, Nichols M A, Xiong Y, Mudryj M. Cell Growth Differ. 1996;7:979–988. [PubMed] [Google Scholar]
- 32.Dimri G P, Hara E, Campisi J. J Biol Chem. 1994;269:16180–16186. [PubMed] [Google Scholar]
- 33.Nobori T, Miura K, Wu D J, Lois A, Takabayashi K, Carson D A. Nature (London) 1994;368:753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 34.Hirama T, Koeffler H P. Blood. 1995;86:841–854. [PubMed] [Google Scholar]
- 35.Marchetti A, Buttitta F, Pellegrini S, Bertacca G, Lori A, Bevilacqua G. Int J Oncol. 1995;6:187–189. doi: 10.3892/ijo.6.1.187. [DOI] [PubMed] [Google Scholar]
- 36.Terry L, Boyd J, Alcorta D, Lyon T, Solomon G, Hannon G, Berchuk A, Beach D, Barrett J C. Mol Carcinogen. 1996;16:221–228. doi: 10.1002/(SICI)1098-2744(199608)16:4<221::AID-MC6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 37.Quelle D E, Zindy F, Ashmun R A, Sherr C J. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]